 图 图式1
环化/Stille串联反应
Figure 图式1.
Tandem cyclization/Stille coupling
图 图式1
环化/Stille串联反应
Figure 图式1.
Tandem cyclization/Stille coupling
引用本文:
丁刚, 王泽宇, 殷中琼, 乐贵洲. 钯催化的串联反应构建苯并五元杂环的研究进展[J]. 有机化学,
2015, 36(1): 43-59.
doi:
10.6023/cjoc201508012

Citation: Ding Gang, Wang Zeyu, Yin Zhongqiong, Yue Guizhou. Progress in Palladium-Catalyzed Tandem Reaction of Constructing Benzo-Five-Membered Heterocycles[J]. Chinese Journal of Organic Chemistry, 2015, 36(1): 43-59. doi: 10.6023/cjoc201508012
Citation: Ding Gang, Wang Zeyu, Yin Zhongqiong, Yue Guizhou. Progress in Palladium-Catalyzed Tandem Reaction of Constructing Benzo-Five-Membered Heterocycles[J]. Chinese Journal of Organic Chemistry, 2015, 36(1): 43-59. doi: 10.6023/cjoc201508012
钯催化的串联反应构建苯并五元杂环的研究进展
English
Progress in Palladium-Catalyzed Tandem Reaction of Constructing Benzo-Five-Membered Heterocycles
Abstract:
The benzo-five-membered heterocycles existed extensively in many important natural products and also showed excellent bioactivites. Therefore, organic chemists around the world made efforts to develop the highly efficient methods of constructing these heterocycles. Recently, the syntheses of them via transition metal-catalyzed tandem reaction, especially palladium-catalyzed reaction, have been reported widely. This review emphasizes on the palladium-catalyzed tandem reaction of the formation of benzo-five-membered heterocycles and their derivatives reported since 2000.
-
Key words:
- heterocycle
- / tandem reaction
- / palladium catalyst
- / synthesis
-
具有苯并五元杂环骨架结构的天然产物大多具有重要的生理活性[1, 2], 从而被广泛应用于医药、农药等领域.因此, 发展高效、快速的构建苯并五元环骨架的方法势在必行, 这也是当前有机化学研究热点之一.
近年来, 过渡金属催化形成碳碳键及碳杂键的偶联反应得到蓬勃发展[3~9].其中有机卤代物或拟卤代物(I, Br, Cl, OTf, NTf等)在钯等过渡金属催化下可以与烯烃(Heck)、炔烃(Sonogashira)、有机硼(Suzuki)、有机锡(Stille)、有机硅(Hiyama)、有机锌(Negishi)及格氏试剂(Kumada)等反应来有效地构筑碳碳键, 同样形成碳氮键的Buchwald-Hartwig偶联反应也受到广泛的关注[10~13].近年来, 羰基α位的芳基化[14]及芳基的C—H活化[15, 16]也得到快速发展.关于构建苯并五元杂环骨架的方法已有广泛研究, 近年来有关于这方面的文献有较多报道, 所用催化剂有Cu[17], Ti[18], Ag[19], Ru[20], Pd[21]等.而钯催化串联合成苯并五元杂环化合物的研究只有少量综述涉及[22~25].相比其它过渡金属, 钯具有与众多官能团的兼容性, 优异的立体、区域选择性以及高效的催化活性等优点.因此, 本文选择从钯参与的串联反应构建苯并五元杂环的角度出发, 对最近15年的相关文献按反应类型加以分类, 并给予总结及评论.
1 钯催化的环化/偶联反应
1.1 环化/Stille偶联
钯催化炔烃与三丁基锡氢在室温下便可制备各种有机锡试剂, 并且它能够在空气中存放, 因此有机锡试剂参与的Stille偶联在合成中被广泛应用.
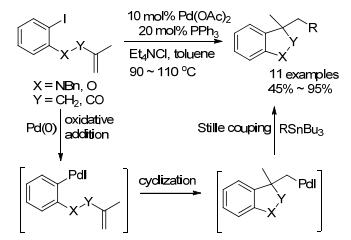
2000年, Grigg小组[26]以邻碘烯基苯和有机锡试剂作为反应底物, 通过钯参与的环化/Stille串联反应得到3-取代二氢吲哚及苯并二氢呋喃衍生物(Scheme 1).反应的可能机理是:原位生成的Pd(0)对C—I键进行氧化加成, 接着对双键进攻并环化形成钯复合物, 最后该复合物再与有机锡试剂完成转金属化, 还原消除形成最终产物.同年, 该小组[27]将含有核苷、糖基、嘌呤等基团的有机锡试剂, 进行相同反应也得到15个类似产物.在反应中添加季铵盐可以提高产率, 主要是由于季铵阳离子对环化过程起加速作用, 同时其阴离子可稳定钯中间体[28].
图 图式1
环化/Stille串联反应
Figure 图式1.
Tandem cyclization/Stille coupling
接着, 该小组[29]尝试将接有Bu3Sn基团的二烯碘代物进行这类反应.反应经两次环化后, 也成功地得到一系列含苯并五元杂环的螺环(Eq. 1)及桥环衍生物(Eq. 2).相比Et4NCl, 添加Ag2CO3更有利于反应.这可能是因为银盐能抑制双键的异构化.随后, 该小组[30]以Wang树脂作为载体实现了这类反应的固相合成(SPOS).相比传统液相合成, 固相合成操作简便、产物易分离提纯、产率较高、环境污染较少, 且容易实现制动化控制等.
2006年, 他们[31]又研究了钯催化氨基甲酰氯与炔基的分子内反应来构建二氢吲哚衍结构(Eq. 3).反应经历5-exo-dig环化/Stille偶联得到α, β-酰胺化合物.
2007年, Lamaty小组[32]报道了缺电子烯烃的串联反应(Eq. 4).他们发现添加1, 3-二甲基-2-咪唑啉酮(DMI)能有效地抑制有机锡试剂的自身偶联.
1.2 环化/Suzuki偶联
尽管有机基锡试剂应用广, 但造价高、毒性大, 逐渐被低毒性的试剂所取代, 其中有机硼试剂就是一个不错的选择.它毒性小、易制备、生成易溶于水的副产物, 大多数对水、空气和热都很稳定.目前已有许多商品化的有机硼试剂, 因此相比有机锡它具有更加广阔的应用前景.
2001年, Grigg小组[33]报道了多组分的钯催化串联反应(Scheme 2).他们以2-碘噻吩、丙二烯、邻碘苯胺为原料, 在四三苯基膦钯的催化下, 2-碘噻吩首先被氧化加成, 接着形成的钯中间体与联烯反应得到烯丙基钯物种, 它再与邻碘苯胺发生胺化反应形成叔胺, 最后添加有机硼试剂后, 发生环化/Suzuki偶联得到最终产物.
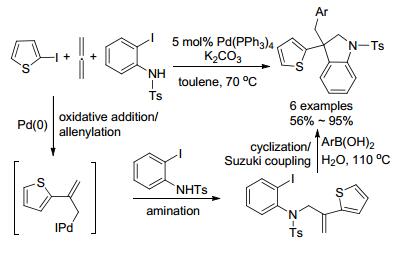 图 图式2
环化/Suzuki串联反应
Figure 图式2.
Tandem cyclization/Suzuki coupling
图 图式2
环化/Suzuki串联反应
Figure 图式2.
Tandem cyclization/Suzuki coupling
2003年, Lamaty小组[34]研究了另一类多组分体系.邻碘苯酚对2-溴烯丙酸酯的亲核取代, 得到缺电子烯烃, 该中间体经历环化/Suzuki偶联, 产率仅有57%. 2007年, 该小组[32]在原有基础上进行了更深入的研究, 将中间产物分离出来进行反应(Eq. 5), 总产率比之前有所增加.值得注意的是, 加入PPh3配体反而会降低反应速率.
2005年, Takemoto小组[35]研究了芳基碘与叁键环化/Suzuki偶联的反应(Eq. 6).底物以5-exo-dig方式环化得烯基钯复合物后, 再与有机硼试剂进行Suzuki偶联形成单一的E式构型产物.研究发现, 添加CsF有助于Suzuki偶联, 芳基硼酸中可以带有吸电子、供电子及大位阻的基团, 此外该反应还具有底物兼容性好等优点.
2014年, Sridharan小组[36]使用Cu/Pd双金属体系催化N-炔基酰胺合成N-三唑二氢吲哚酮(Scheme 3).首先, Cu(I)催化叁键与叠氮化物的Click反应, 生成三唑类化合物; 随后, 钯催化三唑化合物发生分子内环化/ Suzuki偶联得终产物.
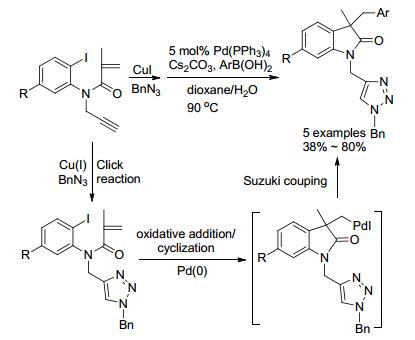 图 图式3
铜/钯双金属催化的串联反应
Figure 图式3.
Cu/Pd bimetal-catalyzed tandem reaction
图 图式3
铜/钯双金属催化的串联反应
Figure 图式3.
Cu/Pd bimetal-catalyzed tandem reaction
1.3 环化/Heck反应
Takemoto小组[35]报道了炔酰胺与烯烃的环化/Heck反应(Scheme 4).与之前报道的环化/Suzuki偶联反应类似(Eq. 6), 经历相同的环化步骤, 接着与烯烃发生Heck反应生成α, δ-不饱和酰胺.
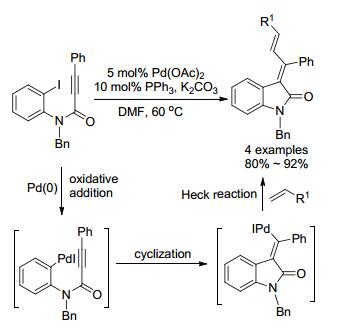 图 图式4
环化/Heck串联反应
Figure 图式4.
Tandem cyclization/Heck coupling
图 图式4
环化/Heck串联反应
Figure 图式4.
Tandem cyclization/Heck coupling
2007年, Liu等[37]报道了有水存在、不需要配体的还原Heck反应(Eq. 7).水在这个反应中是必要的, 但过多的水(>5%)则会抑制环化反应, 从而产生更多副产物.正是由于不怕水的特点, 该反应为进行放大试验提供了可行性.
2009年, Kim小组[38]报道的还原Heck反应, 产率为47%~80% (Eq. 8). 2015年, Day等[39]报道的这类反应不仅可以在苯环上引入卤素, 而且氮原子上不需要取代基.此外含氮的芳杂环也可发生反应.
1.4 环化/胺化反应
2002年, Grigg小组[40]研究了环化/联烯化/胺化的多组分反应(Scheme 5和Eq. 9).反应以丙二烯、取代碘苯、胺等为原料, 在Pd(OAc)2的催化作用下, 取代碘苯经环化后接着对丙二烯的插入得到烯丙基钯, 最后它通过胺化反应形成C—N键而得产物.
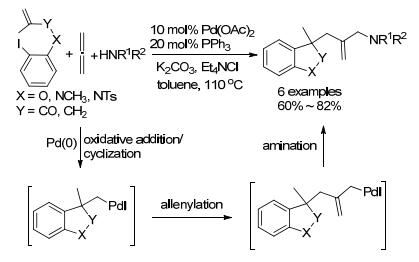 图 图式5
环化/胺化串联反应
Figure 图式5.
Tandem cyclization/amination
图 图式5
环化/胺化串联反应
Figure 图式5.
Tandem cyclization/amination
2010年, 祝介平小组[41]对钯催化串联合成吲哚螺环衍生物的反应进行了报道, 反应包括环化及分子内的胺化反应(Eqs. 10和11).研究发现, 配体的选择至关重要, t-BuMephos配体不仅可以促进螺环的形成, 而且使反应具有好的选择性, 得到单一的非对映体异构体.该方法为合成螺环结构的二氢吲哚类天然产物提供了一种很好的途径.
1.5 环化/羰基化反应
2010年, Somfai-Ludlow等[42]报道了钯催化环化/羰基甲氧基化的反应(Scheme 6).作者发现, 当氮原子上没有取代基时, 反应不发生, 这是因为芳基碘优先发生羰基甲氧基化形成酯, 抑制了环化过程; 当含有大体积取代基时, 由于空间位阻的作用抑制了上述副反应, 从而促进环化反应的顺利进行.值得一提的是, 尽管先前已有类似的反应, 但是通过控制CO压力、溶剂等因素而选择性地合成具有非对映选择性的例子鲜有报道.
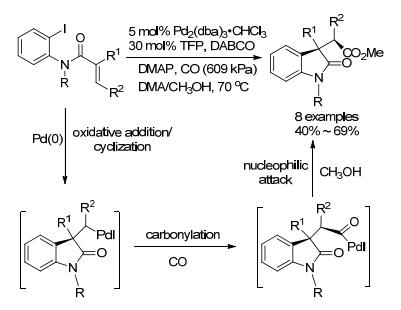 图 图式6
环化/羰基化串联反应
Figure 图式6.
Tandem cyclization/carbonylation
图 图式6
环化/羰基化串联反应
Figure 图式6.
Tandem cyclization/carbonylation
Sridharan小组[36]使用Cu/Pd双金属催化的环化/ Suzuki偶联取得很好的效果(Scheme 3), 随后也进行了环化/羰基胺化反应研究, 得到中等收率的酰胺类衍生物(Scheme 7).
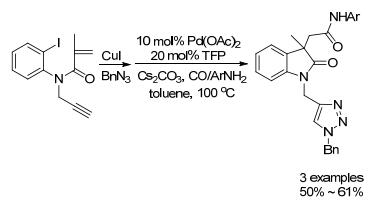 图 图式7
铜/钯双金属催化的串联反应
Figure 图式7.
Cu/Pd bimetal-catalyzed reaction
图 图式7
铜/钯双金属催化的串联反应
Figure 图式7.
Cu/Pd bimetal-catalyzed reaction
2015年, 顾振华小组[43]报道了以氯仿作为羰基前体合成羧酸的反应(Eq. 12).氯仿在强碱作用下脱去HCl, 形成二氯卡宾, 该中间体然后再与环化后形成的烷基钯复合物还原消除, 最后氯代物经水解得到羧酸衍生物, 最高产率可达98%.用CHCl3代替CO进行羰基化反应更加安全, 且易操作.
1.6 环化/C—H活化
2009年, Fagnou小组[44]首次报道了噻吩α位的C—H活化研究(Scheme 8).溴代物环化后形成的钯复合物可直接活化噻吩α位的C—H键, 通过还原消除构建C(sp3)—C(sp2)键.反应体系中生成的特戊酸盐会促进噻吩α位C(sp2)—H的活化, 从而可提高反应的产率.
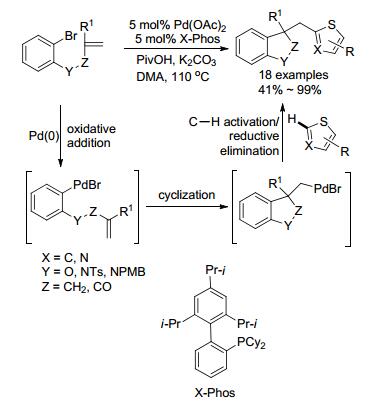 图 图式8
环化/C—H活化串联反应
Figure 图式8.
Tandem cyclization/C-H activation
图 图式8
环化/C—H活化串联反应
Figure 图式8.
Tandem cyclization/C-H activation
2008年, Kim小组[45]报道了钯催化分子内环化/C—H活化构建3/5并环的方法(Eq. 13).底物首先环化得到烷基钯物种, 由于缺乏β-H, 该物种不能进行消除, 而是直接活化氧α位C(sp3)—H键, 最后形成具环丙烷骨架的苯并二氢呋喃衍生物.
2008年, Ruck等[46]报道了涉及活化芳基C—H键合成螺环衍生物的研究(Eq. 14).钯催化溴代烯酰胺发生分子内的环化, 紧接着对另一个苯环C—H键活化, 同时环化完成螺环的构建.
2012年, 祝介平小组[47]通过底物的设计实现了钯催化另一种串联反应合成螺环化合物的方法(Eq. 15).与祝介平小组(Eq. 14)不同之处在于活化位点, Ruck小组活化的是C(sp2)—H, 而祝介平小组活化的是C(sp3)—H.同年, 祝介平小组[48]在此基础上又报道类似反应(Eq. 16).但反应启动顺序相反, 并且对卤代物及拟卤代物有一定限制, 当X=Cl或OTf时, 反应不能发生.
接着, 祝介平小组[49]运用类似方法又报道了四并环化合物的合成, 该方法涉及钯迁移(Scheme 9).底物环化形成的烷基钯经过1, 4-迁移得到芳基钯物种, 最后再通过活化苯亚甲基上C(sp3)—H键还原消除而成四环化合物. 2015年, 该小组[50]再对该类反应进行了研究, 将底物中苯环的α位碳去掉, 直接实现了芳基C—H键的活化(Eq. 17).这一系列反应都具有很好的底物适应性及高收率等优点, 但酰胺中氮原子上需要有取代基来抑制副反应的发生.
 图 图式9
环化/钯迁移/C-H活化串联反应
Figure 图式9.
Tandem cyclization/Pd-migration/C—H activation
图 图式9
环化/钯迁移/C-H活化串联反应
Figure 图式9.
Tandem cyclization/Pd-migration/C—H activation
2015年, 李瑞祥小组[51]首次报道纳米级钯催化分子间串联反应, 得到苯并二氢呋喃二聚体的研究(Scheme 10).反应可能机理是钯催化溴代物环化形成烷基钯, 该物种可被还原; 接着钯物种直接活化还原产物的C(sp3)—H, 最后还原消除得到二聚体.正如之前提到, 季铵盐四正丁基溴化铵(TBAB)提高了反应的产率.反应不仅适用于自身的偶联, 还可以适用于不同分子间的偶联.这是首例发现钯活化分子间C(sp3)—H形成二聚的反应, 为今后的研究提供了重要参考价值.
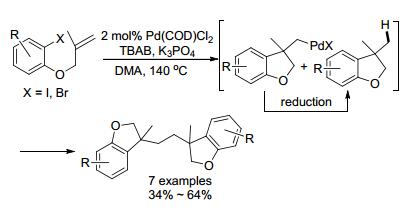 图 图式10
串联的环化/二聚化反应
Figure 图式10.
Tandem cyclization/dimerization
图 图式10
串联的环化/二聚化反应
Figure 图式10.
Tandem cyclization/dimerization
1.7 环化/其他偶联
2007年, 祝介平小组[52]报道了钯作用下碘代烯酰胺的串联反应(Scheme 11).首先钯插入到底物C-I中, 紧接着环化产生五元钯复合物, 然后氰基置换钯物种的碘原子, 最后发生还原消除得到氰基吲哚.产物经过几步简单的转化便可得到能够抑制乙酰胆碱酶的天然产物esermethole和physostigmine.另外, 他们也研究了该反应的不对称版本, 当加入手性配体(S)-Difluorphos, 对映选择性最高为72% ee. 2011年, Kim小组[53]以Pd(OAc)2/TBAC/Na2CO3作为最佳反应条件, 将该方法扩展到了氰基苯并二氢呋喃的合成.
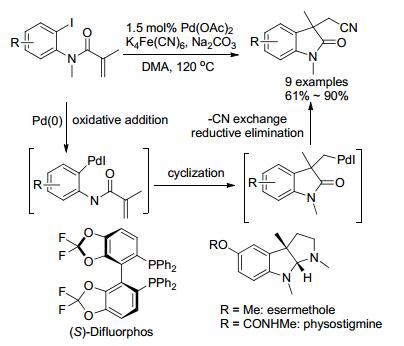 图 图式11
环化/氰基化串联反应
Figure 图式11.
Tandem cyclization/cyanation
图 图式11
环化/氰基化串联反应
Figure 图式11.
Tandem cyclization/cyanation
2009年, Takemoto小组[54]报道氨基甲酰卤与分子内共轭二烯的分子内反应(Eq. 18).酰卤环化生成烯丙基钯, 随后它被亲核试剂进攻生成末端取代的产物.除了外加的亲核试剂以外, 还可以被自身脱除的Cl-进攻. 2011年, 作者[55]在原有基础上发展了构建C—Si键的反应(Scheme 12).收率整体有所提高, 而且该产物可通过Sakurai反应, 得到含有多个手性中心的环状化合物.
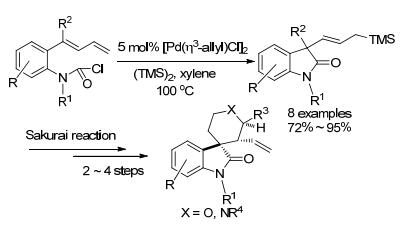 图 图式12
`环化/形成碳硅键的串联反应
Figure 图式12.
Tandem cyclization/formation of C—Si bond
图 图式12
`环化/形成碳硅键的串联反应
Figure 图式12.
Tandem cyclization/formation of C—Si bond
2011年, Lautens小组[56]首次研究了钯催化下, 芳基碘环化/还原消除得到碘代苯并五元杂环的反应(Eq. 19).研究表明, 大位阻Q-phos配体起着决定性作用, 它能够加速烷基钯复合物还原消除形成C—I键.该方法避免了传统方法中对卤素的浪费, 符合绿色化学的原则, 且产率均能达到92%以上.但是该方法仅限于芳基碘化合物, 芳基溴、氯不能进行类似反应.
2013年, 顾振华小组[57]研究了钯催化下芳基卤化物与对甲苯磺酰腙的串联反应(Eq. 20).芳基卤代物在钯作用下首先环化得到烷基钯, 它能够与对甲苯磺酰腙形成的卡宾顺利进行偶联形成烯基吲哚.延长酰胺烷基链的长度可以得到更复杂的螺环化合物, 该方法为合成复杂的吲哚烯烃提供了一种新的方法.
1.8 C—H活化环化/其他偶联
2009年, 李金恒小组[58]报道了C—H活化/烯基钯还原的方法(Scheme 13).首先Pd(OAc)2对炔酰胺的苯环进行C—H活化, 接着再与炔配位、加成得到烯基钯, 随后该物种被氢还原形成产物.作者也提出了另一种反应机理则, 先对炔的氢钯化反应, 随后活化C—H及还原消除得到五元环.该反应受热力学控制, 化合物的构型可以通过调控温度而得到.当温度为80 ℃时, (Z)-产物为主; 温度为140 ℃时, (E)-产物为主.
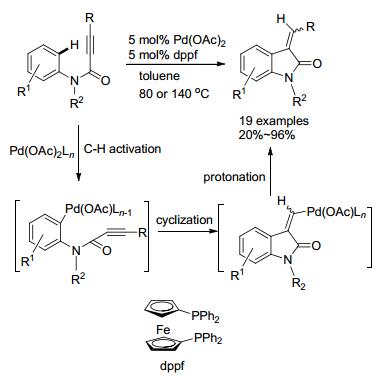 图 图式13
C—H活化/环化/质子化串联反应
Figure 图式13.
Tandem C—H activation/cyclization/protonation
图 图式13
C—H活化/环化/质子化串联反应
Figure 图式13.
Tandem C—H activation/cyclization/protonation
2010年, 祝介平小组[59]研究了钯催化不饱和烯酰胺的反应(Eqs. 21和22).首先钯活化烯酰胺邻位的C—H键, 随后环化得到钯复合物, 最后该物种发生乙酰氧基化形成C—O键或发生分子内的胺化反应形成C—N键.当使用不同催化剂及溶剂, 则得到不同的产物. Pd(OAc)2/AcOH体系主要发生乙酰氧基化反应, PdCl2/MeCN体系主要发生胺化反应.
2011年, 刘国生小组[60]报道了腈类化合物的合成.反应依次对芳基及乙腈中的C—H键活化而形成两个C—C键(Eq. 23).添加剂AgF对C—H的活化扮演重要的角色, 在扩展这类反应时, 乙腈的活性比其它腈类高. 2012年, 他们[61]以相同的起始物, 又报道三氟甲基化的反应(Eq. 24).
1.9 异氰酸苯酯的环化/其他偶联
2005年, Yamamoto小组[62]报道了钯催化异氰酸苯酯与末端炔化物的串联反应(Scheme 14).首先炔在钯作用下形成钯炔, 随后钯炔与异氰酸苯酯的叁键配位, 紧接着碳钯化形成烯基钯, 该物种对异氰酸苯酯中的羰基亲核进攻, 最后还原消除得(Z)-产物, 同时可以异构化形成(E)-产物.该反应立体选择性好, 以(E)式为主要构型, 大部分产物的(E)/(Z)比例>99:1.
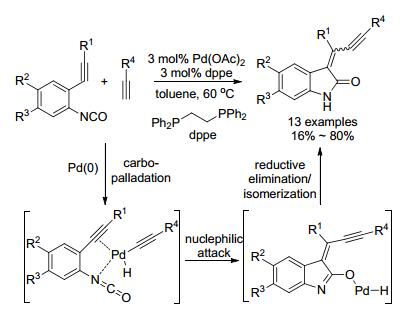 图 图式14
碳钯化/亲核加成串联反应
Figure 图式14.
Tandem carbopalldation/nuclephilic reaction
图 图式14
碳钯化/亲核加成串联反应
Figure 图式14.
Tandem carbopalldation/nuclephilic reaction
2009年, Murakami小组[63]用硫醇(Scheme 15)或醇(Eq. 15)代替末端炔进行了这类反应.首先钯与异氰酸苯酯配位, 接着氧化环化后得到烯基钯复合物, 随后醇置换配体, 最后还原消除和异构化得吲哚的烯炔产物.同样, (E)-产物为主, 多数产物(E)/(Z)>90:1, 反应只需1 mol%催化量便可顺利进行.同年, 他们又成功地将这类反应拓展到了胺类化合物的合成[64].
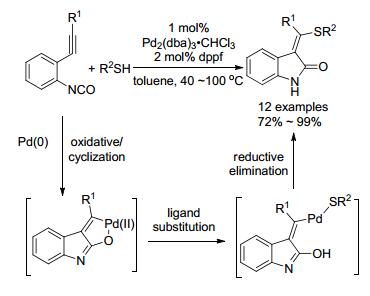 图 图式15
环化/羟基化串联反应
Figure 图式15.
Tandem cyclization/hydroxylation
图 图式15
环化/羟基化串联反应
Figure 图式15.
Tandem cyclization/hydroxylation
2010年, 他们[65]进一步研究, 将普通醇换成苯甲醇(Scheme 16)或烯丙醇(Eq. 26), 使用CpPd(π-allyl)/dppf作催化体系, 没有得到预期的产物, 却生成3, 3-双取代衍生物.这是因为在这类催化体系下, 烯醇式中间体的烯丙基或苯亚甲基从氧迁移到碳上, 经历1, 3-迁移.而且增加烯丙醇的用量时(>10 equiv.), 氮原子会被烯丙基化.
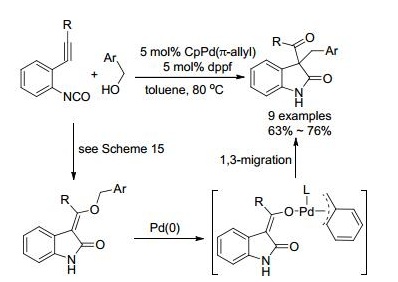 图 图式16
环化/羟基化/迁移串联反应
Figure 图式16.
Tandem cyclization/ hydroxylation/migration
图 图式16
环化/羟基化/迁移串联反应
Figure 图式16.
Tandem cyclization/ hydroxylation/migration
2 钯催化的偶联/环化反应
2004年, Wolfe小组[66]报道了钯催化2-烯丙基苯胺与芳基溴的反应(Schemes 17和18).钯首先催化芳基溴与取代苯胺进行Buchwald-Hartwig反应形成仲胺, 接着催化另一分子芳基溴与烯基胺发生胺钯化形成C—N键, 最后还原消除形成C—C键.研究表明, 双齿配体DPE-phos的电子效应及空间位阻能有效地避免形成叔胺, 同时也能促进C—N键的形成及最后的还原消除, 使用不同的芳基溴可得到多种衍生物.
 图 图式17
Buchwald-Hartwig/胺钯化串联反应
Figure 图式17.
Tandem Buchwald-Hartwig/aminopadalladation
图 图式17
Buchwald-Hartwig/胺钯化串联反应
Figure 图式17.
Tandem Buchwald-Hartwig/aminopadalladation
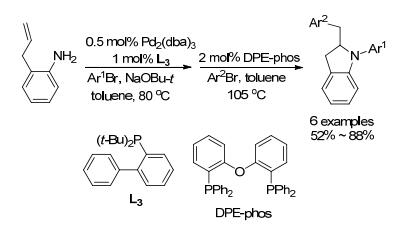 图 图式18
Buchwald-Hartwig/胺钯化串联反应
Figure 图式18.
Tandem Buchwald-Hartwig/aminopadalladation
图 图式18
Buchwald-Hartwig/胺钯化串联反应
Figure 图式18.
Tandem Buchwald-Hartwig/aminopadalladation
2010年, 该小组[67]在相同底物的氮上引入烯丙基进行类似反应, 又合成了三环含氮化合物(Eq. 27).与上面机理不同在于先形成胺钯化中间体, 再形成碳钯化中间体, 最后还原消除得到6/5/5三环化合物.该方法具有高的非对映体选择性, dr最高可达20:1, 芳基上能够容忍三氟甲基、甲氧基、甲基等基团.当氮上的取代基为o-烯丙基苯基时, 芳基钯物种在此条件下会经历1, 3-钯迁移形成6/5/5/6四环化合物(Eq. 28).
2009年, Lautens小组[68]以苯胺和降冰片二烯为底物, Pd(OAc)2/P(t-Bu)3•HBF4/Cs2CO3为催化体系, 执行碳钯化/胺化反应形成多环化合物(Scheme 19).产物即可通过Diles-Alder反应得到更复杂的多环化合物; 又可在低温下经臭氧氧化得到二羟基化合物.
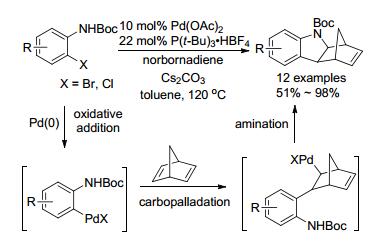 图 图式19
碳钯化/胺化串联反应
Figure 图式19.
Tandem carbopalladation/amination
图 图式19
碳钯化/胺化串联反应
Figure 图式19.
Tandem carbopalladation/amination
2011年, Cacchi小组[69]研究了Heck/Heck环化反应(Scheme 20).首先在Pd2(dba)3催化下, 苯基重氮四氟硼酸盐与烯丙基胺的Heck反应得到中间体, 紧接着加入Pd(OAc)2/P(o-tol)3, 再发生分子内的Heck环化得到(Z)-二氢吲哚产物.
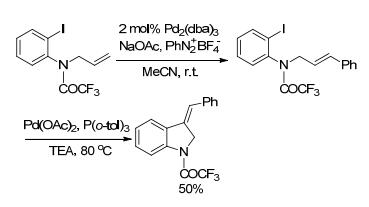 图 图式20
Heck/还原Heck串联反应
Figure 图式20.
Tandem carbopalladation/amination
图 图式20
Heck/还原Heck串联反应
Figure 图式20.
Tandem carbopalladation/amination
2008年, Booker-Miburm小组[70]研究了取代脲与共轭二烯为底物的反应(Scheme 21).脲的芳基邻位C—H被活化, 接着与共轭烯烃碳钯化形成C—C键, 并得到烯丙基钯, 随后钯物种发生分子内胺化反应, 最后得到烯基二氢吲哚产物.芳基中间位有强吸电子基时会抑制C—H键的活化, 从而降低反应的速率及产率.
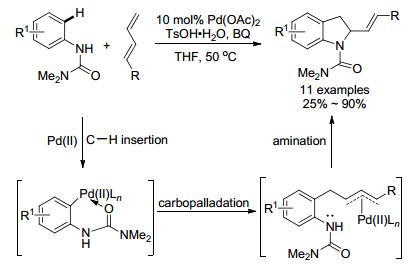 图 图式21
C—H插入/碳钯化/胺化串联反应
Figure 图式21.
Tandem C—H insertion/carbopalladation/amination
图 图式21
C—H插入/碳钯化/胺化串联反应
Figure 图式21.
Tandem C—H insertion/carbopalladation/amination
2015年, Maiti小组[71]报道了取代苯酚与不饱和烯酸的反应(Scheme 22, Eq. 29).邻二氮杂菲(1, 10-phen)与钯的配位增加了钯盐的溶解性与亲电性, 快速活化苯酚邻位C—H键, 接着对不饱和烯酸双键的插入, 随后脱羧、环化得到环状钯物种, 最后β-H消除得产物.值得一提的是, 在体系中加入D2O, 在脱羧得到烯丙基钯后发生H/D交换, 直接形成较高收率的氘代化合物.
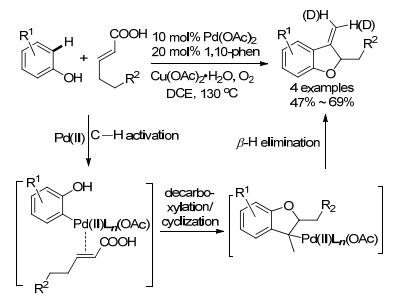 图 图式22
C—H活化/脱羧/环化串联反应
Figure 图式22.
Tandem C—H activation/decaoxylation/cyclization
图 图式22
C—H活化/脱羧/环化串联反应
Figure 图式22.
Tandem C—H activation/decaoxylation/cyclization
2007年, 祝介平小组[72]使用Cu/Pd催化三组分构建二氢吲哚结构(Scheme 23).酰胺与炔基进行Sonogashira偶联得到内炔, 接着另一分子芳基碘被氧化加成, 形成的钯物种对酰胺炔基的碳钯化反应, 最后活化芳基邻位C—H键及还原消除形成最终产物.
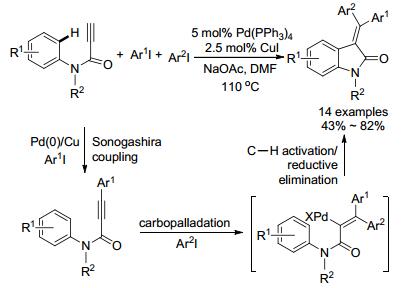 图 图式23
Sonogashira偶联/碳钯化/C—H活化串联反应
Figure 图式23.
Tandem Sonogashira coupling/carbopalladation/ C—H activation
图 图式23
Sonogashira偶联/碳钯化/C—H活化串联反应
Figure 图式23.
Tandem Sonogashira coupling/carbopalladation/ C—H activation
2008年, 李金恒小组也研究了炔酰胺的反应(Scheme 24和Eq. 30), 但该反应涉及Pd(II)与Pd(IV)的氧化还原.反应过程包括胺钯化[73]或乙酰氧基化[74]、C—H活化及还原消除.产物以(E)-构型为主, 但随着苯环上基团位阻的加大, 会降低产率.
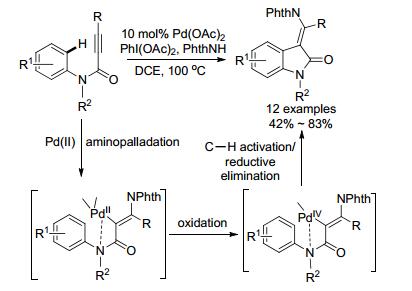 图 图式24
胺钯化/碳钯化/C—H活化串联反应
Figure 图式24.
Tandem aminopalladation/carbopalladation/C—H activation
图 图式24
胺钯化/碳钯化/C—H活化串联反应
Figure 图式24.
Tandem aminopalladation/carbopalladation/C—H activation
2010年, 潘毅小组[75]报道了在空气中进行钯催化合成二氢吲哚过氧化物的反应(Scheme 25).反应可能机理为在钯作用下, 过氧叔丁醇对烯酰胺的双键以自由基方式氧化加成, 得到过氧中间体, 接着依次活化C(sp2)—H, C(sp3)—H而成环, 形成稳定的双过氧基产物.
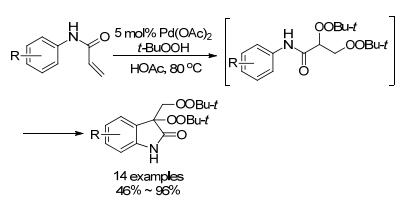 图 图式25
过氧化/C—H活化串联反应
Figure 图式25.
Tandem peroxidation/C—H activation
图 图式25
过氧化/C—H活化串联反应
Figure 图式25.
Tandem peroxidation/C—H activation
2014年, 王细胜小组[76]也对钯催化自由基的反应进行了研究(Scheme 26). PhSO2CF2I与钯作用容易形成二氟甲基自由基, 该自由基对烯酰胺双键的插入形成叔碳自由基, 接着它对苯环进行自由基取代完成反应.
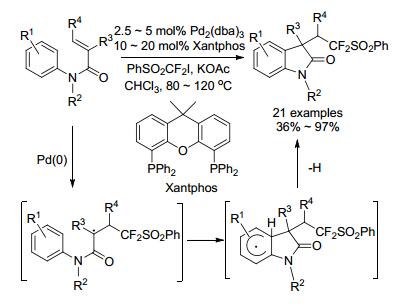 图 图式26
自由基环化串联反应
Figure 图式26.
Tandem radical cyclization
图 图式26
自由基环化串联反应
Figure 图式26.
Tandem radical cyclization
3 钯催化Wacker型反应
Wacker反应是将烯烃变成醛、酮的有效方法之一, 使用CuCl2/PdCl2作催化剂, 空气或氧气作氧化剂, 发现至今一直被广泛应用于有机合成中.
3.1 Wacker/炔基化反应
2010年, Waser小组[77]首次报道了2-烯基苯酚与有机高价碘的串联反应(Scheme 27).钯催化2-烯基苯酚进行Wacker型环化, 接着被高价碘化物氧化形成四价钯中间体, 最后还原消除.与其它钯催化串联反应相比, 该反应实现了C(sp3)—C(sp)键的形成, 合成炔基苯并二氢呋喃.
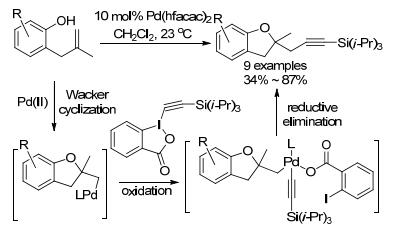 图 图式27
Wacker环化/炔基化串联反应
Figure 图式27.
Tandem Wacker cyclization/alkynylation
图 图式27
Wacker环化/炔基化串联反应
Figure 图式27.
Tandem Wacker cyclization/alkynylation
3.2 aza-Wacker/串联反应
2006年, 杨丹小组[78]利用吡啶作为配体, 进行了aza-Wacker反应研究(Scheme 28).不饱和酰胺化合物依次通过胺钯化反应、对双键的插入及β-H消除一步实现了两个环、一根C—N键及一根C—C键的形成.反应以Pd(II)来启动反应, 氧气作为氧化剂实现了Pd(0)到Pd(II)的循环.
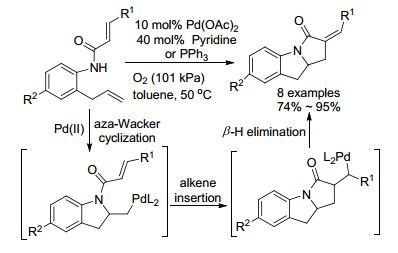 图 图式28
aza-Wacker环化/烯烃插入串联反应
Figure 图式28.
Tandem aza-Wacker cyclization/alkene insertion
图 图式28
aza-Wacker环化/烯烃插入串联反应
Figure 图式28.
Tandem aza-Wacker cyclization/alkene insertion
2009年, 该小组[79]进一步研究, 增加了氮原子上取代基的长度, 使用了简单、易得的喹啉或异喹啉作为配体, 经历上述相似历程得到螺环化合物, 其非对映选择性高(dr>24:1)(Eq. 31), 另外配体喹啉或异喹啉还能够抑制烯烃的异构化.
2006年, 他们[78]也研究了不对称催化版本的反应(Eq. 32), 最后发现使用手性配体(-)-sparteine及分子筛, 会得到较高ee的产物.
然而, 由于sparteine配体制备困难, 限制了其应用. 2009年, 她们[80]又发展了一种能够在空气中稳定存在的手性钯催化体系Pd(OAc)2/t-Bu-QUOX. R1无论接吸电子基还是供电子基都能得到具有更高的对映(ee>80%)及非对映体选择性(dr>24:1)的产物(Eq. 33).
2015年, 该小组[81]再次报道烯基胺或酰胺的串联反应.反应经过aza-Wacker环化/苯亚甲基C(sp3)—H活化(Eq. 34)或酰胺α位C(sp3)—H活化(Eq. 35).虽然特戊酸有利于活化C(sp3)—H, 但是不同的催化体系, 会活化不同位点的C(sp3)—H.作者还观察到苯亚甲基
C(sp3)—H活化是决速步骤, 而酰胺α位C(sp3)—H活化却不是决速步骤.
4 钯催化Catellani型反应
Catellani反应是以芳基碘代物、碘代脂肪烃、取代烯烃为原料, 加入降冰片烯(norbornene)在钯催化作用下, 芳基碘代物经过多次邻位的烷基化反应, 最后再发生Heck反应, 生成多取代的芳烃的反应.该反应牵涉到Pd(0), Pd(II), Pd(IV)的循环过程, 降冰片烯起到了关键的接力作用.
2006年, Lautens小组[82]报道钯催化Catellani型反应合成苯并五元杂环化合物的研究(Scheme 29).首先, Pd(0)对芳基卤的氧化加成, 接着对降冰烯片的碳钯化反应, 形成的钯物种作为亲电试剂活化芳基中C—H键, 再次钯化形成钯环, 它接着对分子内C—Br键的插入形成Pd(IV)物种, 该物种通过还原、去碳钯化得到Pd(II)物种, 最后再与Zn(CN)2发生Negishi偶联得到氰基二氢吲哚.
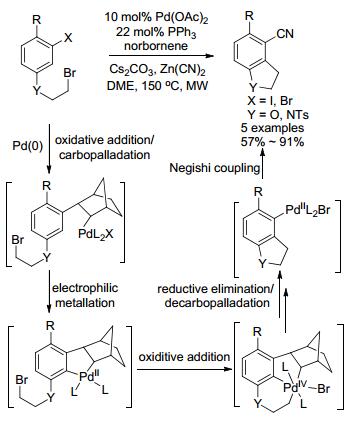 图 图式29
Catellani/Negishi串联反应
Figure 图式29.
Tandem Catellani/Negishi reaction
图 图式29
Catellani/Negishi串联反应
Figure 图式29.
Tandem Catellani/Negishi reaction
2009年, 该小组[83]在已有研究基础上作了进一步探索, 又成功地报道了与活泼烯烃的反应(Eq. 36).在经历相似的历程后, 最后一步发生Heck反应, 得到苯并二氢呋喃及二氢吲哚的烯基化合物.
2014年, 梁永民小组[84]以Pd(OAc)2作催化剂, 以芳基磺酰腙代替Zn(CN)2, 一步实现了4-取代烯基的二氢吲哚的合成(Eq. 37).反应具有产率高、良好的官能团兼容性和底物适用广的特点.
2007年, Lautens小组[85]又以取代碘苯和仲胺为底物, 报道钯催化Catellani型反应, 反应经过连续C—C键和C—N键的形成(Eq. 38).与上面不同在于(Scheme 29), 该反应是由分子间的溴代物进攻形成Pd(IV)物种, 最后发生胺化反应并关环.选择合适的N-取代基可避免直接发生Buchwald-Hartwig偶联形成叔胺.
5 其他反应
2007年, Muñiz[86]报道钯催化烯基二胺分子内合成二氢吲哚二聚体(Scheme 30).首先二价钯催化仲胺以不利的5-endo-trig方式成环, 接着二价钯物种被二醋酸碘苯氧化成烷基四价钯, 最后还原去钯化形成第二根C—N键, 同时构筑第二个吡咯环.该二聚体可进一步用于磺胺类药物的合成.
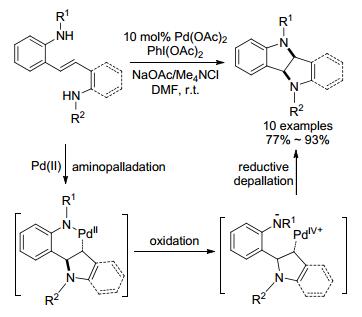 图 图式30
胺钯化/去钯化串联反应
Figure 图式30.
Tandem aminopalladation/depalladation
图 图式30
胺钯化/去钯化串联反应
Figure 图式30.
Tandem aminopalladation/depalladation
离子液体作为一种绿色环保型溶剂, 可有效地稳定过渡金属催化剂, 促进反应的进行, 是常规有机溶剂理想的替代品[87]. 2009年, Radhakrishnan小组[88]使用离子液体1-丁基-3-甲基咪唑六氟磷酸盐(bmim-PF6)作为溶剂, 邻碘苯酚和桥环肼在钯催化下发生碳钯化作用, 钯复合物与氧配位并断裂C—N键打开桥环, 最后通过β-H消除非对映选择性得到三环化合物(Scheme 31).稳定剂TBAC不可缺少, 而当加入三苯基膦配体, 却不能发生氧钯化反应, 而是直接被还原得到取代苯酚.
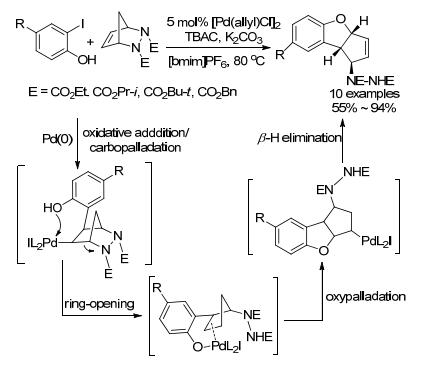 图 图式31
碳钯化/氧钯化串联反应
Figure 图式31.
Tandem carbopalladation/oxypalladation
图 图式31
碳钯化/氧钯化串联反应
Figure 图式31.
Tandem carbopalladation/oxypalladation
2013年, 该小组[89]进行了更深入研究, 当桥环肼的桥头碳换成环丙基时, 在相似的反应条件下合成四环骨架的苯并二氢呋喃衍生物(Scheme 32).与上述反应不同, β-H消除时环丙烷结构被打开, 消除得到共轭二烯中间体, 随后该中间体发生氢钯化反应形成π-烯丙基钯, 最后发生分子内的亲核取代反应.
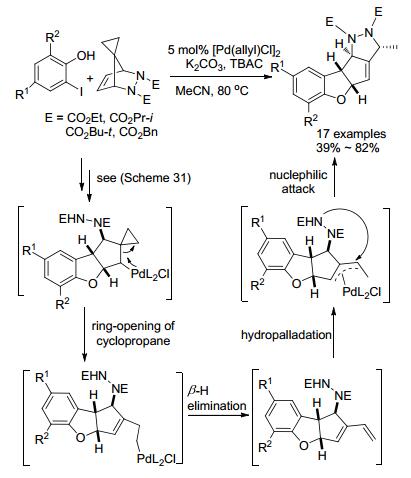 图 图式32
环丙烷开环/氢钯化/亲核取代的串联反应
Figure 图式32.
Tandem ring-opening of cyclopropane/hydro-palladation/nuclephilic subsitution
图 图式32
环丙烷开环/氢钯化/亲核取代的串联反应
Figure 图式32.
Tandem ring-opening of cyclopropane/hydro-palladation/nuclephilic subsitution
同年, 该小组[90]又将桥环肼的桥头碳换成烯基, 以邻碘苯胺为起始物, 路易斯酸和钯共同作催化剂(Scheme 33).首先路易斯酸催化促进苯胺对环肼的C—N键亲核进攻, 同时打开环形成二级胺, 接着Pd(0)对二级胺中C—I的氧化加成, 随后立即对双键的插入形成烯丙基钯中间体, 最后由亲核试剂进攻.这类方法非对映选择性好, 一步能产生多个手性中心的分子.
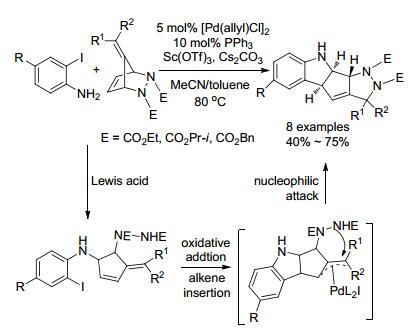 图 图式33
路易斯酸及钯催化的串联反应
Figure 图式33.
Lewis acid and Pd catalyzed reaction
图 图式33
路易斯酸及钯催化的串联反应
Figure 图式33.
Lewis acid and Pd catalyzed reaction
2013年, 史一安小组[91]报道钯催化α-甲基苯乙烯和二叔丁基环状脲得到螺环化合物的反应(Scheme 34). Pd(0)插入到二叔丁基环状脲N—N键中, 形成四元环, 接着它与α-甲基苯乙烯的双键配位, 然后接受烯丙位的氢后立即还原消除; 钯再次插入到另一分子二叔丁基环状脲中N—N键, 然后由第一分子脲上氮原子进攻成环, 该中间体随后活化邻位C—H键并释放一分子脲; 第三分子二叔丁基环状脲的N—N被插入形成四价钯物种, 通过脱去叔丁基异氰酸酯得到含钯的乃春中间体, 它经历两次还原消除得到最终产物.该反应一次可形成两个环和四根C—N键, 具有底物适应广、反应条件温和等优点.
 图 图式34
芳基及烯丙基C—H活化的串联反应
Figure 图式34.
Tandem allylic and aromatic C—H activation
图 图式34
芳基及烯丙基C—H活化的串联反应
Figure 图式34.
Tandem allylic and aromatic C—H activation
随后, 他们[92]以取代碘苯、二叔丁基环状脲和降冰片烯为底物进行了类似研究.在Pd(PPh3)4/Cs2CO3的催化体系下, 碘苯氧化加成后立即发生碳钯化, 接着它活化芳基中C—H键形成五元环钯物种, 最后钯物种插入到二叔丁环状脲的N—N键中, 随后经历相似历程非对映选择性地形成多环化合物(Scheme 35).用降冰片二烯代替降冰片烯, 也适用于该反应, 而且还研究了分子内的反应.
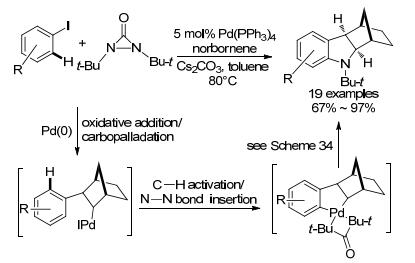 图 图式35
碳钯化/C—H活化/胺化的串联反应
Figure 图式35.
Tandem carbopalladation/C—H activation/amination
图 图式35
碳钯化/C—H活化/胺化的串联反应
Figure 图式35.
Tandem carbopalladation/C—H activation/amination
2014年, Anderson小组[93]报道了合成三环类二氢吲哚的方法(Scheme 36).链状的二炔胺在钯的催化作用下经连续碳钯化反应, 再以β-H消除同时构建5/6/5并环体系.
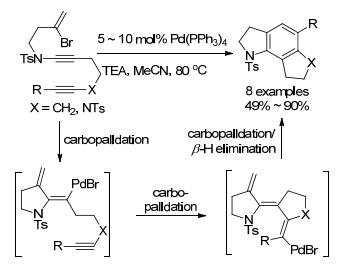 图 图式36
连续的碳钯化反应
Figure 图式36.
Consecutive carbopalladation
图 图式36
连续的碳钯化反应
Figure 图式36.
Consecutive carbopalladation
2015年, Wang等[94]对邻碘苯胺与异腈在钯催化下进行了深入研究(Scheme 37).异腈先后两次插入到邻碘苯胺中, 随后胺化关环, 接着还原消除得到类似于席夫碱的钯环, 最后它经酸水解得到取代靛红.该方法反应试剂简单、易得, 反应条件温和, 且不需要含膦配体.
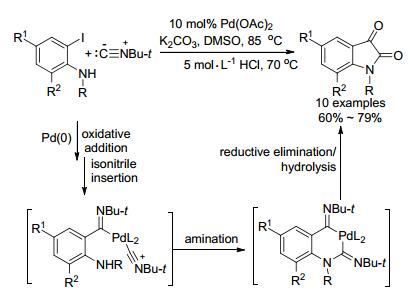 图 图式37
异腈连续插入的串联反应
Figure 图式37.
Consecutive insertion of isonitrile
图 图式37
异腈连续插入的串联反应
Figure 图式37.
Consecutive insertion of isonitrile
5 总结与展望
总结了近15年来钯催化串联反应合成二氢吲哚及苯并二氢呋喃衍生物的方法.其中应用最广泛的是钯催化环化/偶联反应, 而且已经被应用于天然产物的合成[52].另外, 钯催化的偶联/环化、Wacker型和Catellani型等反应类型也成功地用于构造取代苯并五元杂环及其并环.除此之外, 最近发展的新颖方法一步可形成多个环, 为这类化合物的研究提供了新的途径.
尽管构建苯并五元杂环的方法很多, 但反应可靠性及广谱性、底物的适应性还有待改进; 其次, 只有少数例子涉及手性合成.因此, 找到新的、有效的手性配体, 实现不对称合成是当前面临的主要问题; 最后, 由于传统偶联会浪费卤原子, 另一方面溶剂的分离、产物纯化十分繁琐, 这些均不符合“绿色、经济化学”原则.离子液体、聚合物支载、微波促进反应等有利于产物分离提纯和减少环境污染, 为解决上述问题提供了参考.相信随着时间的推移、研究的深入, 这些难题将逐一被解决, 为这类化合物的合成提供更加有效的选择.
-
-
[1]
Luize, P. S.; Ueda-Nakamura, T.; Filho, B. P. D.; Cortez, D. A. G.; Nakamura, C. V. Biol. Pharm. Bull. 2006, 29, 2126. doi: 10.1248/bpb.29.2126
-
[2]
Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748. doi: 10.1002/(ISSN)1521-3773
-
[3]
李浩, 丁昌华, 许斌, 侯雪龙, 化学学报, 2014, 72, 765. doi: 10.6023/A14040329Li, H.; Ding, C.; Xu, B.; Hou, X. Acta Chim. Sinica 2014, 72, 765 (in Chinese). doi: 10.6023/A14040329
-
[4]
刘杰, 朱庆仁, 杜鹃, 张袖丽, 有机化学, 2015, 35, 15. doi: 10.6023/cjoc201408014Liu, J.; Zhu, Q.; Du, J.; Zhang, X. Chin. J. Org. Chem. 2015, 35, 15 (in Chinese). doi: 10.6023/cjoc201408014
-
[5]
倪晨, 沈安, 曹育才, 叶晓峰, 有机化学, 2014, 34, 278. doi: 10.6023/cjoc201308031Ni, C.; Shen, A.; Cao, Y.; Ye, X. Chin. J. Org. Chem. 2014, 34, 278 (in Chinese). doi: 10.6023/cjoc201308031
-
[6]
倪传法, 朱林桂, 胡金波, 化学学报, 2015, 73, 90. doi: 10.6023/A14110758Ni, C.; Zhu, L.; Hu, J. Acta Chim. Sinica 2015, 73, 90 (in Chinese). doi: 10.6023/A14110758
-
[7]
肖玉兰, 潘强, 张新刚, 化学学报, 2015, 73, 383. doi: 10.6023/A15010042Xiao, Y.; Pan, Q.; Zhang, X. Acta Chim. Sinica 2015, 73, 383 (in Chinese). doi: 10.6023/A15010042
-
[8]
张剑, 陆庆全, 刘超, 雷爱文, 有机化学, 2015, 35, 743. doi: 10.6023/cjoc201411028Zhang, J.; Lu, Q.; Liu, C.; Lei, A. Chin. J. Org. Chem. 2015, 35, 743 (in Chinese). doi: 10.6023/cjoc201411028
-
[9]
张艳, 冯柏年, 有机化学, 2014, 34, 2406. doi: 10.6023/cjoc201408030Zhang, Y.; Feng, B. Chin. J. Org. Chem. 2014, 34, 2406 (in Chinese). doi: 10.6023/cjoc201408030
-
[10]
白东虎, 李春举, 李健, 贾学顺, 有机化学, 2012, 32, 994. doi: 10.6023/cjoc1202073Bai, D.; Li, C.; Li, J.; Jia, X. Chin. J. Org. Chem. 2012, 32, 994 (in Chinese). doi: 10.6023/cjoc1202073
-
[11]
方晒, 吕梅香, 龙玉华, 杨定乔, 有机化学, 2011, 31, 1573. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract340478.shtmlFang, S.; Lü, M.; Long, Y.; Yang, D. Chin. J. Org. Chem. 2011, 31, 1573 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract340478.shtml
-
[12]
唐石, 梁云, 林文杰, 李金恒, 有机化学, 2004, 24, 1133.Tang, S.; Liang, Y.; Liu, W.-J.; Li, J.-H. Chin. J. Org. Chem. 2004, 24, 1133 (in Chinese).
-
[13]
王乃兴, 有机化学, 2011, 31, 1319. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract340304.shtmlWang, N. Chin. J. Org. Chem. 2011, 31, 1319 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract340304.shtml
-
[14]
Culkin, D. A.; Hartwig, J. F. Acc. Chem. Res. 2003, 36, 234. doi: 10.1021/ar0201106
-
[15]
Ritleng, V.; Sirlin, C.; Pfeffer, M. Chem. Rev. 2002, 102, 1731. doi: 10.1021/cr0104330
-
[16]
刚芳莉, 徐光利, 董涛生, 杨丽, 杜正银, 有机化学, 2015, 35, 1428. doi: 10.6023/cjoc201502020Gang, F.; Xu, G.; Dong, T.; Yang, L.; Du, Z. Chin. J. Org. Chem. 2015, 35, 1428 (in Chinese). doi: 10.6023/cjoc201502020
-
[17]
Sheng, R.-L.; Tang, S.; Zhou, D.; Li, Z.-H.; Fu, M.-J.; Jie, L.; Li, S.-H. Synthesis 2015, 47, 1567. doi: 10.1055/s-00000084
-
[18]
Gansauer, A.; Hildebrandt, S.; Michelmann, A.; Dahmen, T.; von Laufenberg, D.; Kube, C.; Fianu, G. D.; Flowers, R. A. Angew. Chem., Int. Ed. 2015, 54, 7003. doi: 10.1002/anie.201501955
-
[19]
Wei, X.-H.; Wu, Q.-X.; Yang, S.-D. Synlett 2015, 26, 1417. doi: 10.1055/s-00000083
-
[20]
Fu, W.; Xu, F.; Fu, Y.; Zhu, M.; Yu, J.; Xu, C.; Zou, D. J. Org. Chem. 2013, 78, 12202. doi: 10.1021/jo401894b
-
[21]
Grigg, R.; Sansano, J. M.; Santhakumar, V.; Sridharan, V.; Thangavelanthum, R.; Thornton-Pett, M.; Wilson, D. Tetrahedron 1997, 53, 11803. doi: 10.1016/S0040-4020(97)00754-0
-
[22]
Grigg, R.; Sridharan, V. Tetrahedron 1999, 576, 65.
-
[23]
韦海龙, 博士论文, 兰州大学, 兰州, 2010.Wei, H.-L. Ph.D. Dissertation, Lanzhou University, Lanzhou, 2010 (in Chinese).
-
[24]
沈金海, 程国林, 崔秀灵, 化学进展, 2012, 24, 1324.Shen, J.; Cheng, G.; Cui, X. Prog. Chem. 2012, 24, 1324 (in Chinese).
-
[25]
Li, J.-H.; Song, R.-J.; Liu, Y.; Xie, Y.-X. Synthesis 2015, 47, 1195. doi: 10.1055/s-00000084
-
[26]
Fretwell, P.; Grigg, R.; Sansano, J. M.; Sridharan, V.; Sukirthalingam, S.; Wilsona, D.; Redpathb, J. Tetrahedron 2000, 56, 7525. doi: 10.1016/S0040-4020(00)00659-1
-
[27]
Casaschi, A.; Grigg, R.; Sansano, J. M. Tetrahedron 2000, 56, 7553. doi: 10.1016/S0040-4020(00)00661-X
-
[28]
Jeffery, T. Tetrahedron 1996, 52, 10113. doi: 10.1016/0040-4020(96)00547-9
-
[29]
Casaschi, A.; Grigg, R.; Sansano, J. M.; Wilsona, D.; Redpathb, J. Tetrahedron 2000, 56, 7541. doi: 10.1016/S0040-4020(00)00660-8
-
[30]
Grigg, R.; MacLachlan, W. S.; MacPherson, D. T.; Sridharan, V.; Suganthan, S. Tetrahedron 2001, 57, 10335. doi: 10.1016/S0040-4020(01)01061-4
-
[31]
Anwar, U.; Fielding, M. R.; Grigg, R.; Sridharan, V.; Urch, C. J. J. Org. Chem. 2006, 691, 1476. doi: 10.1016/j.jorganchem.2005.11.045
-
[32]
Szlosek-Pinaud, M.; Diaz, P.; Martinez, J.; Lamaty, F. Tetrahedron 2007, 63, 3340. doi: 10.1016/j.tet.2007.02.035
-
[33]
Grigg, R.; Mariani, E.; Sridharan, V. Tetrahedron Lett. 2001, 42, 8677. doi: 10.1016/S0040-4039(01)01811-1
-
[34]
Szlosek-Pinaud, M.; Diaz, P.; Martinez, J.; Lamaty, F. Tetrahedron Lett. 2003, 44, 8657. doi: 10.1016/j.tetlet.2003.09.169
-
[35]
Yanada, R.; Obika, S.; Inokuma, T.; Yanada, K.; Yamashita, M.; Ohta, S.; Takemoto, Y. J. Org. Chem. 2005, 70, 6972. doi: 10.1021/jo0508604
-
[36]
Packer, G.; Lepre, K.; Kankanala, J.; Sridharan, V. RSC Adv. 2014, 4, 3457. doi: 10.1039/C3RA46906A
-
[37]
Liu, P.; Huang, L.; Lu, Y.; Dilmeghani, M.; Baum, J.; Xiang, T.; Adams, J.; Tasker, A.; Larsen, R.; Faul, M. M. Tetrahedron Lett. 2007, 48, 2307. doi: 10.1016/j.tetlet.2007.01.156
-
[38]
Kim, H. S.; Lee, H. S.; Kim, S. H.; Kim, J. N. Tetrahedron Lett. 2009, 50, 3154. doi: 10.1016/j.tetlet.2008.11.127
-
[39]
Day, J.; Frederickson, M.; Hogg, C.; Johnson, C.; Meek, A.; Northern, J.; Reader, M.; Reid, G. Synlett 2015, 26, 2570. doi: 10.1055/s-00000083
-
[40]
Grigg, R.; Savic, V.; Sridharan, V.; Terrier, C. Tetrahedron 2002, 58, 8613. doi: 10.1016/S0040-4020(02)00763-9
-
[41]
Jaegli, S.; Erb, W.; Retailleau, P.; Vors, J. P.; Neuville, L.; Zhu, J. Chem. Eur. J.2010, 16, 5863. doi: 10.1002/chem.201000312
-
[42]
Seashore-Ludlow, B.; Somfai, P. Org. Lett. 2010, 12, 3732. doi: 10.1021/ol1009703
-
[43]
Liu, X.; Li, B.; Gu, Z. J. Org. Chem.2015, 80, 7547. doi: 10.1021/acs.joc.5b01126
-
[44]
René, O.; Lapointe, D.; Fagnou, K. Org. Lett. 2009, 11, 4560. doi: 10.1021/ol901799p
-
[45]
Kim, H. S.; Gowrisankar, S.; Kim, S. H.; Kim, J. N. Tetrahedron Lett. 2008, 49, 3858. doi: 10.1016/j.tetlet.2008.04.080
-
[46]
Ruck, R. T.; Huffman, M. A.; Kim, M. M.; Shevlin, M.; Kandur, W V.; Davies, I. W. Angew. Chem., Int. Ed. 2008, 47, 4711. doi: 10.1002/(ISSN)1521-3773
-
[47]
Piou, T.; Neuville, L.; Zhu, J. Angew. Chem., Int. Ed. 2012, 51, 11561. doi: 10.1002/anie.201206267
-
[48]
Piou, T.; Neuville, L.; Zhu, J. Org. Lett. 2012, 14, 3760. doi: 10.1021/ol301616w
-
[49]
Piou, T.; Bunescu, A.; Wang, Q.; Neuville, L.; Zhu, J. Angew. Chem., Int. Ed. 2013, 52, 12385. doi: 10.1002/anie.201306532
-
[50]
Bunescu, A.; Piou, T.; Wang, Q.; Zhu, J. Org. Lett.2015, 17, 334. doi: 10.1021/ol503442n
-
[51]
Wang, W.; Zhou, R.; Jiang, Z.-J.; Wang, X.; Fu, H.-Y.; Zheng, X.-L.; Chen, H.; Li, R.-X. Eur. J. Org. Chem.2015, 2015, 2579.
-
[52]
Pinto, A.; Jia, Y.; Neuville, L.; Zhu, J. Eur. Chem. J.2007, 13, 961. doi: 10.1002/(ISSN)1521-3765
-
[53]
Lee, H.-S.; Kim, K.-H.; Lim, J.-W.; Kim, J.-N. Bull. Korean Chem. Soc. 2011, 32, 1083. doi: 10.5012/bkcs.2011.32.3.1083
-
[54]
Kamisaki, H.; Yasui, Y.; Takemoto, Y. Tetrahedron Lett. 2009, 50, 2589. doi: 10.1016/j.tetlet.2009.03.100
-
[55]
Hande, S. M.; Nakajima, M.; Kamisaki, H.; Tsukano, C.; Takemoto, Y. Org. Lett. 2011, 13, 1828. doi: 10.1021/ol2003447
-
[56]
Newman, S. G.; Lautens, M. J. Am. Chem. Soc. 2011, 133, 1778. doi: 10.1021/ja110377q
-
[57]
Liu, X.; Ma, X.; Huang, Y.; Gu, Z. Org. Lett.2013, 15, 4814. doi: 10.1021/ol402210a
-
[58]
Jiang, T. S.; Tang, R. Y.; Zhang, X. G.; Li, X. H.; Li, J. H. J. Org. Chem. 2009, 74, 8834. doi: 10.1021/jo901963g
-
[59]
Jaegli, S.; Dufour, J.; Wei, H. L.; Piou, T.; Duan, X. H.; Vors, J. P.; Neuville, L.; Zhu, J. Org. Lett. 2010, 12, 4498. doi: 10.1021/ol101778c
-
[60]
Wu, T.; Mu, X.; Liu, G. Angew. Chem., Int. Ed. 2011, 50, 12578. doi: 10.1002/anie.201104575
-
[61]
Mu, X.; Wu, T.; Wang, H. Y.; Guo, Y. L.; Liu, G. J. Am. Chem. Soc. 2012, 134, 878. doi: 10.1021/ja210614y
-
[62]
Kamijo, S.; Sasaki, Y.; Kanazawa, C.; Schüßeler, T.; Yamamoto, Y. Angew. Chem., Int. Ed.2005, 44, 7718. doi: 10.1002/(ISSN)1521-3773
-
[63]
Miura, T.; Toyoshima, T.; Ito, Y.; Murakami, M. Chem. Lett. 2009, 38, 1174. doi: 10.1246/cl.2009.1174
-
[64]
Miura, T.; Toyoshima, T.; Takahashi, Y.; Murakami, M. Org. Lett. 2009, 11, 2141. doi: 10.1021/ol900759f
-
[65]
Toyoshima, T.; Mikano, Y.; Miura, T.; Murakami, M. Org. Lett. 2010, 12, 4584. doi: 10.1021/ol101892b
-
[66]
Lira, R.; Wolfe, J. P. J. Am. Chem. Soc. 2004, 126, 13906. doi: 10.1021/ja0460920
-
[67]
Schultz, D. M.; Wolfe, J. P. Org. Lett. 2010, 12, 1208. doi: 10.1021/ol902974j
-
[68]
Thansandote, P.; Hulcoop, D. G.; Langer, M.; Lautens, M. J. Org. Chem.2009, 74, 1673. doi: 10.1021/jo802604g
-
[69]
Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Sferrazza, A. Org. Biomol. Chem. 2011, 9, 1727. doi: 10.1039/c0ob01052a
-
[70]
Houlden, C. E.; Bailey, C. D.; Ford, J. G.; Gagne, M. R.; Lloyd-Jones, G. C.; Booker-Milburn, K. I. J. Am. Chem. Soc. 2008, 130, 10066. doi: 10.1021/ja803397y
-
[71]
Agasti, S.; Maity, S.; Szabo, K. J.; Maiti, D. Adv. Synth. Catal. 2015, 357, 2331. doi: 10.1002/adsc.v357.10
-
[72]
Pinto, A.; Neuville, L.; Zhu, J. Angew. Chem., Int. Ed.2007, 46, 3291. doi: 10.1002/(ISSN)1521-3773
-
[73]
Tang, S.; Peng, P.; Pi, S. F.; Liang, Y.; Wang, N. X.; Li, J. H. Org. Lett. 2008, 10, 1179. doi: 10.1021/ol800080w
-
[74]
Tang, S.; Peng, P.; Wang, Z.-Q.; Tang, B.-X.; Deng, C.-L.; Li, J.-H.; Zhong, P.; Wang, N.-X. Org. Lett. 2008, 10, 1875. doi: 10.1021/ol8006315
-
[75]
An, G.; Zhou, W.; Zhang, G.; Sun, H.; Han, J.; Pan, Y. Org. Lett. 2010, 12, 4482. doi: 10.1021/ol101664y
-
[76]
Wang, J. Y.; Su, Y. M.; Yin, F.; Bao, Y.; Zhang, X.; Xu, Y. M.; Wang, X. S. Chem. Commun. 2014, 50, 4108. doi: 10.1039/c3cc49315f
-
[77]
Nicolai, S.; Erard, S.; Gonzalez, D. F.; Waser, J. Org. Lett. 2010, 12, 384. doi: 10.1021/ol9027286
-
[78]
Yip, K.-T.; Yang, M.; Law, K.-L.; Zhu, N.-Y.; Yang, D. J. Am. Chem. Soc. 2006, 128, 3130. doi: 10.1021/ja060291x
-
[79]
Yip, K. T.; Zhu, N. Y.; Yang, D. Org. Lett. 2009, 11, 1911. doi: 10.1021/ol900355h
-
[80]
He, W.; Yip, K.-T.; Zhu, N.-Y.; Yang, D. Org. Lett. 2009, 11, 5626. doi: 10.1021/ol902348t
-
[81]
Du, W.; Gu, Q.; Li, Z.; Yang, D. J. Am. Chem. Soc. 2015, 137, 1130. doi: 10.1021/ja5102739
-
[82]
Mariampillai, B.; Alberico, D.; Bidau, V.; Lautens, M. J. Am. Chem. Soc. 2006, 128, 14436. doi: 10.1021/ja064742p
-
[83]
Rudolph, A.; Rackelmann, N.; Turcotte-Savard, M.-O.; Lautens, M. J. Org. Chem. 2009, 74, 289. doi: 10.1021/jo802180h
-
[84]
Wu, X. X.; Zhou, P. X.; Wang, L. J.; Xu, P. F.; Liang, Y. M. Chem. Commun. 2014, 50, 3882. doi: 10.1039/c4cc00809j
-
[85]
Thansandote, P.; Raemy, M.; Rudolph, A.; Lautens, M. Org. Lett. 2007, 9, 5255. doi: 10.1021/ol702472u
-
[86]
Muñiz, K. J. Am. Chem. Soc. 2007, 129, 14543.
-
[87]
应安国, 叶伟东, 刘泺, 吴国峰, 陈新志, 钱胜, 张秋萍, 有机化学, 2008, 28, 2081.Ying, A.-G.; Ye, W.-D.; Liu, L.; Wu, G.-F.; Chen, X.-Z.; Qian, S.; Zhang, Q.-P. Chin. J. Org. Chem. 2008, 28, 2081 (in Chinese).
-
[88]
John, J.; Indu, U.; Suresh, E.; Radhakrishnan, K. V. J. Am. Chem. Soc.2009, 131, 5402. doi: 10.1021/ja900306m
-
[89]
Prakash, P.; Jijy, E.; Preethanuj, P.; Pihko, P. M.; Sarath Chand, S.; Radhakrishnan, K. V. Chem. Eur. J. 2013, 19, 10473. doi: 10.1002/chem.v19.32
-
[90]
Chand, S. S.; Jijy, E.; Prakash, P.; Szymoniak, J.; Preethanuj, P.; Dhanya, B. P.; Radhakrishnan, K. V. Org. Lett. 2013, 15, 3338. doi: 10.1021/ol401374d
-
[91]
Ramirez, T. A.; Wang, Q.; Zhu, Y.; Zheng, H.; Peng, X.; Cornwall, R. G.; Shi, Y. Org. Lett. 2013, 15, 4210. doi: 10.1021/ol401935c
-
[92]
Zheng, H.; Zhu, Y.; Shi, Y. Angew. Chem., Int. Ed. 2014, 53, 11280. doi: 10.1002/anie.201405365
-
[93]
Campbell, C. D.; Greenaway, R. L.; Holton, O. T.; Chapman, H. A.; Anderson, E. A. Chem. Commun. 2014, 50, 5187. doi: 10.1039/C3CC45634J
-
[94]
Senadi, G. C.; Hu, W. P.; Boominathan, S. S.; Wang, J. J. Chem. Eur. J. 2015, 21, 998. doi: 10.1002/chem.201405933
-
[1]
-

图式23 Sonogashira偶联/碳钯化/C—H活化串联反应
Scheme 23 Tandem Sonogashira coupling/carbopalladation/ C—H activation

图式24 胺钯化/碳钯化/C—H活化串联反应
Scheme 24 Tandem aminopalladation/carbopalladation/C—H activation

图式32 环丙烷开环/氢钯化/亲核取代的串联反应
Scheme 32 Tandem ring-opening of cyclopropane/hydro-palladation/nuclephilic subsitution
-


 下载:
下载:




































 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 2708
- HTML全文浏览量: 363
 下载:
下载:
