Received Date:
29 July 2020 Available Online:
15 December 2020
Fund Project:
Project supported by the National Natural Science Foundation of China(Nos.21931001, 21922105), the lnnovation Platform of Rare-earth Funcional Maeri.als of Gansu Province (No.2019ZX-04) and the 111 Project (No.B20027)
Abstract:
The large-scale use of coal, oil, and natural gas will cause environmental pollution and resource shortages, which is incompatible with the sustainable development. Therefore, it is essential to the development of renewable energy. H2 has a high heat of combustion, and it's combustion products do not include greenhouse gases, so it is considered as an ideal clean energy carrier. Industrial hydrogen production methods will bring CO2 inevitably. As an emerging energy conversion device, hydrogen produced by water splitting has simple equipment and little pollution, making it the first choice for clean energy in the future. Generally, the adsorption energy of hydrogen on the surface of precious metal catalysts is close to zero, and its hydrogen evolution reaction (HER) performance is the most prominent. Pt is an excellent HER catalyst. Commercial Pt/C has high alkaline HER performance, but its higher cost, material instability and resource scarcity limit its widespread applications. Therefore, this research is devoted to the development of high-activity, low-cost transition metal catalysts for HER by water splitting. Firstly, CoMoO4 nanorods were synthesized by hydrothermal method. Then, using the precursor morphology oriented strategy method, the activated Co9S8/MoS2 heterostructure catalyst was successfully prepared by sulfurating CoMoO4 into CoS2/MoS2 and further, calcining CoS2/MoS2 nanorod in hydrogen atmosphere. X-rays diffraction (XRD), transmission electron microscopy (TEM), electron spin resonance (ESR), Raman spectra, X-ray photoelectron spectra (XPS) and synchrotron-based X-ray absorption fine structure (XAFS) characterizations exhibit that the Co coordination mode change from octahedron in CoS2 to tetrahedron in Co9S8, leading to the activation of inert basal plane in MoS2. Owing to this activation, the interlayer spacing of MoS2 is reduced and thus generate abundant defects. Meanwhile, the increased electrochemical surface area (ECSA) and roughness of the catalysts are more conducive to the adsorption of H*. The test of the contact angle data show that the electrode has good hydrophilicity, which can facilitate the penetration of electrolyte and diffusion of gas molecules quickly. When the current density is at 10 mA·cm-2 in 1 mol·L-1 KOH solution, an overpotential of 84 mV and a Tafel slope of 93 mV·dec-1 can be achieved. Due to the strong interaction between different components of the heterostructures, the nanorods possess good structural stability in alkaline solutions. This work highlights the vital role of the sulfides heterostructure construction in HER, opening a new way to advanced alkaline HER catalysts.
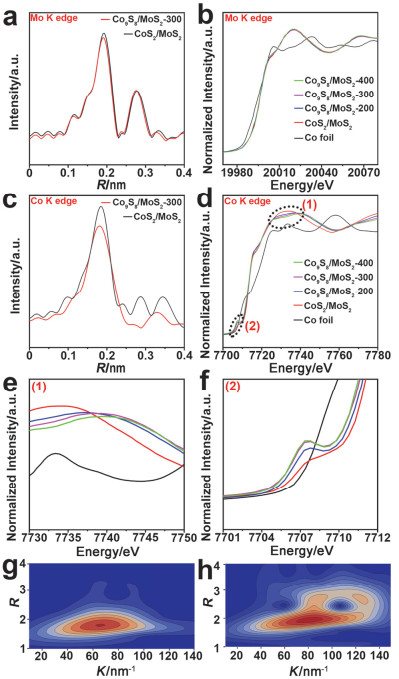
图 4.
Co9S8/MoS2-300和CoS2/MoS2在R空间(a) Mo的K-edge和(c) Co的K-edge的傅里叶转换EXAFS光谱. Co9S8/MoS2-X在(b) Mo的K-edge和(d) Co的K-edge的XANES光谱. (e)和(f)为(d)的XANES高分辨谱图. Co9S8/MoS2-300的(g) Co K-edge和(h) Mo K-edge的EXAFS光谱小波转换图
Figure 4.
Fourier-transformed EXAFS spectra of Co9S8/MoS2-300 and CoS2/MoS2 in R-space at (a) Mo K-edge and (c) Co K-edge. The XANES spectra of Co9S8/MoS2-X at (b) Mo K-edge and (d) Co K-edge. (e) and (f) are the high resolution of XANES spectra in (d). Wavelet transforms for (g) Co K-edge and (h) Mo K-edge EXAFS signals of Co9S8/MoS2-300
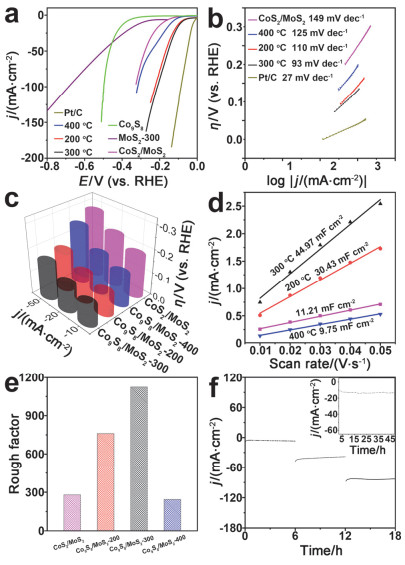
Figure 5.
(a) LSV, (b) Tafel, (c) summary of HER overpotentials in different current densities, (d) Cdl images and (e) Rf of Co9S8/MoS2-X and controls. (f) i-t curves of Co9S8/MoS2-300 recorded at current density of 10, 50 and 100 mA•cm-2 for 6 h. Insert is i-t curve of Co9S8/MoS2-300 recorded at current density of 10 mA•cm-2 for 48 h
黄刚, 陈玉贞, 江海龙, 化学学报, 2016, 74, 113.Huang, G.; Chen, Y. Z.; Jiang, H. L. Acta Chim. Sinica2016, 74, 113(in Chinese).
[5]
宋芳源, 丁勇, 赵崇超, 化学学报, 2014, 72, 133.Song, F. Y.; Ding, Y.; Zhao, C. C. Acta Chim. Sinica2014, 72, 133(in Chinese).
[6]
Staszak-Jirkovský, J.; Malliakas, C. D.; Lopes, P. P.; Danilovic. N.; Kota, S. S.; Chang, K.-C.; Genorio, B.; Strmcnik, D.; Stamenkovic, V. R.; Kanatzidis, M. G.; Markovic, N. M. Nat. Mater.2016, 15, 197.
Podjaski, F.; Weber, D.; Zhang, S. Y.; Diehl, L.; Eger, R.; Duppel, V.; Alarcón-Lladó, E.; Richter, G.; Haase, F.; Morral, A. F. i.; Scheu, C.; Lotsch, B. V. Nat. Catal.2019, 3, 55.
[11]
Men, Y. N.; Li, P.; Zhou, J. H.; Cheng, G. Z.; Chen, S. L.; Luo, W. ACS Catal.2019, 9, 3744.
[12]
Qi, K.; Cui, X. Q.; Gu, L.; Yu, S. S.; Fan, X. F.; Luo, M. C.; Xu, S.; Li, N. B.; Zheng, L. R.; Zhang, Q. H.; Ma, J. Y.; Gong, Y.; Lv, F.; Wang, K.; Huang, H. H.; Zhang, W.; Guo, S. J.; Zheng, W. T.; Liu, P. Nat. Commun.2019, 10, 5231.
[13]
Li, H.; Tsai, C.; Koh, A. L.; Cai, L. L.; Contryman, A. W.; Fragapane, A. H.; Zhao, J. H.; Han, H. S.; Manoharan, H. C.; Abild-Pedersen, F.; Nørskov, J. K.; Zheng, X. L. Nat. Mater.2016, 15, 48.
[14]
Li, G. Q.; Chen, Z. H.; Li, Y. F.; Zhang, D.; Yang, W. T.; Liu, Y. Y.; Cao, L. Y. ACS Nano2020, 14, 1707.
[15]
Lu, Q.; Yu, J.; Zou, X. H.; Liao, K. M.; Tan, P.; Zhou, W.; Ni, M.; Shao, Z. P. Adv. Funct. Mater.2019, 29, 1904481.
[16]
Wu, H. B.; Xia, B. Y.; Yu, L.; Yu, X.-Y.; Lou, X. W. Nat. Commun. 2015, 6, 6512.
[17]
Mak, K. F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T. F. Phys.Rev.Lett. 2010, 105, 136805.
[18]
Smith, R. J.; King, P. J.; Lotya, M.; Wirtz, C.; Khan, U.; De, S.; O'Neill, A.; Duesberg, G. S.; Grunlan, J. C.; Moriarty, G.; Chen, J.; Wang, J. Z.; Minett, A. I.; Nicolosi, V.; Coleman, J. N. Adv.Mater. 2011, 23, 3944.
[19]
Xiong, Q. Z.; Wang, Y.; Liu, P.-F.; Zheng, L.-R.; Wang, G. Z.; Yang, H.-G.; Wong, P.-K.; Zhang, H. M.; Zhao, H. J. Adv. Mater.2018, 30, 1801450.
Xie, J. F.; Zhang, H.; Li, S.; Wang, R. X.; Sun, X.; Zhou, M.; Zhou, J. F.; Lou, X. W.; Xie, Y. Adv. Mater.2013, 25, 5807.
[22]
Cai, L.; He, J. F.; Liu, Q. H.; Yao, T.; Chen, L.; Yan, W. S.; Hu, F. C.; Jiang, Y.; Zhao, Y. D.; Hu, T. D.; Sun, Z. H.; Wei, S. Q. J. Am. Chem. Soc.2015, 137, 2622.
[23]
Toh, R. J.; Sofer, Z.; Luxa, J.; Sedmidubsky, D.; Pumera, M. Chem. Commun.2017, 53, 3054.
[24]
Wang, S.; Zhang, D.; Li, B.; Zhang, C.; Du, Z. G.; Yin, H. M.; Bi, X. F.; Yang, S. B. Adv. Energy Mater.2018, 8, 1801345.
[25]
Chen, Y. P.; Wang, X. Y.; Lao, M. M.; Rui, K.; Zheng, X. B.; Yu, H. B.; Ma, J.; Dou, S. X.; Sun, W. P. Nano Energy2019, 64, 103918.
[26]
Li, Y. G.; Wang, H. L.; Xie, L. M.; Liang, Y. Y.; Hong, G. S.; Dai, H. J. J.Am.Chem.Soc. 2011, 133, 7296.
[27]
Uddin, N.; Zhang, H. Y.; Du, Y. P.; Jia, G. H.; Wang, S. B.; Yin, Z. Y. Adv. Mater.2020, 32, 1905739.
[28]
Hinnemann, B.; Moses, P. G.; Bonde, J.; Jørgensen, K. P.; Nielsen, J. H.; Horch, S.; Chorkendorff, I.; Nørskov, J. K. J. Am. Chem. Soc.2005, 127, 5308.
[29]
Jaramillo, T. F.; Jørgensen, K. P.; Bonde, J.; Nielsen, J. H.; Horch, S.; Chorkendorff, I. Science 2007, 317, 100.
[30]
Yin, J.; Li, Y. X.; Lv, F.; Lu, M.; Sun, K.; Wang, W.; Wang, L.; Cheng, F. Y.; Li, Y. F.; Xi, P. X.; Guo, S. J. Adv. Mater.2017, 29, 1704681.
[31]
An, L.; Li, Y. X.; Luo, M. C.; Yin, J.; Zhao, Y.-Q.; Xu, C. L.; Cheng, F. Y.; Yang, Y.; Xi, P. X.; Guo, S. J. Adv. Funct. Mater.2017, 27, 1703779.
[32]
Yin, J.; Li, Y. X.; Lv, F.; Fan, Q. H.; Zhao, Y.-Q.; Zhang, Q. L.; Wang, W.; Cheng, F. Y.; Xi, P. X.; Guo, S. J. ACS Nano2017, 11, 2275.
[33]
Zhang, H. B.; Yu, L.; Chen, T.; Zhou, W.; Lou, X. W. Adv. Funct. Mater.2018, 28, 1807086.
[34]
Deng, J.; Li, H. B.; Wang, S. H.; Ding, D.; Chen, M. S.; Liu, C.; Tian, Z. Q.; Novoselov, K. S.; Ma, C.; Deng, D. H.; Bao, X. H. Nat. Commun. 2017, 8, 14430.
[35]
Yang, R.; An, L.; Zhang, Y.; Zhang, N.; Dai, T. Y.; Xi, P. X. ChemCatChem2019, 11, 6002.
[36]
Cai, Z.; Li, L. D.; Zhang, Y. W.; Zhao, Y.; Yang, J.; Guo. Y. J.; Guo, L. Angew. Chem. Int. Ed.2019, 58, 4189.
[37]
Bai, J. M.; Meng, T.; Guo, D. L.; Wang, S. G.; Mao, B. G.; Cao, M. H. ACS Appl. Mater. Interfaces2018, 10, 1678.
图 4
Co9S8/MoS2-300和CoS2/MoS2在R空间(a) Mo的K-edge和(c) Co的K-edge的傅里叶转换EXAFS光谱. Co9S8/MoS2-X在(b) Mo的K-edge和(d) Co的K-edge的XANES光谱. (e)和(f)为(d)的XANES高分辨谱图. Co9S8/MoS2-300的(g) Co K-edge和(h) Mo K-edge的EXAFS光谱小波转换图
Figure 4
Fourier-transformed EXAFS spectra of Co9S8/MoS2-300 and CoS2/MoS2 in R-space at (a) Mo K-edge and (c) Co K-edge. The XANES spectra of Co9S8/MoS2-X at (b) Mo K-edge and (d) Co K-edge. (e) and (f) are the high resolution of XANES spectra in (d). Wavelet transforms for (g) Co K-edge and (h) Mo K-edge EXAFS signals of Co9S8/MoS2-300
Figure 5
(a) LSV, (b) Tafel, (c) summary of HER overpotentials in different current densities, (d) Cdl images and (e) Rf of Co9S8/MoS2-X and controls. (f) i-t curves of Co9S8/MoS2-300 recorded at current density of 10, 50 and 100 mA•cm-2 for 6 h. Insert is i-t curve of Co9S8/MoS2-300 recorded at current density of 10 mA•cm-2 for 48 h

 下载:
下载:


 下载:
下载:






