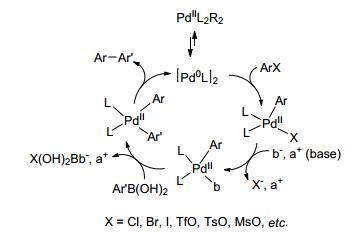
 图 图式1
金属钯催化的Suzuki-Miyaura交叉偶联反应机理
Figure 图式1.
Catalytic cycle for the Suzuki-Miyaura cross-coup-ling
图 图式1
金属钯催化的Suzuki-Miyaura交叉偶联反应机理
Figure 图式1.
Catalytic cycle for the Suzuki-Miyaura cross-coup-ling
引用本文:
李清寒, 丁勇, 张刚, 张震, 莫松. 微波技术在钯催化Suzuki-Miyaura交叉偶联反应中的应用研究进展[J]. 有机化学,
2015, 36(1): 83-104.
doi:
10.6023/cjoc201507008

Citation: Li Qinghan, Ding Yong, Zhang Gang, Zhang Zhen, Mo Song. Advance on Applications of Microwave Technique in Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling Reaction[J]. Chinese Journal of Organic Chemistry, 2015, 36(1): 83-104. doi: 10.6023/cjoc201507008
Citation: Li Qinghan, Ding Yong, Zhang Gang, Zhang Zhen, Mo Song. Advance on Applications of Microwave Technique in Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling Reaction[J]. Chinese Journal of Organic Chemistry, 2015, 36(1): 83-104. doi: 10.6023/cjoc201507008
微波技术在钯催化Suzuki-Miyaura交叉偶联反应中的应用研究进展
摘要:
与传统的加热方图式相比, 微波加热具有加热速度快、热效率高、节约能源、洁净、操作简单等优点, 已成为重要的有机合成工具之一.钯催化的Suzuki-Miyaura交叉偶联反应提供了一种合成各种联芳烃的温和方法, 具有较好的选择性, 受到了合成化学家的广泛关注.综述了近年来微波技术在钯催化的Suzuki-Miyaura交叉偶联反应中的应用研究进展, 包括多种反应体系, 并对其在天然产物和生物活性分子合成中的应用作简要概述.
-
关键词:
- 微波技术
- / 钯
- / Suzuki-Miyaura交叉偶联反应
- / 催化
English
Advance on Applications of Microwave Technique in Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling Reaction
Abstract:
Compared with conventional heating, microwave heating is one of the most useful tools in organic synthesis because of its obviously advantages of fast heating, thermal efficiency, saving energy, clean, and easy operation. Palladium catalyzed Suzuki-Miyaura cross-coupling reaction could tolerate a broad range of functional groups with high stereoselectivity to provide a mild method in preparation of kinds of substituted biaryls. In this paper, the recent research results about the microwave technique applied in Suzuki-Miyaura cross-coupling reaction are reviewed, involving various reaction systems. The applications of the methodology on the synthesis of natural products and bioactive molecules have also been introduced.
-
Key words:
- microwave technique
- / palladium
- / Suzuki-Miyaura cross-coupling reaction
- / catalyze
-
在许多的天然产物、药物、染料及功能材料中都含有联芳烃结构单元, 因此, 分子中联芳烃键的构建是现代有机合成最重要的手段之一.过渡金属催化的Suzuki-Miyaura交叉偶联反应是构建联芳烃键的最有效方法之一[1].该反应具有反应条件温和、试剂毒性小、底物普适性大、反应后处理简单、高度的区域选择性和立体选择性等优点, 特别是可以有效合成不对称的联苯类化合物[2, 3].自20世纪80年代发现这类反应以来, 由于其在天然产物、医药、农药、功能高分子、精细化学等方面的合成有着广泛的应用前景, 而日益受到人们的广泛关注[4].在过去的几十年中, 已发展了大量的构建联芳烃键的Suzuki-Miyaura交叉偶联反应方法, 其中钯催化的Suzuki-Miyaura交叉偶联反应是最重要和最有效的方法之一[5~9], 金属钯催化的Suzuki-Miyaura交叉偶联反应的催化循环反应机理如Scheme 1所示.
图 图式1
金属钯催化的Suzuki-Miyaura交叉偶联反应机理
Figure 图式1.
Catalytic cycle for the Suzuki-Miyaura cross-coup-ling
通常情况下, Suzuki-Miyaura交叉偶联反应大多使用常规的加热方式进行, 一般需要几个小时到几十个小时才能完成反应. 1986年, Gedye[10]和Giguere等[11]首次将微波加热方式应用于有机合成, 使得常规有机合成反应时间大大缩短, 受到了合成化学家的青睐.在此之后, 便出现了大量的有关微波合成化学的文献和综述文章[12~14].随后, 应用微波技术促进Suzuki-Miyaura交叉偶联反应的研究在化学界掀起了一股热潮.
本文按照催化体系的不同, 将Suzuki-Miyaura交叉偶联反应分为两大类, 即是传统的和新的Suzuki-Miyaura交叉偶联反应, 其中传统的Suzuki-Miyaura交叉偶联反应按照使用的催化剂结构特点又分为均相的和非均相的Suzuki-Miyaura交叉偶联反应.下面我们对微波辅助的金属钯催化的Suzuki-Miyaura交叉偶联反应进行介绍.
1 传统的Suzuki-Miyaura交叉偶联反应
1.1 均相Suzuki-Miyaura交叉偶联反应
在现代有机合成中, 过渡金属催化的均相Suzuki-Miyaura交叉偶联反应是最重要的构建C—C及C—N键的反应类型之一.在均相反应体系中, 催化剂具有较高的催化活性.
1996年Larhed和Hallberg[15]首次将微波技术应用于Suzuki-Miyaura交叉偶联反应中(Eq. 1), 在均相条件下, 使用3 mol%的Pd(PPh3)4为催化剂, Na2CO3作碱, DME:H2O:C2H5OH (V:V:V=4:2:1)为溶剂, 在55 W的微波功率辐射下, 仅用2.8 min便以55%的收率获得了芳基溴与苯硼酸的交叉偶联产物.
1999年, Schotten等[16]首先采用PEG-6000键合的芳香卤化物与芳基硼酸进行Suzuki-Miyaura交叉偶联反应, 合成了PEG键合的联芳烃类化合物(Scheme 2).他们以10 mol%的Pd(OAc)2为催化剂, 在75 W的微波功率作用下, 仅需2~4 min即可完成芳(杂环)基卤化物与芳基硼酸的交叉偶联反应, 其偶联产物收率可达59%~86%.同时, 在干态下交叉偶联反应亦可进行, 但需要更高的微波能量. PEG在900 W的微波作用下仍然稳定, 而在常规加热下, 水解率高达45%.
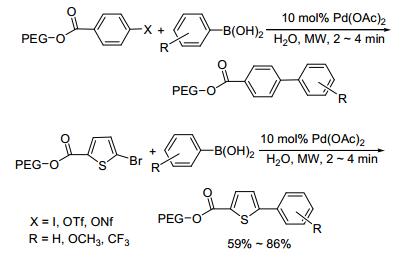 图 图式2
微波辅助Pd(OAc)2催化PEG支载的芳(杂环)卤化物与芳基硼酸的Suzuki-Miyaura交叉偶联反应
Figure 图式2.
Microwave-mediated Suzuki-Miyaura cross-coup-ling of PEG supported (hetero)aryl halide with arylboronic acid using Pd(OAc)2
图 图式2
微波辅助Pd(OAc)2催化PEG支载的芳(杂环)卤化物与芳基硼酸的Suzuki-Miyaura交叉偶联反应
Figure 图式2.
Microwave-mediated Suzuki-Miyaura cross-coup-ling of PEG supported (hetero)aryl halide with arylboronic acid using Pd(OAc)2
PEG除了作载体, 也可作为相转移催化剂参与反应. 2001年, Varma课题组[17]以PEG-400的水溶液为反应介质, 使用28 mol% PdCl2作催化剂, KF为碱, 溴/碘代芳烃与芳基硼酸在240 W的微波功率作用下加热至100~105 ℃反应50 s, 以74%~90%的收率获得偶联产物(Eq. 2), 不过氯代苯只有15%的收率.虽然油浴加热15 min也可获得相同的收率, 但是不如微波炉方便、洁净.实验结果表明, 碘代芳烃的活性比溴代芳烃高, 含有吸电基的苯硼酸和卤代芳烃的反应比含供电基的活性高.
2002年, Leadbeater小组[18]使用0.4 mol%的Pd(OAc)2为催化剂, 水为溶剂, 在60 W的微波功率辐射下加热至150~175 ℃反应5~10 min, 溴/碘/氯代芳烃均可与芳基硼酸发生Suzuki-Miyaura交叉偶联反应(Eq. 3).但在实验中发现氯代芳烃需要更高的反应温度, 且收率较低, 同时出现了苯硼酸的脱硼作用.后来, 他们研究发现该体系成功的关键是相转移催化剂四丁基溴化铵的加入.它有两个作用:一是解决了底物难溶的问题, 二是生成了活性更高的[ArB(OH)3]-[R4N]+, 加速了反应.用同样的条件, 他们做了10 mmol的放大实验, 并比较了微波辐射与常规加热方式对反应的影响.实验表明在相同的条件下, 无论是1或10 mmol的反应规模, 常规加热都比微波加热效果好, 并未发现与反应速度有关的非热微波效应[19].
2006年, 该课题组[20]又报道了在敞口反应体系中, 使用超低含量的Pd (0.0009 mol%)催化剂, Na2CO3作碱, EtOH和H2O为溶剂, 用微波辐射至80~83 ℃反应20 min, 可有效催化带有供电子基及吸电子基的溴代芳烃与苯硼酸的Suzuki-Miyaura交叉偶联反应, 其目标化合物的收率可达50%~99% (Eq. 4).该方法可适用于多种反应底物, 对于目标物的大量制备来说, 敞口反应体系更加安全, 而且特别是在需要连续的微波辐射反应中, 对于固体、高粘性液体及非均相反应体系的处理操作更容易.该反应条件无需重新优化, 就可完成毫摩尔级到摩尔级的放大反应.
由于氯代芳烃的活性较低, 其与苯硼酸的Suzuki-Miyaura交叉偶联反应的产物收率受到极大的影响.因此寻找高活性的催化剂, 提高氯代芳烃的反应收率成为亟待解决的问题. 2004年, Bedford和Butts等[21]发现在三环己基膦(PCy3)的存在下, Pd(OAc)2具有较高的催化活性.用微波加热至150 ℃反应30 min, 各种取代的氯代芳烃与苯硼酸可顺利的进行Suzuki-Miyaura交叉偶联反应(Eq. 5), 偶联产物收率可高达96%, 对于空间位阻较大的邻位取代联苯收率也可高达94%.这为低活性的氯代芳烃进行偶联反应提供了一种高效的合成方法.
为了发展更多更有效的催化氯代芳烃与苯硼酸的Suzuki-Miyaura交叉偶联反应催化体系, 2005年, Buchwald和Anderson课题组[22]发现使用大体积2-环己基膦-2', 6'-二甲氧基联苯-3-磺酸钠(1)为配体, Pd(OAc)2为催化剂, 以H2O为溶剂及K2CO3作碱的条件下, 用微波加热至150 ℃反应10 min, 可有效催化氯代芳烃与芳基硼酸的Suzuki-Miyaura交叉偶联反应, 其偶联产物的收率可达94%~98% (Eq. 6).该方法适合于多种氯代芳烃, 其中包括杂环化合物.而且在微波辐射下, 能显著的缩短反应时间, 降低催化剂用量, 可从常规方法催化剂用量的2 mol%降低至0.1 mol%, 而对反应活性没有影响.
2007年, Espinet小组[23]报道了微波辅助钯催化α-溴萘与α-萘硼酸的不对称Suzuki-Miyaura交叉偶联反应, 目标化合物收率可达20%~95%, 对映选择性可达40%~70% ee (Eq. 7).该交叉偶联反应最佳条件是:以0.8 mol%的Pd(OAc)2或0.4 mol%的Pd2(dba)3•CHCl3为催化剂, 3.2 mol%的(R, Sp)-(-)-PFENMe为配体, CsF作碱, THF或DME作溶剂, 250 W微波功率加热至100~120 ℃反应60 min.这为手性联萘衍生物的合成提供了一种十分有效的合成方法.相比于常规合成法, 微波合成法具有反应时间短, 目标物收率高的优点.
2013年, Zhang小组[24]报道了使用5 mol%的PdCl2(PPh3)2为催化剂, K2CO3作碱, DMF-H2O (V:V=5:1)为溶剂, 在75 W微波功率辐射下加热至130 ℃反应13 min, 取代苄溴及氯与苯基硼酸可有效的发生Suzuki-Miyaura交叉偶联反应, 同时α-溴甲基萘及β-溴甲基萘也都可以顺利的进行Suzuki-Miyaura交叉偶联反应, 偶联产物的收率为82%~92% (Eq. 8).该方法具有简单、快速和收率高的优点.
均相反应虽然具有较高的催化活性, 但也存在易生成难以分离的钯黑、昂贵的金属钯不易回收再利用的缺点.这些原因造成了制备成本的增加及对产物的污染, 尤其是合成纯度要求较高的药物时, 更是如此, 这在一定程度上限制了均相反应的应用.
1.2 非均相Suzuki-Miyaura交叉偶联反应
与均相反应体系相比, 非均相反应体系中催化剂的催化效率略低.但近年来也出现了不少活性较高的催化剂, 如大体积磷钯、环状钯、氮杂环碳卡宾钯配合物(NHC-Pd)、Pd/C及Pd/Al2O3等.非均相体系的最大优点是通过简单的过滤即可将催化剂与反应液分离, 使价格昂贵的金属钯能够回收再利用, 降低了制备成本; 同时也减少了对产物的污染, 易于纯化.
1.2.8 肟钯配合物
2004年, Kirschning小组[55]用吡啶醛肟和NaPdCl4制备了一种不溶性的肟钯配合物14.在微波加热下, 用1 mol%的该配合物为催化剂, 可有效催化芳基卤与芳基硼酸的Suzuki-Miyaura交叉偶联反应, 微波作用5 min即可以48%~100%的收率获得偶联产物(Eq. 33);该催化剂的优点是可回收循环再利用14次, 且活性没有明显降低, 偶联产物的收率仍然高达93%.由于氯代芳烃价格低廉, 更适合于工业化生产, 但是其与碘、溴代芳烃相比, 活性太低, 使其应用受到一定的限制.
2009年, Pombeiro小组[56]合成了2-吡啶酰胺钯(Ⅱ)及酮亚胺钯(Ⅱ)络合物15, 16和17.并以硅胶为载体进行固相反应, 在微波辐射下, 考察了该类配合物催化Suzuki-Miyaura交叉偶联反应的效果(Eq. 34).结果表明, 络合物15, 16和17均能有效的催化芳基溴与芳基硼酸的Suzuki-Miyaura交叉偶联反应, 偶联产物收率为27%~85%, 当使用0.25 mol%的15为催化剂时, 其偶联产物收率可达85%, 而当使用0.025 mol%的15为催化剂时, 其偶联产物收率有所下降, 仅为51%, 但在该条件下, 催化剂15的最大TON可达2.0×105, TOF为1.3×104 min-1.
2011年, Nájera小组[57]用4, 4'-二氯二苯甲酮肟和PdCl2合成了不溶性的肟钯配合物18, 使用0.1~0.5 mol%的该配合物为催化剂, {[HP(t-Bu)3]BF4}为配体, 四叔丁基氢氧化铵(TBAOH)为助催化剂, DMF为溶剂, K2CO3作碱, 微波加热至130 ℃反应20 min, 可有效催化芳基氯、烯基氯及苄氯与烯基硼酸及氟硼酸钾的Suzuki-Miyaura交叉偶联反应, 得到取代单烯烃和取代二烯烃化合物, 其目标物的收率分别可达31%~95%, 63%~86%和63%~95% (Scheme 5), 其中芳基氯与烯基硼酸的Suzuki-Miyaura交叉偶联反应的区域选择性高达99/1.
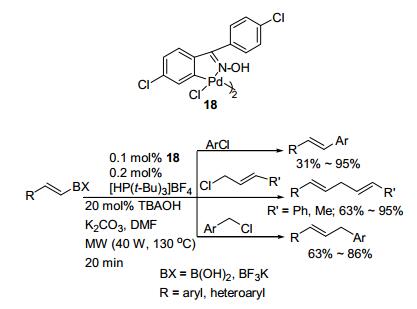 图 图式5
微波辅助催化剂18催化芳基氯与芳基硼酸的Suzuki-Miyaura交叉偶联反应
Figure 图式5.
Suzuki-Miyaura cross-coupling of alkylboronic acid with arylhalides using catalyst 18 under microwave irradiation
图 图式5
微波辅助催化剂18催化芳基氯与芳基硼酸的Suzuki-Miyaura交叉偶联反应
Figure 图式5.
Suzuki-Miyaura cross-coupling of alkylboronic acid with arylhalides using catalyst 18 under microwave irradiation
2007年, Dawood小组[58]应用苯并噻唑酮肟与PdCl2制备了肟钯配合物19和20, 并应用常规加热和微波辐射两种加热的方法, 以水为溶剂, 评价了苯并噻唑肟钯络合物19和20催化溴/氯代芳烃与芳(杂环)硼酸的Suzuki-Miyaura交叉偶联反应的催化活性(Eq. 35).结果表明, 这些络合物无论是在常规加热还是微波辐射下, 对于活化的芳基溴和杂环溴均具有高的催化活性, 而对于没有活化的芳基卤化物, 其溴化物活性大于氯化物.特别是使用0.02 mol%的肟钯配合物19催化对硝基氯苯与苯硼酸的偶联反应时, 在NaOH做碱及水作溶剂的体系中, 微波辐射7 min, 其偶联产物的收率可达90%, 其TON值可达5000, TOF可达42850 h-1, 而常规加热7 h, 其偶联产物的收率为85%, 此时TON值仅为4250, TOF仅为607 h-1.
1.2.7 桥环钯配合物
2005年, Yu小组[54]发现在常规加热作用下, 用桥环钯配合物POPd2 (13)为催化剂可以有效地催化氯代芳烃与芳基硼酸的Suzuki-Miyaura交叉偶联反应, 但是偶联产物的收率很低, 仅有10%~30%, 其主要原因是芳基硼酸容易发生自身交叉偶联反应.为了抑制该副反应, 提高交叉偶联产物收率, 他们使用微波辐射, 对反应条件进行了优化(Eqs. 31, 32).实验表明, 在150 ℃芳基硼酸与氯代芳烃的物质的量比为1.5:1时, 可以有效地避免芳基硼酸的自偶与脱硼反应.在最佳条件下, 桥环钯催化剂与均相钯催化剂如Pd(OAc)2、Pd(PPh3)4进行对比实验, 结果表明使用3 mol% POPd2, 微波辐射15 min便可以获得76%~85%的Suzuki-Miyaura偶联产物, 其催化活性明显地比均相催化剂的活性高.同时, 对空间位阻较大的邻位取代反应底物也有较高的收率.
1.2.6 氮杂环卡宾钯配合物
2004年, Nolan等[51]合成了一类氮杂环卡宾钯配合物[NHC-Pd(allyl)Cl]2 (5), 他们使用该配合物为催化剂, 以二噁烷为溶剂, NaOBu-t为碱, 在微波辐射的条件下考察了它催化氯代芳烃与苯硼酸的交叉偶联反应.该方法与常规加热方式相比, 收率无明显的差别, 但是在专用的单模微波炉加热下需要更高的温度, 反应时间更短, 只需要1.5 min即可完成反应, 偶联产物的收率可以高达88%~99% (Eq. 28).
2009年, Lamaty等[52]合成了有吡啶参与配位的氮杂环卡宾钯配合物PEPPSI-i-Pr-Pd (6).在无溶剂条件下, K2CO3作碱, 微波加热至110 ℃反应10 min, 0.5 mol%的PEPPSI-i-Pr-Pd可有效地催化氯/溴/碘代芳烃与芳基硼酸的Suzuki-Miyaura交叉偶联反应, 其偶联产物的收率为84%~99% (Eq. 29).该方法与其它方法相比, 具有快速、简单和高效的特点, 特别是对于活性较差的氯代芳烃, 也可以得到很好的收率, 其收率高达98%.
2011年, Küçükbay小组[53]以苯并咪唑为原料, 通过与卤代烷作用制备了6种含三甲硅基的苯并咪唑盐类化合物7~12.他们将2 mol%的苯并咪唑盐与1 mol% Pd(OAc)2配位形成络合物, 在微波辐射下以此配合物为催化剂, 有效地催化了苯硼酸与多种取代芳基卤(Br, Cl)的Suzuki-Miyaura交叉偶联反应, 其偶联产物的收率可达39%~99.9% (Eq. 30).如果在该反应体系中不加苯并咪唑盐, 则交叉偶联反应不会发生.同时在该反应体系中, 对于带有吸电子基的苯并咪唑盐配体的催化效率较差, 而对于带有供电子基的苯并咪唑盐配体的催化效率较高.该反应的优点在于无论是芳基卤(Cl, Br)的苯环上带有供电子基还是吸电子基, 都可有效地进行交叉偶联反应, 特别是芳基卤(Cl, Br)的苯环上带有供电子基氨基(NH2)时, 不需要保护即可进行反应.其具体反应条件是: 2 mol%的苯并咪唑盐与1 mol% Pd(OAc)2, DMF-H2O (V:V=1:1)为溶剂, K2CO3作碱, 微波加热至120 ℃反应10 min.
1.2.3 负载型纳米钯催化剂
2004年, Hu小组[37]制备了稳定的聚(N, N'-二己基)酰亚胺负载纳米钯配合物(PDHC-Pd), 其平均尺寸在3 nm左右.他们用该配合物为催化剂, 二噁烷作溶剂, K2CO3作碱, 微波辐射下反应40 min, 碘代苯与苯硼酸可顺利的进行Suzuki-Miyaura交叉偶联反应, 其偶联产物收率可达95% (Eq. 18);然而要达到相同的目标物收率, 常规加热需要20 h.虽然该催化剂可以回收循环5次以上, 但是在每次回收后催化活性都略有降低.他们[38]通过对其进一步的研究发现, 在强烈的微波作用下, 纳米钯微粒的尺寸发生了重新分配, 发现PDHC-Pd由第一次反应后的无定形球状微晶逐渐变为了第5次反应后的针状纳米结晶体.这主要是在催化循环过程中发生了化学刻蚀, 生成游离的Pd(Ⅱ), 然后还原消除所生成的纳米Pd(0)再重新回到载体时发生了团聚的结果.因此要获得高活性纳米催化剂, 聚合物配体的选择是至关重要的, 它可以有效阻止纳米颗粒的团聚.而且他们发现当使用邻(2-环己基膦)联苯为配体时, 催化剂的量可降为0.000001~0.02 mol.
2007年, Sharma小组[39]使用聚脲负载钯催化剂(Pd EnCat 30), 芳基溴与2-甲酰基苯硼酸衍生物在微波辐射下反应20 min, 以76%~93%的较好收率得到Suzuki-Miyaura交叉偶联反应产物(Eq. 19).而要获得相同收率的偶联产物, 常规加热需要24 h, 微波辐射加热反应的速度是常规加热的72倍.同时, 他们应用该偶联反应方法为关键合成步骤, 成功地合成了多环碳氢化合物及其代谢产物, 获得了好的收率.
2008年, Antunes小组[40]使用多聚乙烯吡咯酮负载的纳米钯(Pd/PVP)催化剂, 以EtOH-H2O (V:V=2:3)为溶剂, K2CO3作碱, 在220 W微波功率下加热反应40 min, 溴/碘代芳基与芳(杂环)硼酸能有效地发生Suzuki-Miyaura叉偶联反应, 以63%~100%的收率获得偶联产物(Eq. 20).虽然该反应在常规加热下也可以得到60%~100%的目标偶联产物, 但是需要18 h, 可见微波辐射技术在该偶联反应中表现出了很好的加速反应的作用.
2012年, El-Shall小组[41]将纳米氧化石墨烯和Pd(NO3)2溶于质子性溶剂(水、甲醇、乙醇)中, 使用脉冲激光照射成功地制备了纳米氧化石墨烯负载的纳米钯催化剂(Pd/PRGO).他们应用微波辐射, 以水为溶剂, 考察了该催化剂对Suzuki-Miyaura, Heck和Sonogashira交叉偶联反应的催化效果, 结果表明该催化剂表现出了极好的催化活性.对于芳基溴与芳基硼酸的Suzuki-Miyaura交叉偶联反应, 其偶联产物的收率可达100%, 同时该催化剂具有很好的可回收性, 其TON可达7800, TOF可达230000 h-1 (Eq. 21).
2014年, Smith小组[42]制备了葡萄糖负载的纳米钯催化剂, 并在微波辅助条件下考察了该催化剂对芳基碘与芳基硼酸的Suzuki-Miyaura交叉偶联反应的催化活性, 取得了很好的效果.其具体反应是将1 mol%的Pd(OAc)2和5 mol%的葡萄糖放于反应试管中, 原位生成负载化的纳米钯催化剂, 然后以K2CO3作碱, 异丙醇为反应溶剂, 微波辐射至120 ℃反应2 h, 其偶联产物的收率可高达94% (Eq. 22), 而采用常规加热法需要在100 ℃反应20 h才能获得同样收率的目标物.该反应体系对带供电子基或吸电子基的反应底物均有很好的催化效果, 可有效提高偶联产物的收率和降低反应中残留钯的含量.
2015年, Pastor和Alonso小组[43]制备了石墨烯片负载的纳米钯催化剂PdNPs-G (2), 并在常规加热和微波辅助条件下考察了该催化剂对芳基溴与芳基三氟硼钾的Suzuki-Miyaura交叉偶联反应的催化活性, 取得了很好的效果.他们使用0.1 mol%的PdNPs-G为催化剂, 以K2CO3作碱, 甲醇与水(V:V=3:1)的混合溶液为反应溶剂, 常规加热至80 ℃反应20 h, 其偶联产物的收率可高达96% (Eq. 23), 但是当催化剂回收循环利用4次后, 其催化活性会明显的下降.而采用微波辐射法只需要在40 W微波功率作用下反应2 h便可获得同样收率的目标物, 而催化剂的活性在回收循环利用8次后, 其催化活性仍无明显的下降, 他们认为出现这样的差异主要在常规加热时, 纳米钯颗粒容易团聚, 而在微波辐射下, 可很好地阻止纳米钯颗粒的团聚, 保持较好的纳米状态结构.该反应体系的优点是对带供电子基或吸电子基的反应底物均有很好的催化效果, 催化剂容易回收再利用, 可有效提高偶联产物的收率和降低反应中残留钯的含量, 特别是在放大反应的情况下, 该催化体系依然保持较好的催化效率.
1.2.1 Al2O3负载钯催化剂
Kabalka课题组[25, 26]将5 mol%的钯粉沉积在Al2O3上, 并以此为催化剂, KF作碱, 在微波作用下, 进行芳基卤化物与芳基硼酸的Suzuki-Miyaura交叉偶联反应.实验结果表明, 在该催化体系中, 芳基卤化物的活性顺序是:碘代芳烃>溴代芳烃>氯代芳烃, 而氟代芳烃则完全不反应(Eq. 9).乙烯基硼酸与芳基碘也有较高的收率, 可达72%, 但当乙烯基卤代烃与芳基硼酸进行交叉偶联反应时, 效果较差, 目标物只有很低的收率, 仅为24%, 而对于烷基卤则不发生反应.虽然该催化体系有一定的局限性, 但是该反应体系的催化剂至少能回收再利用6次, 催化活性无明显的降低.
2001年, Villemin等[27]将5 mol%的Pd(OAc)2负载到KF/Al2O3载体上, 在单模微波炉中进行Suzuki-Miyaura、Heck、Stille等交叉偶联反应, 得到了较高的目标物收率(Eq. 10). Suzuki-Miyaura偶联产物的收率可达40%~98%, 其中2-溴吡啶和2-碘吡啶与苯硼酸进行交叉偶联反应时, 可分别得到收率为40%和66%的预期产物.他们认为获得高收率最重要的因素是KF/Al2O3负载钯的分散度.同时, 由于家用微波炉控制功率能力较差, 并且微波辐射分散使实验的重复性较差, 而单模反应器则克服了上述缺点.
2003年, Basu等[28]同样采用4 mol%的Pd(OAc)2/ KF/Al2O3作催化剂, 在家用微波炉加热下反应7~20 min, 成功地催化了多卤代芳烃与苯基硼酸的Suzuki-Miyaura交叉偶联反应(Eq. 11), 制备了一系列多聚芳烃, 其收率可达53%~91%.该方法对于一些荧光材料的合成提供了一类新的合成途径.
Barbarella等[29]将[1, 1-双(二苯基膦基)二茂铁]二氯钯[PdCl2(dppf)2]负载到KF/Al2O3上, 在无溶剂的条件下, 应用微波加热至80 ℃反应6 min, 成功地催化了溴代噻吩的Suzuki-Miyaura交叉偶联反应, 合成了多聚噻吩(四聚噻吩, 五聚噻吩)(Eq. 12).同年该课题组[30]采用Pd(OAc)2作催化剂, 水与甲苯作溶剂, 四丁基溴化铵(TBAB)为相转移催化剂, K2CO3作碱, 用类似的方法, 经过三步反应合成了可溶性的六聚噻吩.为了解决不溶性多聚噻吩与催化剂分离困难的问题, 他们采用可溶性PdCl2(dppf)2/KF作催化剂[31], 合成了取代的五聚噻吩和六聚噻吩(Eq. 13).通过离心分离即可将产物从反应混合物中分离出来.
2009年, Mondal小组[32]使用10 mol%碱性Al2O3负载的Pd(PPh3)4为催化剂, 在微波无溶剂条件下考察了该催化剂催化芳基硼酸与5, 7-二溴香豆素衍生物的双Suzuki-Miyaura交叉偶联反应.结果表明, 在K2CO3存在下, 用180 W的微波功率将反应加热至120 ℃反应3 min, 可以以高达90%的收率获得5, 7-二芳基香豆素的偶联产物(Eq. 14).
2010年, Nelles小组[33]使用5 mol%的KF/Al2O3负载的Pd(PPh3)4为催化剂, 在微波辅助下催化芳基硼酸与硝基氟代苯的Suzuki-Miyaura交叉偶联反应.实验表明, 在K2CO3存在下, DMF作溶剂, 用微波加热至150 ℃反应15 min, 5 mol%的Pd(PPh3)4/KF/Al2O3催化剂可有效催化各种卤代芳烃与不同的芳基硼酸酯的Suzuki-Miyaura交叉偶联反应, 偶联产物的收率可达27%~81% (Eq. 15).该催化体系对具有吸电子基或供电子基的苯硼酸酯均有好的效果.
2013年, Qi小组[34]制备了γ-Al2O3-Pd催化剂, 该催化剂对卤代芳烃与芳基硼酸的Suzuki-Miyaura交叉偶联反应有非常高的催化效率.当使用0.2 mol%的γ-Al2O3-Pd为催化剂, DMF-H2O作溶剂, 微波加热至150 ℃反应45 min, 芳基卤与芳基硼酸的Suzuki-Miyaura交叉偶联反应的目标物收率可以达到81%~95% (Eq. 16).该催化剂可以通过简单的过滤回收再次利用, 循环5次未见催化剂失活.
1.2.9 缩氨基硫脲钯配合物
2006年Loupy小组[59]制备了一种能在空气中稳定存在的缩氨基硫脲钯配合物21.常规加热时, 该配合物对需氧条件下的Suzuki-Miyaura交叉偶联反应是惰性的.然而, 微波辐射下, DMF-H2O作为溶剂, Na2CO3作碱, 该配合物能有效的催化芳基溴与苯硼酸的交叉偶联反应, 其偶联产物的收率可达85%, 其催化剂可循环37000次(Eq. 36).而对于活化的1-氯-4-硝基苯与苯硼酸的交叉偶联在常规加热至155 ℃反应1 h, 其偶联产物收率仅为25%.他们认为该配合物能有效加速该反应是由于微波效应而非热效应.
1.2.4 碳负载钯(Pd/C)
Pd/C是一种价格低廉、性质稳定的催化剂, 在常规与微波两种加热方式下均有较高的催化活性, 且易于回收再利用, 是最适合于C—C交叉偶联反应(如Suzuki、Heck、Sonogashira等反应)的候选催化剂之一, 得到了广泛的应用[44, 45].
2005年, Arvela和Leadbeater[46]以Pd/C为催化剂, 水作溶剂, 在Na2CO3和TBAB存在下, 采用微波加热与同步冷却相结合的方法, 成功地催化了氯代芳烃与苯硼酸的Suzuki-Miyaura交叉偶联反应.在微波辐射条件下, 其交叉偶联反应目标物收率为21%~96%.他们对反应结果进行了分析, 认为在高温微波作用下氯代芳烃的分解是限制目标物收率提高的主要因素.为了防止氯代芳烃在高温下分解, 他们采用在微波作用的同时加以同步冷却, 则偶联产物的整体收率有明显的提高, 可达48%~94%.同时, 他们证明了在反应过程中, 采用微波加热与同步冷却相结合的方法能显著延长氯代芳烃的使用寿命, 提高目标物的收率以及原料的回收, 其中共溶剂的选择也是影响反应收率的重要因素.具体反应是在密封管内用微波加热至120 ℃反应10 min (Eq. 24).
2013年, Schmidt小组[47]使用直接购买的Pd/C为催化剂, 水为溶剂, 在K2CO3和TBAF存在下, 用微波加热至150 ℃反应30 min, 同样成功地催化了3-甲氧基-4-羟基-5-溴苯甲醛与邻羟基苯硼酸的Suzuki-Miyaura交叉偶联反应, 制备了2, 2'-联苯酚衍生物(Eq. 25).该反应体系的优点在于不需要在反应体系中加入配体或其它添加剂.
2014年, 该小组[48]又使用Pd/C为催化剂, 水为溶剂, KOH或NBu4F作碱, 用微波加热至150 ℃反应0.5 h, 成功地催化了邻/间/对溴苯酚/苯甲醚类化合物与邻/间/对羟基(甲氧基)苯硼酸类化合物的Suzuki-Miyaura交叉偶联反应, 以46%~96%的较好收率制备了多种联苯酚类植物抗毒素衍生物, 特别是对于常规方法难以制备的2, 4'-联苯酚类化合物的合成非常有效.该反应中, 无需对反应底物中所带的敏感基团-羟基进行预先保护(Eq. 26).
2015年, 该小组[49]又使用2 mol%的Pd/C为催化剂, 水为溶剂, KOH或NBu4F作碱, 用微波加热至150 ℃反应0.5 h, 成功地催化了3-溴-4-羟基苯甲醛/甲酸酯类化合物与对羟基苯硼酸类化合物的Suzuki-Miyaura交叉偶联反应, 分别以81%和87%的较好收率制备了多种联苯酚类衍生物.该反应中, 无需对反应底物中所带的敏感基团羟基进行预先保护(Eq. 27).并以此方法为关键合成步骤合成了具有抗肿瘤活性的化合物3 (magnaldehyde B)和4 (4'-Methoxymagnaldehyde B).
1.2.5 硫改性的金负载钯配合物
2013年, Arisawa小组[50]合成了用硫改性的金负载钯配合物(SAPd).他们应用微波辐射技术, 以单通道和多通道相结合的模式考察了该配合物催化未活化的溴/氯代芳烃与芳基硼酸的Suzuki-Miyaura交叉偶联反应, 并以96%~99%的收率得到了偶联产应, 其反应时间比常规加热显著缩短.要达到相同的收率, 微波辐射只需要2 h, 而常规加热需要12 h (Scheme 4).其具体反应过程是:先在单通道模式下, 以DMF为溶剂, K2CO3为碱, 微波加热至90 ℃反应50 min, 然后再在多通道模式下, 以甲苯/H2O (V:V=3:1)为溶剂, 微波加热至102 ℃反应60 min.
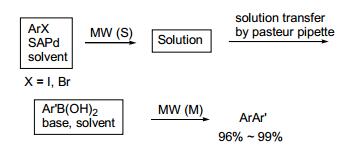 图 图式4
微波辅助SAPd催化芳基溴和碘与芳基硼酸的Suzuki-Miyaura交叉偶联反应.
Figure 图式4.
Suzuki-Miyaura coupling reactions of aryl bromides and iodoium with arylboronic acid catalyzed by SAPd under microwave irraditions
图 图式4
微波辅助SAPd催化芳基溴和碘与芳基硼酸的Suzuki-Miyaura交叉偶联反应.
Figure 图式4.
Suzuki-Miyaura coupling reactions of aryl bromides and iodoium with arylboronic acid catalyzed by SAPd under microwave irraditions
1.2.2 高聚物负载Pd催化剂
2004年, Sauer等[35]用功能化的纤维负载钯(Fibre-Cat)和聚苯乙烯负载钯(PS-PPh3-Pd)作催化剂(图 1), 在K2CO3存在下, EtOH为溶剂, 用微波辐射10~25 min, 成功地催化了多种卤代芳烃与不同的芳基硼酸的Suzuki-Miyaura交叉偶联反应, 以50%~99%的收率获得偶联产物, 有的甚至是定量反应, 并且在偶联反应中没有发现与磷有关的副产物, 该反应体系对于带吸电子基或供电子基的反应底物均有效(Eq. 17).
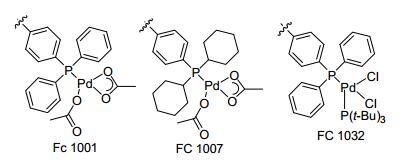 图 1
聚乙烯纤维负载钯催化剂
Figure 1.
Polyethylene-supported Pd catalysts
图 1
聚乙烯纤维负载钯催化剂
Figure 1.
Polyethylene-supported Pd catalysts
2004年, Wang小组[36]也合成了聚苯乙烯负载钯配合物, 并以1 mol%的PS-Pd(Ⅱ)为催化剂, K2CO3作碱, 甲苯-水(V:V=10:1)为溶剂, 微波辐射反应10 min, 芳基硼酸与芳基溴的Suzuki-Miyaura交叉偶联反应产物的收率可达95%.同时, 在相同条件下, 他们尝试了苯基硼酸及四苯硼钠与各种溴代芳烃的反应, 其目标物的收率高达84%~96%, 不过对四苯硼钠的4个苯基并未全部参与反应(Scheme 3).该反应体系的催化剂可回收再利用5次, 催化活性并未降低.在相同条件下, 要达到同样的收率, 常规加热方法则需要反应3 h.
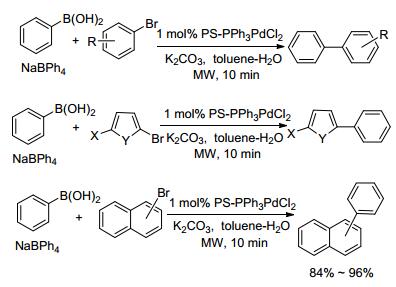 图 图式3
微波辅助PS-PPh3PdCl2催化Suzuki-Miyaura交叉偶联反应
Figure 图式3.
Suzuki-Miyaura cross-coupling reactions catalyzed by PS-PPh3PdCl2 under microwave irradiation
图 图式3
微波辅助PS-PPh3PdCl2催化Suzuki-Miyaura交叉偶联反应
Figure 图式3.
Suzuki-Miyaura cross-coupling reactions catalyzed by PS-PPh3PdCl2 under microwave irradiation
2 新的Suzuki-Miyaura交叉偶联反应
通常情况下, Suzuki-Miyaura交叉偶联反应的亲核部分常用硼酸或者酯, 但近年来, 出现了不少新颖的反应底物, 其中硼试剂在Suzuki-Miyaura交叉偶联反应中已经得到了很好的应用.
2.1 四苯硼钠参与的交叉偶联反应
2001年, Villemin等[60]首先报道了微波促进的四苯硼钠与卤代杂环芳烃的Suzuki-Miyaura交叉偶联反应(Eq. 37).他们用均相Pd(OAc)2为催化剂, Na2CO3为碱, H2O或MMF(单甲基甲酰胺)为溶剂, 微波加热至105~195 ℃反应8~12 min, 预期产物的收率可达60%~85%.以上反应均在氮气保护下的密闭聚四氟乙烯反应管内进行.
2011年, Guo小组[61]报道了以5 mol%的Pd(PPh3)2为催化剂, Na2CO3作碱, 水或乙醇为反应溶剂, 微波辐射30 min, 成功地催化了6-氯嘌呤核苷衍生物与四芳基硼化钠的Suzuki-Miyaura交叉偶联反应, 他们研究了18个反应底物, 其偶联产物的收率最高可达98% (Eq. 38).
2.2 磺酸酯参与的交叉偶联反应
2004年, Zhang小组[62]用芳基的全氟代辛烷磺酸酯22来代替卤代芳烃, 以甲苯-丙酮-水作溶剂, 用10 mol%的PdCl2(dppf)作催化剂, 采用专用微波炉加热至100~130 ℃反应10 min, 可顺利的完成Suzuki-Miyaura交叉偶联反应, 偶联产物收率为75%~95% (Eq. 39).该法最大的优点是过量的全氟代辛烷磺酸酯与产物易于分离.
2012年, Nájera小组[63]又合成了不溶性的肟钯配合物23和24.他们使用0.5 mol%的配合物23或24为催化剂, 添加15 mol%的表面活性剂CTAB, 三乙胺作碱, 水为溶剂, 在微波加热下反应30 min, 可有效催化咪唑磺酸酯与芳基硼酸及三氟硼化钾的Suzuki-Miyaura交叉偶联反应, 其偶联产物的收率可达55%~91% (Eq. 40).该反应体系优点在于反应体系中无需使用膦配体, 就可制得各种联苯及苯乙烯类化合物, 且具有很高的区域和立体选择性.
2.3 苯二酚硅的三乙铵盐参与的交叉偶联反应
芳基-双邻苯二酚硅的三乙铵盐25可以用来代替芳基硼酸与卤代芳烃或三氟磺酸酯反应.在常规加热下, 以二(二亚苄基丙酮)钯[Pd(dba)2]作催化剂, 碘代芳烃和芳基三氟磺酸酯参与的偶联反应收率较高, 而芳溴则收率较低, 最高收率也只有40%[64].但是在微波加热下, 以2-(二环己基膦)基联苯钯作催化剂, THF作溶剂, 四丁基氟化胺作为助催化剂, 在50 W微波功率辐射至120 ℃反应10 min, 对多种取代的芳基溴均能给出74%~93%的较高收率(Eq. 41)[65].
2.4 2-取代苯并-1, 3, 2-双氮杂硼烷参与的交叉偶联反应
2011年, Hadebe小组[66]报道了2-取代苯并-1, 3, 2-双氮杂硼烷参与的Suzuki-Miyaura交叉偶联反应.在该反应中, 他们以Pd(OAc)2/PCy3为催化剂, Na2CO3作碱, 水或乙醇为反应溶剂, 微波辐射5 min或常规加热反应48 h, 2-取代苯并-1, 3, 2-双氮杂硼烷与芳基溴能有效地进行Suzuki-Miyaura交叉偶联反应, 偶联产物的收率可达35%~89% (Eq. 42).他们发现在反应中使用三甲硅基对2-取代苯并-1, 3, 2-双氮杂硼烷的氮原子进行修饰可提高其反应活性.
2.5 芳基三氟硼钾参与的交叉偶联反应
芳基三氟硼钾盐相比于硼酸和酯更容易制备、储存和处理, 可以替代苯硼酸参与Suzuki-Miyaura交叉偶联反应, 其优点是对水稳定, 具有高度的构象与立构选择性, 且易与无机副产物分离. Al-Masum小组[67]用2 mol%的PdCl2(dppf)•CH2Cl2作催化剂, 采用专用微波炉加热至100 ℃, 多种取代的碘代苯与芳基三氟硼钾在异丙醇的水溶液中均能顺利的进行偶联反应, 以50%~100%的收率得到偶联产物, 其中邻、对位碘代苯甲醚可以定量反应.与常规加热相比, 可以将几个小时的反应在10 min内完成(Eq. 43).
2006年, Leadbeater小组[68]使用芳基三氟硼钾与多种取代的碘代及溴代苯在低剂量钯催化下, 以乙醇和水为溶剂, Na2CO3作碱, TBAB为相转移催化剂, 采用专用微波炉加热至150 ℃反应5 min, 可顺利地完成Suzuki-Miyaura交叉偶联反应, 偶联产物收率为24%~98% (Eq. 44).然而, 这一合成策略并非十分有效, 如对于非活化的芳基氯其收率很低.
同年, Naravane小组[69]报道了芳基磺酸酯和芳基三氟硼钾在微波辅助下进行的Suzuki-Miyaura交叉偶联反应.在该反应体系中不需要使用膦配体或相转移催化剂, 只需Pd(OAc)2作为催化剂, 乙醇和水作为反应溶剂, 用微波加热至95 ℃反应15 min, 可以顺利的进行芳基磺酸酯与芳基三氟硼钾的Suzuki-Miyaura交叉偶联反应, 并以50%~99%的收率获得目标产物(Eq. 45).
2007年, Crouch小组[70]报道了与Leadbeater相类似的合成策略, 他们使用微波辅助成功地促进了芳基三氟硼钾与各种取代氯代苯的Suzuki-Miyaura交叉偶联反应.实验结果表明, 在PdCl2催化下, 以甲醇和水为溶剂, K2CO3作碱, 采用专用微波炉加热至125 ℃反应20 min, 可完成各种取代溴代苯的Suzuki-Miyaura交叉偶联反应, 偶联产物收率为71%~99% (Eq. 46).这种合成方法的优点在于同样不需要使用膦配体或相转移催化剂, 对于带吸电子基或供电子基的反应底物均有很好的效果.
2009年, Ham小组[71]使用二卤苯化合物与硒粉、烷基溴、三异丙氧基硼、正丁基锂及氟氢化钾反应合成了一系列的含烷基硒的芳基三氟硼钾.然后使用3.0 mol%的Pd(PPh3)4为催化剂, 3.0倍量的K2CO3作碱, 二噁烷和水为溶剂, 微波辐射至130 ℃反应20 min, 可有效地催化带有芳基或烯基的芳基溴/碘化物与含烷基硒的芳基三氟硼钾的Suzuki-Miyaura交叉偶联反应, 并以54%~91%的收率获得了含烷基硒的联芳基衍生物(Scheme 6).该方法具有简单、快速和收率高的优点.
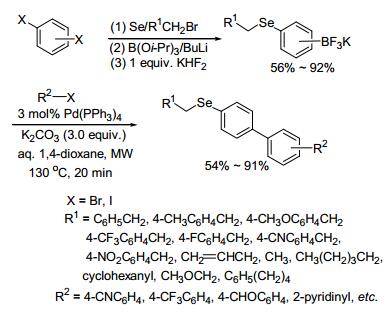 图 图式6
微波辅助芳基三氟硼钾的Suzuki-Miyaura交叉偶联反应
Figure 图式6.
Microwave-accelerated Suzuki-Miyaura cross-coupling reactions using potassium arytrifluoroborates
图 图式6
微波辅助芳基三氟硼钾的Suzuki-Miyaura交叉偶联反应
Figure 图式6.
Microwave-accelerated Suzuki-Miyaura cross-coupling reactions using potassium arytrifluoroborates
2010年, 该小组[72]又使用3 mol%的PdCl2(TPP)2及1 mol%的CuI为催化剂, 通过Sonogashira交叉偶联反应合成了一系列的含炔基的芳基三氟硼钾.他们再使用5.0 mol%的Pd(TPP)4为催化剂, 3.0倍量的Cs2CO3作碱, 二噁烷为溶剂, 微波辐射至150 ℃反应1 h, 有效地催化了带炔基的芳基三氟硼钾与芳基溴及烯基溴化物的Suzuki-Miyaura交叉偶联反应, 偶联产物收率可达62%~80% (Scheme 7).
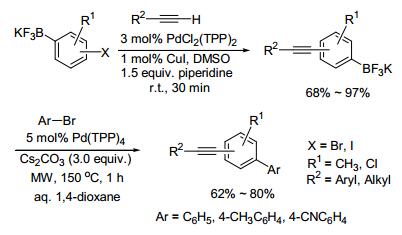 图 图式7
微波辅助芳基三氟硼钾的Suzuki-Miyaura交叉偶联反应
Figure 图式7.
Microwave-accelerated Suzuki-Miyaura cross-coupl-ing reactions using potassium arytrifluoroborates
图 图式7
微波辅助芳基三氟硼钾的Suzuki-Miyaura交叉偶联反应
Figure 图式7.
Microwave-accelerated Suzuki-Miyaura cross-coupl-ing reactions using potassium arytrifluoroborates
3 微波加热方式下Suzuki-Miyaura反应的应用
3.1 半导体聚合物的合成
2004年, Farrell和Scherf等[73]采用单模微波反应器, 通过Suzuki-Miyaura交叉偶联反应为关键步骤, 合成了一系列全共轭的梯形半导体聚合物, 其反应条件为: 4 mol% Pd(PPh3)Cl2为催化剂, THF-水溶液为反应溶剂, 反应温度130 ℃, 并考察了微波功率对聚合物分子量的影响.反应在密封管内进行, 微波辐射11 min, 便可获得8000以上的分子量, 而在常规加热方式下, 需要1~3 d.使用该方法[74], 他们又合成了类似的半导体聚合物, 其平均分子量高达29900 (Scheme 8).
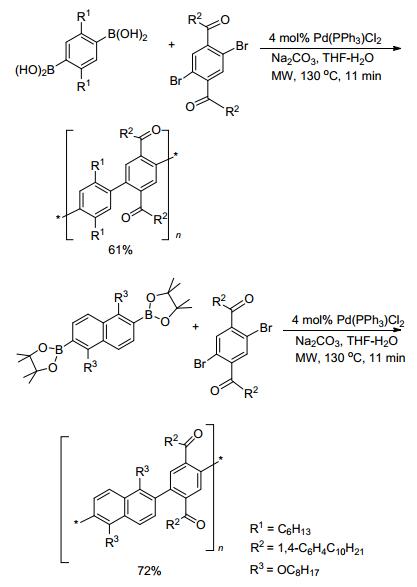 图 图式8
微波辅助芳基硼酸的Suzuki-Miyaura交叉偶联反应
Figure 图式8.
Microwave-accelerated Suzuki-Miyaura cross-coupl-ing reactions using arylboronic acid
图 图式8
微波辅助芳基硼酸的Suzuki-Miyaura交叉偶联反应
Figure 图式8.
Microwave-accelerated Suzuki-Miyaura cross-coupl-ing reactions using arylboronic acid
3.2 具有生物活性的杂环化合物的合成
Kappe等[75]报道了使用Pd(PPh3)4为催化剂催化的Suzuki-Miyaura及Heck反应为关键合成步骤, 在微波辐射下反应30 min, 便可以高达91%的收率获得具有生物活性的化合物26. 2006年, 该课题组[76]又报道了使用4-氯喹啉2-(1H)酮为原料, 使用微波辐射技术, 经一锅连续的硼烷化和Suzuki-Miyaura交叉偶联反应制备了具有生物活性的4, 4'-联喹啉酮类衍生物, 其目标物收率可达56%~94% (Eq. 47).其最佳条件是: 2 mol% PdCl2(dppf)为催化剂, KOH作碱, 微波加热至130~145 ℃反应35 min.
2005年, Tyte等[77]应用微波辐射技术, 通过Suzuki-Miyaura交叉偶联反应合成了一系列的抗真菌药物3-芳基-5-甲基-2, 5-二氢-2-呋喃酮, 其收率为17%~63%, 而使用常规加热则需要1.5 h才能获得相同的收率(Eq. 48).其最佳反应条件是: Pd(PPh3)4作催化剂, K2CO3作碱, 甲苯为溶剂, 微波加热至150 ℃反应3 min.
Heo课题组[78, 79]也采用微波加热方式, 以4 mol%的Pd(PPh3)4为催化剂, Na2CO3作碱, 二噁烷和水作反应溶剂, 于150 W微波功率加热至150 ℃反应5 min, 成功地催化了芳基硼酸与4-甲氧基-3-溴-2(5H)-呋喃酮的Suzuki-Miyaura交叉偶联反应, 以52%~85%收率得到了一系列具有生物活性的药物中间体2-芳基-3-甲氧基-2-环戊(己)烯酮(Eq. 49). 2006年, 该课题组[80]又采用相同的方法, 以2, 6-二氯苯并噻唑和芳基硼酸为原料, 通过Suzuki-Miyaura交叉偶联反应的关键合成步骤, 分别以77%和96%的较好收率合成了具有生物活性的药物中间体2-芳基-5-吗啉基苯并噻唑和2-芳基-5-苯基苯并噻唑(Scheme 9).
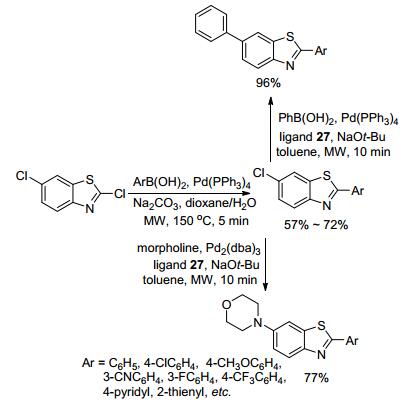 图 图式9
微波辅助芳基硼酸的Suzuki-Miyaura交叉偶联反应
Figure 图式9.
Microwave-accelerated Suzuki-Miyaura cross-coupl-ing reactions using arylboronic acid
图 图式9
微波辅助芳基硼酸的Suzuki-Miyaura交叉偶联反应
Figure 图式9.
Microwave-accelerated Suzuki-Miyaura cross-coupl-ing reactions using arylboronic acid
2008年, Hammond小组[81]采用Suzuki-Miyaura交叉偶联反应为关键合成步骤, 以二氟代炔丙醇为原料, 经碘环化制得碘代呋喃衍生物28, 然后以2 mol%的PdCl2(PPh3)2为催化剂, K2CO3作碱, DMF-H2O (V:V=5:1)为溶剂, 与芳基硼酸发生Suzuki-Miyaura交叉偶联反应, 在微波辐射下反应13 min, 以82%~92%的收率获得目标产物2, 4, 5-三取代-3-氟呋喃衍生物29 (Scheme 10).
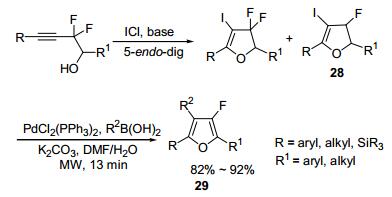 图 图式10
微波辅助2, 3, 5-三取代-3-氟呋喃的合成
Figure 图式10.
Synthesis of 2, 3, 5-trisubstituted-3-fluorofurans under microwave irradiation
图 图式10
微波辅助2, 3, 5-三取代-3-氟呋喃的合成
Figure 图式10.
Synthesis of 2, 3, 5-trisubstituted-3-fluorofurans under microwave irradiation
同年, Cao小组[82]使用5, 6-二氯-2-苄基吡嗪酮与不同的仲胺在乙醇中经微波辐射25 min制得6-氯-2-苄基-5-取代氨基-3(2H)吡嗪酮, 然后使用3 mol%的Combiphos-Pd或5 mol%的Pd-SPhos为催化剂, Cs2CO3作碱, 二噁烷为溶剂, 微波辐射至135~140 ℃反应30 min, 成功地催化了6-氯-2-苄基-5-取代氨基-3(2H)吡嗪酮与芳基硼酸的Suzuki-Miyaura交叉偶联反应, 以61%~100%的收率获得偶联目标产物(Eq. 50).
2009年, Vanelle小组[83]报道了微波辅助钯催化4-溴咪唑衍生物与芳基硼酸的Suzuki-Miyaura交叉偶联反应.用3~4 mol%的Pd(PPh3)4为催化剂, Na2CO3作碱, DMF-EtOH为溶剂, 150 W微波功率加热至150 ℃反应1 min, 其偶联反应产物收率可达60%~98%.他们合成了18种偶联产物(Eq. 51).
同年, 该小组[84]又报道了使用低剂量的钯(5 mol%)为催化剂, Na2CO3作碱, TBAB为相转移催化剂, 水为溶剂, 微波辐射至150 ℃反应1 min, 有效地催化5-溴-2-甲基-4-(对甲苯磺酰甲基)噻唑与芳基硼酸的Suzuki-Miyaura交叉偶联反应, 并以73%~98%的收率获得5-芳基噻唑衍生物(Eq. 52).该方法具有简单、快速和收率高的优点.
2007年, St. Jean小组[85]使用2 mol%的Pd(PPh3)4为催化剂, 以K2CO3作碱, DME-H2O (V:V=4:1)作溶剂, 微波辐射至140 ℃反应15 min, 成功的催化了磺酰胺基芳基硼酸酯与芳基溴/碘的Suzuki-Miyaura交叉偶联反应, 得到了功能化的咔唑衍生物, 其收率为47%~96% (Eq. 53).该反应体系可适用于含有醛基、酯基及砜基在内的各种吸电子基的反应底物.同时, 他们使用该方法经四步反应合成了glycosinine (30), 其总收率达50%.
2010年, Makriyannis小组[86]采用微波促进的Suzuki-Miyaura交叉偶联反应, 以邻羟基苯硼酸和邻溴代苯甲酸酯为原料经一步高效地合成了香豆素类化合物.其最佳反应条件是: 10 mol%的Pd(PPh3)4为催化剂, Cs2CO3作碱, 乙二醇二甲醚与水(V:V=20:3)为溶剂, 微波加热至125 ℃反应15 min, 目标物的收率可高达98% (Eq. 54).而且, 在该反应体系中, 如果使用邻-(双环己基膦)联苯为配体, 其催化剂用量可低至0.000001~0.02 mol%.同时在其进行的内酯化过程中, 其收率不受芳环上所带取代基的电性效应的影响.
2009年, Vanelle小组[87]报道了使用微波辅助Suzuki-Miyaura交叉偶联反应为关键合成步骤, 合成了一系列的具有抗疟疾作用的喹唑啉化合物(Eq. 55).他们采用2.5 mol%的Pd(PPh3)4为催化剂, Na2CO3作碱, DME或EtOH为溶剂, 在300 W微波功率辐射至80 ℃反应3 h, 2, 6-二取代-4-氯喹唑啉与芳(杂环)基硼酸可有效的发生Suzuki-Miyaura交叉偶联反应, 以63%~94%的收率得到2, 6-二取代-4-芳基喹唑啉化合物.该反应体系对于带吸电子基及供电子基的反应底物均有好的催化效果.
2015年, Vanelle小组[88]又使用微波辅助Suzuki-Miyaura交叉偶联反应为关键合成步骤, 合成了一系列的四取代的喹唑啉化合物(Scheme 11).他们采用5 mol%的PdCl2(PPh3)2催化6, 8-二溴-2-氯-4-二乙基氨基喹唑啉与芳(杂环)基硼酸进行Suzuki-Miyaura交叉偶联反应, 用微波辐射至90 ℃反应1.5 h, 经过双Suzuki-Miyaura交叉偶联反应以61%~82%的收率得到6, 8-双取代的喹唑啉衍生物31.同时, 他们采用类似的反应条件, 通过一锅煮, 成功地进行了以6, 8-二溴-2-氯-4-二乙基氨基喹唑啉为原料的三Suzuki-Miyaura交叉偶联反应, 2, 6, 8-三取代的喹唑啉衍生物32的收率可达66%~90%, 具有非常好的化学选择性.为了得到6, 8-二芳基-2, 4-二取代氨基喹唑啉衍生物, 他们又采用上述类似条件, 通过一锅煮, 成功地进行了以6, 8-二溴-2-氯-4-二乙基氨基喹唑啉为原料的C—N偶联和双Suzuki-Miyaura交叉偶联反应, 以69%~92%的收率获得6, 8-二芳基-2, 4-二取代氨基喹唑啉衍生物33, 表现出了优异的化学选择性.
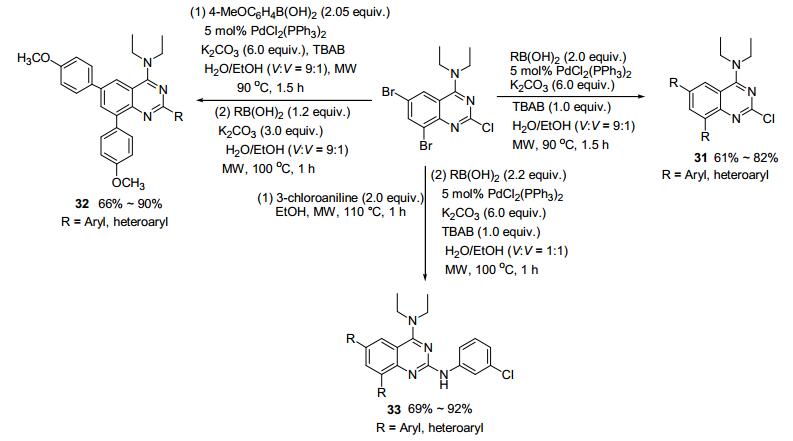 图 图式11
微波辅助一锅法化学选择性三Suzuki-Miyaura交叉偶联反应或芳基取代和双Suzuki-Miyaura交叉偶联反应合成2, 6, 8-三取代-4-氨基奎唑啉
Figure 图式11.
2, 6, 8-Trisubstituted 4-aminoquinazolines through microwave-assisted one-pot chemoselective tris-Suzuki-Miyaura or SNAr/bis-Suzuki-Miyaura reactions
图 图式11
微波辅助一锅法化学选择性三Suzuki-Miyaura交叉偶联反应或芳基取代和双Suzuki-Miyaura交叉偶联反应合成2, 6, 8-三取代-4-氨基奎唑啉
Figure 图式11.
2, 6, 8-Trisubstituted 4-aminoquinazolines through microwave-assisted one-pot chemoselective tris-Suzuki-Miyaura or SNAr/bis-Suzuki-Miyaura reactions
3.3 具有生物活性的buflavine类似物的合成
2005年, Van der Eycken小组[89]报道了应用微波辅助合成buflavine类似物.其中最关键的联芳基生成步骤采用钯催化Suzuki-Miyaura交叉偶联反应在微波照射下进行, 再通过环化得到刚性及中等大小环的buflavine类似物34 (Scheme 12).该反应条件可克服反应进行所需高反应活化能的障碍.反应中, 他们使用5 mol%的Pd(PPh3)4为催化剂, NaHCO3为碱, DMF-H2O为溶剂, 微波加热至150 ℃反应15 min, 偶联产物目标物的收率可以达到91%~93%.
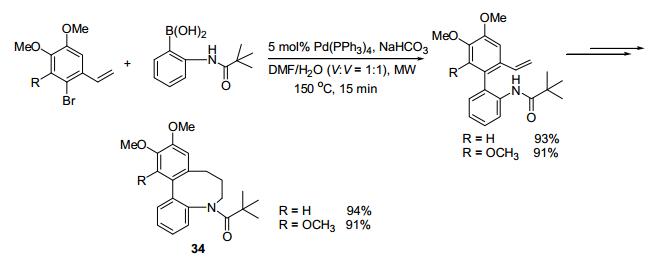 图 图式12
微波辅助buflavine类似物的合成
Figure 图式12.
Microwave-assisted synthesis of N-shifted buflavine analogues
图 图式12
微波辅助buflavine类似物的合成
Figure 图式12.
Microwave-assisted synthesis of N-shifted buflavine analogues
2007年, 该小组[90]又以邻溴苯乙烯衍生物和2-甲酰基苯硼酸为原料, 5 mol% Pd(PPh3)4为催化剂, 通过微波促进的Suzuki-Miyaura交叉偶联反应首先制备得到联苯类化合物, 再通过环化反应成功地合成了9-元中型环系的buflavine类似物35.其Suzuki-Miyaura交叉偶联反应的目标物收率可达90%~92%, 最终产物的收率可达93%~94% (Scheme 13).
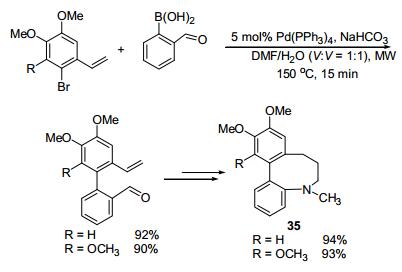 图 图式13
微波辅助buflavine类似物的合成
Figure 图式13.
Microwave-assisted synthesis of N-shifted buflavine analogues
图 图式13
微波辅助buflavine类似物的合成
Figure 图式13.
Microwave-assisted synthesis of N-shifted buflavine analogues
3.4 具有生物活性的多肽类化合物的合成
Zhu小组[91]使用大环膦钯配合物, 在微波辐射下, 以甲苯-水作为溶剂, K2CO3作碱及TBAB为相转移催化剂, 成功地催化了多肽分子内的Suzuki-Miyaura交叉偶联反应, 制备了具有抗菌活性的环状三肽化合物联苯霉菌素B (Biphenomycin B.) (36), 其收率为50% (Eq. 56).当只使用简单的Pd(OAc)2为催化剂, 其偶联产物只有33%.然而在常规加热条件下, 当使用一些效果比较好的催化体系进行催化时, 目标物的收率很低甚至不反应.
2006年, Larhed小组[92]应用微波辅助Suzuki-Miyaura交叉偶联反应成功的合成了24种具有C2对称性的功能化新型多肽化合物37, 该类化合物具有抗HIV病毒的作用.他们使用10 mol%的Pd(OAc)2为催化剂, 20 mol%的[(t-Bu)3PH]BF4为配体, K2CO3作碱, DMF-H2O为溶剂, 微波辐射至120 ℃反应30 min, 可有效催化多肽链上的芳基碘与芳杂环硼酸的Suzuki-Miyaura交叉偶联反应, 目标物收率为19%~65% (Eq. 57).该方法在将来会成为蛋白质功能化的一个非常有效的方法.
2012年, Planas和Feliu小组[93]应用微波辅助固相Suzuki-Miyaura交叉偶联反应成功地合成了21种5-芳基组氨酸多肽衍生物38, 该类化合物具有很好的抗菌作用.他们使用20 mol%的Pd2(dba)3为催化剂, 40 mol% SPhos为配体, KF作碱, DME/EtOH/H2O (V:V:V=9:9:2)为溶剂, 微波辐射至110~130 ℃反应30 min, 有效地催化了5-溴组氨酸多肽与芳基硼酸的Suzuki-Miyaura交叉偶联反应, 目标物收率为35%~95% (Eq. 58).
3.5 核苷类衍生物的合成
核苷衍生物在对抗各种病毒的候选药物中占据举足轻重的地位.微波在药物合成中已表现出了强大的作用, 特别是通过微波辐射促进Suzuki-Miyaura交叉偶联反应来对核苷化合物进行修饰已取得了不少成果[94~97].
Shaughnessy等[98, 99]使用5 mol%的Pd(OAc)2/13 mol%的TPPTS为催化剂, 以乙腈-水为溶剂, 经微波辐射至150 ℃反应1 min, 4-氯嘌呤核苷与4-(3-α-氨基丙酸基)苯硼酸可顺利地进行Suzuki-Miyaura交叉偶联反应, 并以84%的收率得到光学纯的腺嘌呤核苷化合物39 (Eq. 59).同时考察了常规加热和微波辐射两种方法对嘌呤核苷合成的影响, 结果表明, 常规加热对于合成8-取代核苷非常有效, 而微波辐射对于嘌呤碱和6-取代核苷更有效.
在三唑环上带有各种芳香基团的5-芳基三氮唑无环核苷是一类具有重要生物活性的核苷化合物.他们可通过简单高效的一步合成得到. 2007年, Peng小组[100]应用微波辐射技术, 在二噁烷和水作溶剂的条件下, 将未保护的5-溴三唑无环核苷与芳基硼酸进行Suzuki-Miyaura交叉偶联反应, 成功地以66%~99%的高收率得到5-芳基三唑无环核苷目标物40 (Eq. 60).这种偶联合成方法能以良好的收率直接得到相应的目标产物, 并且在反应中不涉及保护和脱保护步骤.
2013年, Len小组[101]报道了在微波辐射下, 以KOH作碱, 水为溶剂, 0.1~0.05 mol%的Na2PdCl4可有效地催化5-碘脲嘧啶-2-脱氧核苷与芳基硼酸的Suzuki-Miyaura交叉偶联反应得到5-芳基尿嘧啶核苷衍生物41, 目标物的收率可达中等以上(Eq. 61). 2014年, 该小组[102]采用相同的条件对5-碘脲嘧啶-2-脱氧核苷和芳基硼酸进行Suzuki-Miyaura交叉偶联反应和去糖基化反应得到5-芳基尿嘧啶衍生物, 目标物收率可达22%~67%.该反应体系无需添加配体, 核苷类化合物无需保护.
3.6 具有生物活性的酰胺类衍生物的合成
2011年, Spencer小组[103]使用微波辅助, 首先合成了一系列带N、S、O取代基的4, 4, 5, 5-四甲基-1, 3, 2-硼杂二噁烷, 然后在Pd(OAc)2或Pd(PPh3)4催化下, 与芳基溴发生Suzuki-Miyaura交叉偶联反应, 以21%~100%的收率制得了联苯类酰胺化合物.他们采用芳基溴与含酰胺基取代的芳基硼酸酯的Suzuki-Miyaura交叉偶联反应为关键反应步骤, 经三步成功地合成了降压药缬沙坦(Valsartan)的前驱物42, 其收率可达89% (Eq. 62).其Suzuki-Miyaura交叉偶联反应的最佳条件是: 0.5 mol%的Pd(OAc)2为催化剂, K2CO3作碱, 四丁基溴化铵为助催化剂, 水为溶剂, 或1 mol%的Pd(PPh3)4为催化剂, Na2CO3作碱, 甲苯-乙醇-水为溶剂, 微波辐射下反应10 min.该反应体系优点是对硼酸酯中的氨甲基、甲硫基、烷氧基及苯氧基官能团均不影响.
2014年, Shuto小组[104]使用片段增长的合成方法, 在微波辐射下, 用硫改性的金负载钯配合物(SAPd)催化烯基碘与硼酸的Suzuki-Miyaura交叉偶联反应及偶联产物在液相中的氨基化反应作为关键步骤, 设计和合成了90种具有潜在生物活性的酰胺类化合物(Scheme 14).
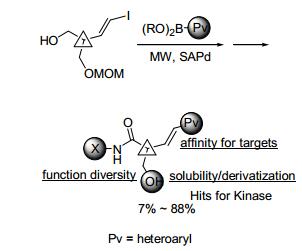 图 图式14
微波辅助SAPd催化烯基碘与芳杂环硼酸的Suzuki-Miyaura交叉偶联反应
Figure 图式14.
SAPd Catalyzed Suzuki-Miyaura cross-coupling of vinyl iodide and heteroarylboronic acid under microwave
图 图式14
微波辅助SAPd催化烯基碘与芳杂环硼酸的Suzuki-Miyaura交叉偶联反应
Figure 图式14.
SAPd Catalyzed Suzuki-Miyaura cross-coupling of vinyl iodide and heteroarylboronic acid under microwave
3.7 其他方面的应用
微波辅助金属钯催化的Suzuki-Miyaura交叉偶联反应除了在上述方面的应用外, 还可以应用于环状金属络合物的制备, 如Aoki小组[105]应用微波辅助钯催化的Suzuki-Miyaura交叉偶联反应成功地制备了对酸碱有响应的三环金属铱络合物[Ir(ppy)3和Ir(ppym)3].
同时, 近年来又出现了一些用新材料制备的反应器用于Suzuki-Miyaura交叉偶联反应中, 并取得了很好的效果. 2014年, Larhed小组[106]报道了使用碳化硅制作的反应器, 在微波辅助下, 使用2 mol%的Pd(dppf)Cl2为催化剂, 成功地催化了芳基卤与芳基硼酸的Suzuki-Miyaura交叉偶联反应, 其目标物收率为70%~87% (Eq. 63).并使用相同的条件, 经三步连续反应, 以72%的总收率成功地制备了NSAID联苯乙酸.该反应体系的优点是每小时可制备14 mmol的Suzuki-Miyaura交叉偶联反应产物, 反应温度容易控制, 反应器坚固不易损坏, 维护容易.
4 结论与展望
微波化学反应操作简单、安全、清洁且节能.在微波作用下, 不但可以提高目标物收率, 而且也可以使一些用常规加热方式不能进行的反应顺利进行.目前, 对Suzuki-Miyaura交叉偶联反应的研究主要集中在两个方面:一是开发更多新颖的底物来替代价格昂贵的苯硼酸、碘代苯、溴代苯; 二是发展更多低廉的、高活性的、可回收循环再利用的催化剂, 以便能够催化惰性的氯代苯的反应.近年来虽然出现了用常规金属(如Ni、Cu、Co等)来代替贵重金属钯催化Suzuki-Miyaura交叉偶联反应的报道[107], 但成功的例子不多, 今后这方面的研究将成为化学工作者的研究目标, 这对从根本上解决催化剂成本过高的问题将具有重要的实际意义.同时, 微波作为一种加热方式, 在有机合成中已被广泛应用, 但是到目前为止, 微波加速化学反应的机理还不十分明确, 微波有机合成的绝大部分反应还处于实验室研究阶段, 主要用于优化反应条件.因此, 深入地研究微波加速化学反应的机理, 设计可多次循环利用的高效催化剂, 开发用水代替有机溶剂的Suzuki-Miyaura交叉偶联反应将是今后研究的一个重点, 同时如何将微波合成化学实验扩大并且用于工业化, 这也是微波化学亟待解决的一个问题.
-
-
[1]
Suzuki, A. J. Organomet. Chem. 1999, 576, 147.
(b) Miyaura, N. Top. Curr. Chem. 2002, 219, 11.
(c) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457.
(d) Hassan, J.; Sevignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Chem. Rev. 2002, 102, 1359.
(e) Kotha, S.; Lahiri, K.; Kashinath, D. Tetrahedron 2002, 58, 9633.
(f) Littke, A. F.; Fu, G. C. Angew. Chem., Int. Ed. 2002, 41, 4176.
(g) Miyaura, N. Top. Curr. Chem. 2002, 219, 11.
(h) Bellina, F.; Carpita, A.; Rossi, R. Synthesis 2004, 2419.
(i) Christmann, U.; Vilar, R. Angew. Chem., Int. Ed. 2005, 44, 366.
(j) Alonso, F.; Beletskaya, I. P.; Yus, M. Tetrahedron 2008, 64, 3047.
(k) de Meijere, A.; Diederich, F. Metal-Catalyzed Cross-Coupling Reactions, Vol. 2, Wiley-VCH, Weinheim, 2004.
(l) Ackermann, L. Modern Arylation Methods, Wiley-VCH, Weinheim, 2009.
(m) Shen, X.; Jones, G. O.; Watson, D. A.; Bhayana, B.; Buchwald, S. L. J. Am. Chem. Soc. 2010, 132, 11278.
(n) Lundin, P. M.; Fu, G. C. J. Am. Chem. Soc. 2010, 132, 11027.
(o) Li, W.-Y.; Zhao, D.-M.; Xiong, X. Q.; Ma, Q. Q.; Cheng, M. S. Chin. J. Org. Chem. 2011, 31, 784 (in Chinese).
李文燕, 赵冬梅, 熊绪琼, 马倩倩, 程卯生, 有机化学, 2011, 31, 784.
(p) Li, Q. H.; Ding, Y.; Yang, X. J. Chin. Chem. Lett. 2014, 25, 1296.
(q) Li, Q. H.; Ding, Y.; Huang, N. W. Chin. Chem. Lett. 2014, 25(11), 1469. -
[2]
徐广庆, 赵庆, 汤文军, 有机化学, 2014, 34, 1919. doi: 10.6023/cjoc201406030Xu, G. Q.; Zhao, Q.; Tang, W. J. Chin. J. Org. Chem. 2014, 34, 1919 (in Chinese). doi: 10.6023/cjoc201406030
-
[3]
赵晓伟, 崔元臣, 化学进展, 2006, 18, 1652.Zhao, X. W.; Cui, Y. C. Prog. Chem. 2006, 18, 1652 (in Chinese).
-
[4]
Bellina, F.; Carpita, A.; Rossi, R. Synthesis 2004, 2419.
-
[5]
For a discussion, see
(a) Grushin, V. V.; Alper, H. In Activation of Unreactive Bonds and Organic Synthesis, Ed.: Murai, S., Springer, Berlin, 1999, p. 193.
(b) Grushin, V. V.; Alper, H. Chem. Rev. 1994, 94, 1047. -
[6]
Polshettiwar, V.; Decottignies, A.; Len, C.; Fihri, A. ChemSusChem 2010, 3, 502. doi: 10.1002/cssc.v3:5
-
[7]
Fihri, A.; Meunier, P.; Hierso, J. C. Coord. Chem. Rev. 2007, 251, 2017. doi: 10.1016/j.ccr.2007.03.020
-
[8]
Ackermann, L.; Vicente, R.; Kapdi, A. R. Angew. Chem., Int. Ed. 2009, 48, 9792. doi: 10.1002/anie.v48:52
-
[9]
刘宁, 刘春, 金子林, 有机化学, 2012, 32, 860. doi: 10.6023/cjoc1109052Liu, L.; Liu, C.; Jin, M. Z. Chin. J. Org. Chem. 2012, 32, 860 (in Chinese). doi: 10.6023/cjoc1109052
-
[10]
Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J. Tetrahedron Lett. 1986, 27, 279. doi: 10.1016/S0040-4039(00)83996-9
-
[11]
Giguere, R. J.; Bray, T. L.; Duncan, S. M.; Majetich, G. Tetrahedron Lett. 1986, 27(41), 4945. doi: 10.1016/S0040-4039(00)85103-5
-
[12]
Kappe, C. O. Angew. Chem., Int. Ed. 2004, 43, 6250. doi: 10.1002/(ISSN)1521-3773
-
[13]
Leadbeater, N. E. Chem. Commun. 2005, 23, 2881.
-
[14]
Dallinger, D.; Kappe, C. O. Chem. Rev. 2007, 107, 2563. doi: 10.1021/cr0509410
-
[15]
Larhed, M.; Hallberg, A. J. Org. Chem. 1996, 61, 9582. doi: 10.1021/jo9612990
-
[16]
Blettner. C. G.; König, W. A.; Stenzel, W.; Schotten, T. J. Org. Chem. 1999, 64(11), 3885. doi: 10.1021/jo982135h
-
[17]
Namboodiri, V. V.; Varma, R. S. Green Chem. 2001, 3(3), 146. doi: 10.1039/b102337n
-
[18]
Leadbeater, N. E.; Marco, M. Org. Lett. 2002, 4, 2973. doi: 10.1021/ol0263907
-
[19]
Leadbeater, N. E.; Marco, M. J. Org. Chem. 2003, 68, 888. doi: 10.1021/jo0264022
-
[20]
Leadbeater, N. E.; Williams, V. A.; Barnard, T. M.; Collins Jr, M. J. Org. Process Res. Dev. 2006, 10, 833. doi: 10.1021/op0600613
-
[21]
Bedford, R. B.; Buts, C. P.; Hurst, T. E.; Lidström, P. Adv. Synth. Catal. 2004, 346, 1627. doi: 10.1002/adsc.200404144
-
[22]
Anderson, K. W.; Buchwald, S. L. Angew. Chem., Int. Ed. 2005, 44, 6173. doi: 10.1002/(ISSN)1521-3773
-
[23]
Genov, M.; Almorín, A.; Espinet, P. Tetrahedron: Asymmetry 2007, 18, 625. doi: 10.1016/j.tetasy.2007.03.001
-
[24]
Zhang, Y. Q. J. Chem. Res. 2013, 375.
-
[25]
Kabalka, G. W.; Pagni, R. M.; Wang, L.; Namboodiri, V.; Hair, C. M. Green Chem. 2000, 3, 120.
-
[26]
Kabalka, G. W.; Wang, L.; Pagni, R. M.; Maxwell, H. C.; Vasudevan, N. Synthesis 2003, 217.
-
[27]
Villemin, D.; Caillot, F. Tetrahedron Lett. 2001, 42, 639. doi: 10.1016/S0040-4039(00)02027-X
-
[28]
Basu, B.; Das, P.; Bhuiyan, M. M. H.; Jha, S. Tetrahedron Lett. 2003, 44(19), 3817. doi: 10.1016/S0040-4039(03)00731-7
-
[29]
Melucci, M.; Barbarella, G.; Sotgiu, G. J. Org. Chem. 2002, 67, 8877. doi: 10.1021/jo026269d
-
[30]
Sotgiu, G.; Zambianchi, M.; Barbarella, G.; Botta, C. Tetrahedron 2002, 58, 2245. doi: 10.1016/S0040-4020(02)00098-4
-
[31]
Melucci, M.; Barbarella, G.; Zambianchi, M.; Pietro, P. D.; Bongini, A. J. Org. Chem. 2004, 69, 4821. doi: 10.1021/jo035723q
-
[32]
Saha, P.; Naskar, S.; Paira, P.; Hazra, A.; Sahu, K. B.; Paira, R.; Banerjee, S.; Mondal, N. B. Green Chem. 2009, 11, 931. doi: 10.1039/b902916h
-
[33]
Cargill, M. R.; Sandford, G.; Tadeusiak, A. J.; Yufit, D. S.; Howard, J. A. K.; Kilickiran, P.; Nelles, G. J. Org. Chem. 2010, 75, 5860. doi: 10.1021/jo100877j
-
[34]
Gao, F. F.; Liu, F. J.; Lin, C.; Li, W. T.; Wang, S. G.; Qi, C. Z. Catal. Commun. 2013, 5, 27.
-
[35]
Wang, Y.; Sauer, D. R. Org. Lett. 2004, 6, 2793. doi: 10.1021/ol048972p
-
[36]
Bai, L.; Zhang, Y. M.; Wang, J. X. QSAR Comb. Sci. 2004, 23, 875. doi: 10.1002/(ISSN)1611-0218
-
[37]
Liu, Y. B.; Khemtong, C.; Hu, J. Chem. Commun. 2004, 398.
-
[38]
Hu, J.; Liu, Y. B. Langmuir 2005, 21, 2121. doi: 10.1021/la0471902
-
[39]
Sharma, A. K.; Gowdahalli, K.; Krzeminski, J.; Amin, S. J. Org. Chem. 2007, 72, 8987. doi: 10.1021/jo701665j
-
[40]
De Souza, A. L. F.; Da Silva, L. C.; Oliveira, B. L.; Antunes, O. A. C. Tetrahedron Lett. 2008, 49, 3895. doi: 10.1016/j.tetlet.2008.04.061
-
[41]
Moussa, S.; Siamaki, A. R.; Gupton, B. F.; El-Shall, M. S. ACS Catal. 2012, 2, 145. doi: 10.1021/cs200497e
-
[42]
Camp, J. E.; Dunsford, J. J.; Cannons, E. P.; Restorick, W. J.; Gadzhieva, A.; Fay, M. W.; Smith, R. J. ACS Sustainable Chem. Eng. 2014, 2, 500. doi: 10.1021/sc400410v
-
[43]
Gómez-Martínez, M.; Buxaderas, E.; Pastor, I. M.; Alonso, D. A. J. Mol. Catal. A: Chem. 2015, 404~405, 1.
-
[44]
Heidenreich, R. G.; Köhler, K.; Krauter, J. G. E.; Pietsch, J. Synlett 2002, 7, 1118.
-
[45]
Lu, G.; Franzén, R.; Zhang, Q.; Xu, Y. J. Tetrahedron Lett. 2005, 46, 4255. doi: 10.1016/j.tetlet.2005.04.022
-
[46]
Arvela, R. K.; Leadbeater, N. E. Org. Lett. 2005, 7, 2101. doi: 10.1021/ol0503384
-
[47]
Schmidt, B.; Riemer, M.; Karras, M. J. Org. Chem. 2013, 78, 8680. doi: 10.1021/jo401398n
-
[48]
Schmidt, B.; Riemer, M. J. Org. Chem. 2014, 79, 4104. doi: 10.1021/jo500675a
-
[49]
Schmidt, B.; Riemer, M. Eur. J. Org. Chem. 2015, 17, 3760.
-
[50]
Al-Amin, M.; Akimoto, M.; Tameno, T.; Ohki, Y.; Takahashi, N.; Hoshiya, N.; Shuto, S.; Arisawa, M. Green Chem. 2013, 15, 1142. doi: 10.1039/c3gc00063j
-
[51]
Navarro, O.; Kaur, H.; Mahjoor, P.; Nolan, S. P. J. Org. Chem. 2004, 69, 3173. doi: 10.1021/jo035834p
-
[52]
Nun, P.; Martinez, J.; Lamaty, F. Synlett 2009, 1761.
-
[53]
Yılmaz, Ü.; Küçükbay, H.; Şireci, N.; Akkurt, M.; Günal, S.; Durmaz, R.; Tahir, M. N. Appl. Organomet. Chem. 2011, 25, 366. doi: 10.1002/aoc.v25.5
-
[54]
Miao, G. B.; Ye, P.; Yu, L. B.; Baldino, C. M. J. Org. Chem. 2005, 70, 2332. doi: 10.1021/jo047975c
-
[55]
Solodenko, W.; Schön, U.; Messinger, J.; Glinschert, A.; Kirschning, A. Synlett 2004, 1699.
-
[56]
Kopylovich, M. N.; Lasri, J.; Guedes da Silva, M. F. C.; Pombeiro, A. J. L. Dalton Trans. 2009, 16, 3074.
-
[57]
Cívicos, J. F.; Alonso, D. A.; Nájera, C. Adv. Synth. Catal. 2011, 353, 1683. doi: 10.1002/adsc.201100019
-
[58]
Dawood, K. M. Tetrahedron 2007, 63, 9642. doi: 10.1016/j.tet.2007.07.029
-
[59]
Kostas, I. D.; Heropoulos, G. A.; Kovala-Demertzi, D.; Yadav, P. N.; Jasinski, J. P.; Demertzis, M. A.; Andreadaki, F. J.; Vo-Thanh, G.; Petit, A.; Loupy, A. Tetrahedron Lett. 2006, 47, 4403. doi: 10.1016/j.tetlet.2006.04.088
-
[60]
Villemin, D.; Escalonilla, M. J. G.; Saint-Clair, J. F. Tetrahedron Lett. 2001, 42(4), 635. doi: 10.1016/S0040-4039(00)02026-8
-
[61]
Qu, G. R.; Xin, P. Y.; Niu, H. Y.; Jin, X.; Guo, X. T.; Yang, X. N.; Guo, H. M. Tetrahedron 2011, 67, 9099. doi: 10.1016/j.tet.2011.09.082
-
[62]
Zhang, W.; Chen, C. H. T.; Lu, Y. M.; Nagashima, T. Org. Lett. 2004, 6, 1473. doi: 10.1021/ol0496428
-
[63]
Cívicos, J. F.; Alonso, D. A.; Nájera, C. Adv. Synth. Catal. 2012, 354, 2771. doi: 10.1002/adsc.v354.14/15
-
[64]
Seganish, W. M.; DeShong, P. J. Org. Chem. 2004, 69, 1137. doi: 10.1021/jo035309q
-
[65]
Seganish, W. M.; DeShong, P. Org. Lett. 2004, 6, 4379. doi: 10.1021/ol048044q
-
[66]
Hadebe, S. W.; Sithebe, S.; Robinson, R. S. Tetrahedron 2011, 67, 4277. doi: 10.1016/j.tet.2011.03.095
-
[67]
Kabalka, G. W.; Al Masum, M. Tetrahedron Lett. 2005, 46, 6329. doi: 10.1016/j.tetlet.2005.07.036
-
[68]
Arvela, R. K.; Leadbeater, N. E.; Mack, T. L.; Kormos, C. M. Tetrahedron Lett. 2006, 47, 217. doi: 10.1016/j.tetlet.2005.10.153
-
[69]
Kabalka, G. W.; Zhou, L. L.; Naravane, A. Tetrahedron Lett. 2006, 47, 6887. doi: 10.1016/j.tetlet.2006.07.042
-
[70]
Harker, R. L.; Crouch, R. D. Synthesis 2007, 25.
-
[71]
Ahn, H. R.; Cho, Y. A.; Kim, D. S.; Chin, J. G.; Gyoung, Y. S.; Lee, S.; Kang, H.; Ham, J. Org. Lett. 2009, 11, 361. doi: 10.1021/ol8025999
-
[72]
Kim, D. S.; Ham, J. Org. Lett. 2010, 12, 1092. doi: 10.1021/ol100081v
-
[73]
Nehls, B. S.; Asawapirom, U.; Füldner, S.; Preis, E.; Farrell, T.; Scherf, U. Adv. Funct. Mater. 2004, 14, 352. doi: 10.1002/(ISSN)1616-3028
-
[74]
Nehls, B. S.; Fldner, S.; Preis, E.; Farrell, T.; Scherf, U. Macromolecules 2005, 38, 687. doi: 10.1021/ma048595w
-
[75]
Glasnov, T. N.; Stadlbauer, W.; Kappe, C. O. J. Org. Chem. 2005, 70, 3864. doi: 10.1021/jo0502549
-
[76]
Hashim, J.; Glasnov, T. N.; Kremsner, J. M.; Kappe, C. O. J. Org. Chem. 2006, 71, 1707. doi: 10.1021/jo052283p
-
[77]
Mathews, C. J.; Taylor, J.; Tyte, M. J.; Worthington, P. A. Synlett 2005, 538.
-
[78]
Song, Y. S.; Kim, B. T.; Heo, J. N. Tetrahedron Lett. 2005, 46, 5987. doi: 10.1016/j.tetlet.2005.07.027
-
[79]
Song, Y. S.; Lee, Y. J.; Kim, B. T.; Heo, J. N. Tetrahedron Lett. 2006, 47, 7427. doi: 10.1016/j.tetlet.2006.08.052
-
[80]
Heo, Y.; Song, Y. S.; Kim, B. T.; Heo, J. N. Tetrahedron Lett. 2006, 47, 3091. doi: 10.1016/j.tetlet.2006.02.152
-
[81]
Arimitsu, S.; Jacobsen, J. M.; Hammond, G. B. J. Org. Chem. 2008, 73, 2886. doi: 10.1021/jo800088y
-
[82]
Cao, P.; Qu, J. Y.; Burton, G.; Rivero, R. A. J. Org. Chem. 2008, 73, 7204. doi: 10.1021/jo801097v
-
[83]
Crozet, M. D.; Zink, L.; Remusat, V.; Curti, C.; Vanelle, P. Synthesis 2009, 3150.
-
[84]
Cohen, A.; Crozet, M. D.; Rathelot, P.; Vanelle, P. Green Chem. 2009, 11, 1736. doi: 10.1039/b916123f
-
[85]
St. Jean, Jr., D. J.; Poon, S. F.; Schwarzbach, J. L. Org. Lett. 2007, 9, 4893. doi: 10.1021/ol702274y
-
[86]
Vishnumurthy, K.; Makriyannis, A. J. Comb. Chem. 2010, 12, 664. doi: 10.1021/cc100068a
-
[87]
Kabri, Y.; Gellis, A.; Vanelle, P. Eur. J. Org. Chem. 2009, 4059.
-
[88]
Kabri, Y.; Crozet, M. D.; Terme, T.; Vanelle, P. Eur. J. Org. Chem. 2015, 3806.
-
[89]
Appukkuttan, P.; Dehaen, W.; Van der Eycken, E. Org. Lett. 2005, 7, 2723. doi: 10.1021/ol050806+
-
[90]
Appukkuttan, P.; Dehaen, W.; Van der Eycken, E. Chem. Eur. J. 2007, 13, 6452. doi: 10.1002/(ISSN)1521-3765
-
[91]
Lépine, R.; Zhu, J. Org. Lett. 2005, 7, 2981. doi: 10.1021/ol050949w
-
[92]
Wannberg, J.; Sabnis, Y. A.; Vrang, L.; Samuelsson, B.; Karlén, A.; Hallberg, A.; Larhed, M. Bioorg. Med. Chem. 2006, 14, 5303. doi: 10.1016/j.bmc.2006.03.045
-
[93]
Ng-Choi, I.; Soler, M.; Cerezo, V.; Badosa, E.; Montesinos, E.; Planas, M.; Feliu, L. Eur. J. Org. Chem. 2012, 4321.
-
[94]
Benali, O.; Deal, M.; Farrant, E.; Tapolczay, D.; Wheeler, R. Org. Process Res. Dev. 2008, 12, 1007. doi: 10.1021/op700225u
-
[95]
De Clercq, E. Nat. Rev. Drug Discovery2002, 1, 13. doi: 10.1038/nrd703
-
[96]
De Clercq, E. Antiviral Res. 2005, 67, 56. doi: 10.1016/j.antiviral.2005.05.001
-
[97]
Capek, P.; Pohl, R.; Hocek, M. Org. Biomol. Chem. 2006, 4, 2278. doi: 10.1039/B604010A
-
[98]
Western, E. C.; Shaughnessy, K. H. J. Org. Chem. 2005, 70, 6378. doi: 10.1021/jo050832l
-
[99]
Western, E. C.; Daft, J. F.; Johnson, E. M.; Gannett, P. M.; Shaughnessy, K. H. J. Org. Chem. 2003, 68, 6767. doi: 10.1021/jo034289p
-
[100]
Zhu, R.; Qu, F.; Quéléver, G.; Peng, L. Tetrahedron Lett. 2007, 48, 2389. doi: 10.1016/j.tetlet.2007.01.154
-
[101]
Gallagher-Duval, S.; Hervé, G.; Sartori, G.; Enderlin, G.; Len, C. New J. Chem. 2013, 37, 1989. doi: 10.1039/c3nj00174a
-
[102]
Lussier, T.; Hervé, G.; Enderlin, G.; Len, C. RSC Adv. 2014, 4, 46218. doi: 10.1039/C4RA04814H
-
[103]
Spencer, J.; Baltus, C. B.; Patel, H.; Press, N. J.; Callear, S. K.; Male, L.; Coles. S. J. ACS Comb. Sci. 2011, 13, 24. doi: 10.1021/co100011g
-
[104]
Arisawa, M.; Sato, T.; Hoshiya, N.; Al-Amin, M.; Kogami, Y.; Shuto, S. ACS Comb. Sci. 2014, 16, 215. doi: 10.1021/co4001138
-
[105]
Nakagawa, K.; Hisamatsu, Y.; Moromizato, S.; Kohno, M.; Aoki, S. Inorg. Chem. 2014, 53, 409. doi: 10.1021/ic402387b
-
[106]
Konda, V.; Rydfjord, J.; Sävmarker, J.; Larhed, M. Org. Process Res. Dev. 2014, 18, 1413. doi: 10.1021/op5001989
-
[107]
Baghbanzadeh, M.; Pilger, C.; Kappe, C. O. J. Org. Chem. 2011, 76, 1507;
(b)Singh, N.; Singh, R.; Raghuvanshi, D. S.; Singh, K. N. Org. Lett. 2013, 15, 5874;
(c) Singh, K. B.; Appukkuttan, P.; Claerhout, S.; Parmar, V. S.; Der Eycken, E. V. Org. Lett. 2006, 8, 1863.
-
[1]
-

图式1 金属钯催化的Suzuki-Miyaura交叉偶联反应机理
Scheme 1 Catalytic cycle for the Suzuki-Miyaura cross-coup-ling

图式2 微波辅助Pd(OAc)2催化PEG支载的芳(杂环)卤化物与芳基硼酸的Suzuki-Miyaura交叉偶联反应
Scheme 2 Microwave-mediated Suzuki-Miyaura cross-coup-ling of PEG supported (hetero)aryl halide with arylboronic acid using Pd(OAc)2

图式3 微波辅助PS-PPh3PdCl2催化Suzuki-Miyaura交叉偶联反应
Scheme 3 Suzuki-Miyaura cross-coupling reactions catalyzed by PS-PPh3PdCl2 under microwave irradiation

图式4 微波辅助SAPd催化芳基溴和碘与芳基硼酸的Suzuki-Miyaura交叉偶联反应.
Scheme 4 Suzuki-Miyaura coupling reactions of aryl bromides and iodoium with arylboronic acid catalyzed by SAPd under microwave irraditions

图式5 微波辅助催化剂18催化芳基氯与芳基硼酸的Suzuki-Miyaura交叉偶联反应
Scheme 5 Suzuki-Miyaura cross-coupling of alkylboronic acid with arylhalides using catalyst 18 under microwave irradiation

图式6 微波辅助芳基三氟硼钾的Suzuki-Miyaura交叉偶联反应
Scheme 6 Microwave-accelerated Suzuki-Miyaura cross-coupling reactions using potassium arytrifluoroborates

图式7 微波辅助芳基三氟硼钾的Suzuki-Miyaura交叉偶联反应
Scheme 7 Microwave-accelerated Suzuki-Miyaura cross-coupl-ing reactions using potassium arytrifluoroborates

图式8 微波辅助芳基硼酸的Suzuki-Miyaura交叉偶联反应
Scheme 8 Microwave-accelerated Suzuki-Miyaura cross-coupl-ing reactions using arylboronic acid

图式9 微波辅助芳基硼酸的Suzuki-Miyaura交叉偶联反应
Scheme 9 Microwave-accelerated Suzuki-Miyaura cross-coupl-ing reactions using arylboronic acid

图式10 微波辅助2, 3, 5-三取代-3-氟呋喃的合成
Scheme 10 Synthesis of 2, 3, 5-trisubstituted-3-fluorofurans under microwave irradiation

图式11 微波辅助一锅法化学选择性三Suzuki-Miyaura交叉偶联反应或芳基取代和双Suzuki-Miyaura交叉偶联反应合成2, 6, 8-三取代-4-氨基奎唑啉
Scheme 11 2, 6, 8-Trisubstituted 4-aminoquinazolines through microwave-assisted one-pot chemoselective tris-Suzuki-Miyaura or SNAr/bis-Suzuki-Miyaura reactions

图式12 微波辅助buflavine类似物的合成
Scheme 12 Microwave-assisted synthesis of N-shifted buflavine analogues

图式13 微波辅助buflavine类似物的合成
Scheme 13 Microwave-assisted synthesis of N-shifted buflavine analogues
-


 下载:
下载:














 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 4199
- HTML全文浏览量: 460
 下载:
下载:
