Received Date:
12 July 2020 Available Online:
15 December 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21973069, 21773169, 21872103), National Key R & D Program (Nos. 2017YFA0204503, 2016YFB0401100), the PEIYANG Young Scholars Program of Tianjin University (No. 2018XRX-0007) and the College Student Innovation and Entrepreneurship Training Program of Tianjin University (No. 201910056451)
Abstract:
Artificial intelligence (AI), especially the machine learning, is playing an increasingly important role in contemporary scientific research. Unlike the traditional computer program, machine learning can analyze a large number of data repeatedly and optimize its own model, a process which is called a "learning process". So that the AI can find the relationship underling the experiments from a large number of data, form a new model with better prediction and decisionmaking ability, and make an optimized strategy. The characteristics of chemical research just hit the strengths of machine learning. Chemical research often faces very complex material system and experimental process, so it is difficult to accurately analyze and making judgment through physical chemistry principles. Artificial intelligence can mine the correlation of massive experimental data generated in chemical experiments, help chemists make reasonable analysis and prediction, and therefore greatly accelerate the process of chemical research. This review presents the modern artificial intelligence method and its basic principles on solving chemical problems, by representative examples with specific machine learning algorithm. The application of artificial intelligence in chemical science is in a period of vigorous rise. Artificial intelligence has initially shown a powerful assist to chemical research. We hope this review can help more domestic chemical workers understand and use this powerful tool.
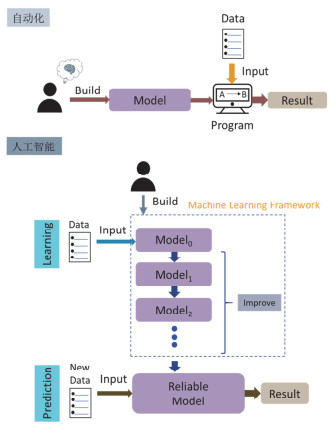
Figure 1.
The development of artificial intelligence concept. The biggest difference between artificial intelligence and automation is the source of the program: the automation program is a fixed model manually set by human, which responds to specific inputs; the artificial intelligence program is a model that can mine laws from existing data and conduct self-learning to improve itself, generate reliable model automatically, this model can also correctly respond to unexpected inputs, and has the ability to predict and analyze new situations.
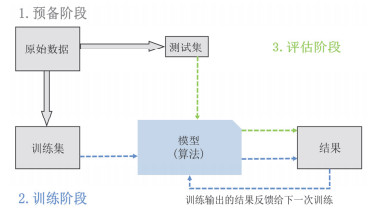
Figure 2.
The machine learning process. (1) Preprocessing: Divide the raw data into a training set and a test set, the training set serves as the model's "learning material", and the test set verifies the model's ability to respond to new situations. (2) Training: Build the structure of training set to model to test set. Adjust the optimization parameters of the algorithm inside the model and iterate multiple times until the model output meets the expected value. (3) Evaluation: Use the test set to evaluate the expressive ability of the model.
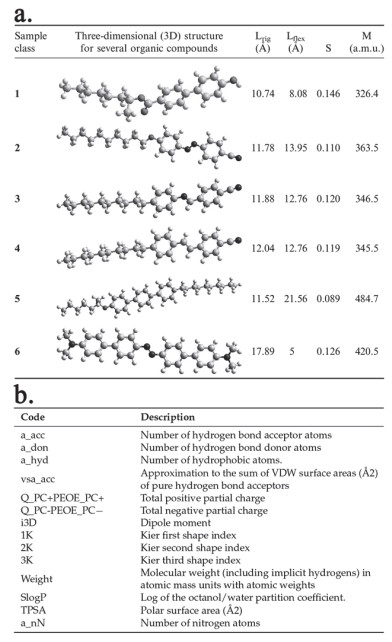
Figure 3.
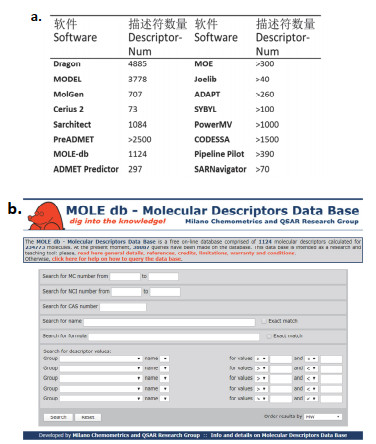
(a) Values of 3D molecular descriptors. (b) Molecular descriptors calculated with MOE[4, 19]. Reprinted with permission from ref. [19]. Copyright [MDPI, Basel, Switzerland].
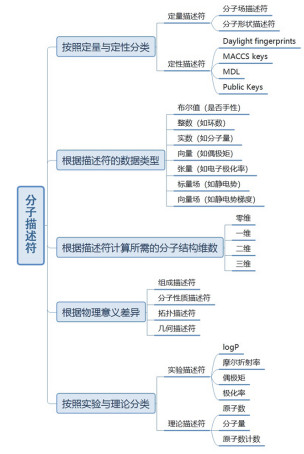
Figure 4.
Rough classification of molecular descriptors. The qualitative descriptor is also called molecular fingerprint, by using a certain code to represent the molecular structure, property, fragment or substructure information[21].
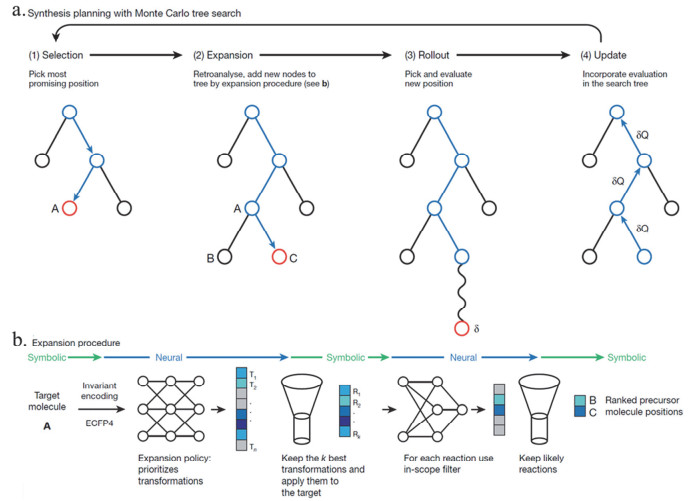
Figure 14.
Synthesis planning with 3N-MCTS. (a) MCTS searches by iterating over four phases. In the selection phase (1), the most urgent node for analysis is chosen on the basis of the current position values. In phase (2) this node may be expanded by processing the molecules of the position A with the expansion procedure (b), which leads to new positions B and C, which are added to the tree. Then, the most promising new position is chosen, and a rollout phase (3) is performed by randomly sampling transformations from the rollout policy until all molecules are solved or a certain depth is exceeded. In the update phase (4), the position values are updated in the current branch to reflect the result of the rollout. (b) Expansion procedure. First, the molecule A to retroanalyse is converted to a fingerprint and fed into the policy network, which returns a probability distribution over all possible transformations (T1 to Tn). Then, only the k most probable transformations are applied to molecule A. This yields the reactants necessary to make A, and thus complete reactions R1 to Rk. For each reaction, the reaction prediction is performed using the in-scope filter, returning a probablity score. Improbable reactions are then filtered out, which leads to the list of admissible actions and corresponding precursor positions B and C[40].
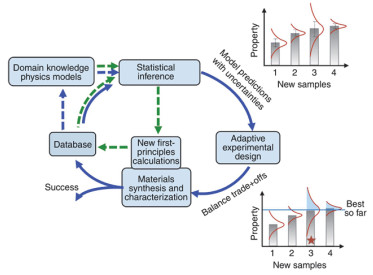
Figure 16.
Closed research route designed by Dezhen Xue et al. Describe materials with data from previous experiments and physical models and related features. This part of the information is input into the machine learning framework for prediction, and the global optimization method is used to optimize the machine learning model. Carrying out new synthetic characterization experiments based on the model prediction results has the dual purpose of improving the model and discovering new materials. The above results are used to update the database. The updated database provides input for the next cycle[49].
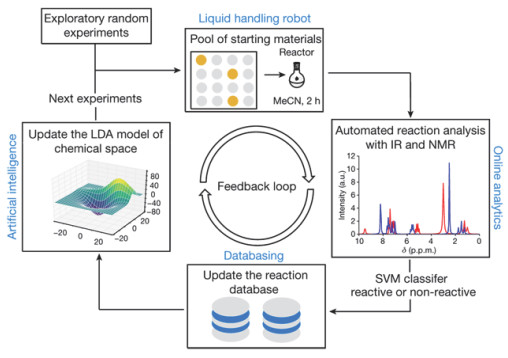
Figure 17.
Experimental process of combining liquid handling robot and artificial intelligence to explore chemical space. The liquid-handling robot performs reactions by choosing reactants from the pool of starting materials. Online analytics is used for real-time interpretation of reaction outcomes as reactive or non-reactive, and the reaction database stores reaction outcomes. Machine learning is used to build a model of the chemical space, recommend the next experiments and control the robot[49].
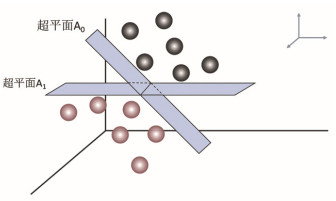
Figure 18.
Schematic diagram of data classification by SVM when the given data is linearly separable. The minimum distance of hyperplane A0 from two groups of points > the minimum distance of hyperplane A1 from two groups of points, so hyperplane P0 is a hyperplane with better classification function.
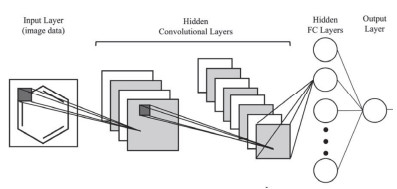
Figure 23.
Schematic diagram of convolutional neural network (CNN). CNN requires the input to be in the form of image data. Each convolutional layer extracts and retains spatial information, and learns a representation, which is then passed to the traditional fully connected feedforward neural network before the output layer[76].
唐振韬, 邵坤, 赵冬斌, 朱圆恒. 控制理论与应用, 2017, 034, 1529. doi: 10.6023/A19020057Tang, Z. T.; Shao, K.; Zhao, D. B.; Zhu, Y. H. Control Theory & Applications2017, 034, 1529 (in Chinese). doi: 10.6023/A19020057
[2]
McKinney, S. M.; Sieniek, M.; Godbole, V.; Godwin, J.; Antropova, N.; Ashrafian, H.; Back, T.; Chesus, M.; Corrado, G. C.; Darzi, A.; Etemadi, M.; Garcia-Vicente F.; Gilbert, F. J.; Halling-Brown, M.; Hassabis, D.; Jansen, S.; Karthikesalingam, A.; Kelly, C. J.; King, D.; Ledsam, J.R.; Melnick, D.; Mostofi, H.; Peng, L.; Reicher, J. J.; Romera-Paredes, B.; Sidebottom, R.; Suleyman, M.; Tse, D.; Young, K. C.; De, Fauw, J.; Shetty, S. Nature2020, 577, 7788.
[3]
Li, J. G.; Gao, Z. K. Acta Biophysica Sinica2009, 25, 51 (in Chinese). (李建更, 高志坤, 生物物理学报, 2009, 25, 51.)
[4]
Leon, F.; Lisa, C.; Curteanu, S. Mol. Cryst. Liq. Cryst.2010, 518, 1542.
Zhong, M.; Tran, K.; Min, Y. M.; Wang, C. H.; Wang, Z. Y.; Ding, C. T.; Luna, P.; Sedighian Rasouli, A.; Brodersen, P.; Sun, S.; Voznyy, O.; Tan, C. S.; Askerka, M.; Che, F. L.; Liu, M.; Seifitokaldani, A.; Pang, Y. J.; Lo, S. C.; Sargent, E. Nature 2020, 581, 178.
Saunders, C.; Stitson, M. O.; Weston, J.; Holloway, R.; Bottou, L.; Scholkopf, B. Comput. Sci.2002, 1, 1.
[10]
Safavian, S. R.; Landgrebe, D. IEEE Trans. Syst., Man, Cybern.1991, 21, 660.
[11]
Hagan, M. T.; Demuth, H. B.; Beale, M. H. Neural Network Design, China Machine Press, Beijing, 2002.
[12]
Browne, C. B.; Powley, E.; Whitehouse, D.; Lucas, S. M.; Cowling, P.I. IEEE Transactions on Computational Intelligence & Ai in Games, 2012, 4, 1.
[13]
Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatic —Second, Revised and Enlarged Edition, Volume I: Alphabetical Listing; Volume Ⅱ: Appendices, Bibliography, 2009.
[14]
Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors, WILEY-VCH, Weinheim, Germany, 2000.
[15]
何冰, 罗勇, 李秉轲, 薛英, 余洛汀, 邱小龙, 杨登贵, 物理化学学报, 2015, 09, 1795.He, B.; Luo, Y.; Li, B. K.; Xue, Y.; Yu, L. T.; Qiu, X. L.; Yang, D. G. Acta Physico-Chimica Sinica2015, 09, 1795 (in Chinese).
[16]
王洁雪, 李瑶, 杨敏, 王琪慧, 邓国伟, 杨帆, 李秉轲. 化学研究与应用, 2019, 031, 1313.Wang, J. X.; Li, Y.; Yang, M.; Wang, Q. H.; Deng, G. W.; Yang, F.; Li, B. K. Chemical Research & Application2019, 031, 1313 (in Chinese).
[17]
王璐, 毛海涛, 张磊, 刘琳琳, 都健, 化工学报, 2019, 70, 4722.Wang, L.; Mao, H. T.; Zhang, L.; Liu, L. L.; Du, J. CIESC J. 2019, 70, 4722 (in Chinese).
[18]
Dai, Y.; Niu, L.; Zou, J.; Liu, D. Y.; Liu, H. J. Cent. South Univ.2018, 25, 1535.
[19]
Ul-Haq, Z.; Ashraf, S.; Al Majid, A.; Barakat, A. Int. J. Mol. Sci. 2016, 17, 657.
[20]
徐优俊, 裴剑锋, 大数据, 2017, 003, 45.Xu, Y. J.; Pei, J. F. Big Data Research2017, 003, 45 (in Chinese).
Mauri, A.; Consonni, V.; Todeschini, R. Molecular Descriptors, Vol. 8, Eds.: Puzyn, T.; Leszczynski, J.; Cronin, M. T. D., Springer, New York, 2009, p. 33.
[23]
Mauri, A.; Consonni, V.; Todeschini, R. Molecular Descriptors, Vol. 8, Eds.: Puzyn, T.; Leszczynski, J.; Cronin, M. T. D., Springer, New York, 2009, p. 34.
[24]
Ren, W.; Kong, D. X. Computers & Applied Chemistry, 2009, 11, 1455 (in Chinese). (任伟, 孔德信. 计算机与应用化学, 2009, 11, 1455.)
[25]
Dickert, F. L.; Hayden, O. Adv. Mater. 2000, 12, 311.
Cova, Tnia F. G. G.; Pais, Alberto A. C. C. Front. Chem.2019, 7, 809.
[33]
Jordan, M. I.; Mitchell, T. M. Science2015, 349, 6245.
[34]
McCulloch, W. S.; Pitts, W. Bull. Math. Biol.1943, 52.
[35]
Gall, J.; Razavi, N.; Van Gool, L. An Introduction to Random Forests for Multi-class Object Detection, Springer-Verlag, Heidelberg, Germany, 2012, pp. 243-263.
[36]
Lim, A.; Breiman, L.; Cutler, A. Computer Science2014 (data package and software).
[37]
Ahneman, D. T.; Estrada, J. G.; Lin, S. S.; Dreher, S. D.; Doyle, A. G. Science2018, 360, 6385.
[38]
Ghosh, A. K.; Feng, T. J. Appl. Phys.1973, 44, 2781.
Segler, M. H. S.; Preuss, M.; Waller, M. P. Nature 2018, 555, 7698.
[41]
Yu, Y. B. M.S. Thesis, Dalian Maritime University, Dalian, 2015 (in Chinese). (于永波, 硕士论文, 大连海事大学, 大连, 2015.)
[42]
Fu, M. C. In 2016 Winter Simulation Conference, Arlington Virginia, 2016, pp. 659-670.
[43]
Xue, Y.; Li, H.; Ung, C. Y.; Yap, C. W.; Chen, Y. Z. Chem. Res. Toxicol.2006, 19, 1030.
[44]
Lü, W. J.; Chen, Y. L.; Ma, W. P.; Zhang, X. Y.; Luan, F.; Liu, M. C.; Chen, X. G.; Hu, Z. D. Eur. J. Med. Chem. 2008, 43, 569.
[45]
Lü, W.; Xue, Y. Acta Phys.-Chim. Sin.2010, 26, 471.
[46]
Li, B. K.; Yong, C.; Yang, X. G; Xue, Y.; Chen, Y. Z. Comput. Biol. Med.43, 395.
[47]
Li, B. K.; Cong Y.; Tian, Z. Y.; Xue, Y. Acta Physico-Chimica Sinica2014, 30, 171 (in Chinese). (李秉轲, 丛湧, 田之悦, 薛英, 物理化学学报, 2014, 30, 171.)
[48]
Barta, T. E.; Becker, D. P.; Bedell, L. J.; Crescenzo, G. A. D.; McDonald, J. J.; Mehta, P.; Munie, G. E.; Villamil, C. I. Bioorg. Med. Chem. Lett.2001, 11, 2481.
[49]
Xue, D. Z.; Balachandran, P. V.; Hogden, J.; Theiler, J.; Xue, D. Q.; Lookman, T. Nat. Commun.2016, 7, 11241.
[50]
Granda, J. M.; Donina, L.; Dragone, V.; Long, D. L.; Cronin, L. Nature2018, 559, 7714.
[51]
Ding, S. F.; Qi, B. J.; Tan, H. Y. Journal of University of Electronic Science and Technology of China, 2011, 40, 1 (in Chinese). (丁世飞, 齐丙娟, 谭红艳, 电子科技大学学报, 2011, 40, 1.)
[52]
Burges, C. J. C. A Tutorial on Support Vector Machines for Pattern Recognition. Data Min. Knowl. Discov. 1998, 2, 121.
[53]
Qi, H. N. Computer Engineering 2004, 30, 10 (in Chinese). (祁亨年, 计算机工程, 2004, 30, 6.)
[54]
Zhang, X. W.; Guo, L. Firepower & Command Control2010, 35, 31 (in Chinese). (张先武, 郭雷, 火力与指挥控制, 2010, 35, 31.)
[55]
Wang, J. F.; Cao, Y. D. Journal of Beijing Institute of Technology2001, 21, 225 (in Chinese). (王建芬, 曹元大, 北京理工大学学报, 2001, 21, 225.)
[56]
Zhang, Q. Y.; Jie, Y.; Li, K. Journal of Computer Applications2008, 28, 3227 (in Chinese). (张秋余, 竭洋, 李凯, 计算机应用, 2008, 28, 3227.)
[57]
Butler, K. T.; Davies, D. W.; Cartwright, H.; Isayev, O.; Walsh, A. Nature2018, 559, 547.
[58]
Schütt, K. T.; Gastegger, M.; Tkatchenko, A.; Müller, K. R.; Maurer, R. J. Nat. Commun.2019, 10, 1.
[59]
Ye, S.; Hu, W.; Li, X.; Zhang, J. X.; Zhong, K.; Zhang, G. Z.; Luo, Y.; Mukamel, S.; Jiang, J. Proc. Natl. Acad. Sci. U. S. A.2019, 116, 11612.
[60]
Grisafi, A.; Wilkins, D. M.; Csányi, G.; Ceriotti, M. Phys. Rev. Lett.2018, 120, 036002.
Goh, G. B.; Hodas, N. O.; Vishnu, A. J. Comput. Chem.2017, 38, 1291.
[77]
Sun, Y. Z. M.S. Thesis, China Medical University, Shengyang, 2009 (in Chinese). (孙也之, 硕士论文, 中国医科大学, 沈阳, 2009.)
[78]
Lusci, A.; Pollastri, G.; Baldi, P. J. Chem. Inf. Model.2013, 53, 1563.
[79]
Markoff, J. New York Times, 2012, 10, pp. 1-71.
[80]
Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Front Environ. Sci. Eng.2016, 3, 80.
[81]
Duvenaud, D.; Dougal, M.; Jorge, A. I.; Rafa, G. B.; Timothy, H.; Alán, A. G.; Ryan, P. A. In Proceedings of Advances in Neural Information Processing Systems 28, MIT Press, Montreal, 2015, pp. 2215-2223.
[82]
Kanal, L. N.; Randall, N. C. Proceedings of the 1964 19th ACM National Conference, Association for Computing Machinery, New York, NY, USA, 1964, pp. 42.501-42.5020.
[83]
Viola, J.; Snow, D.; Jones, M. J. In Proceedings Ninth IEEE International Conference on Computer Vision, Springer-Verlag, Nice, 2003, pp. 734-741.
[84]
Riley, P. Nature2019, 572, 27.
[85]
Baltz, E. A.; Trask, E.; Binderbauer, M.; Dikovsky, M.; Gota, H.; Mendoza, R.; Platt, J. C.; Riley, P. F. Sci. Rep.2017, 7, 6425.
Yosinski, J.; Clune, J.; Bengio, Y.; Lipson, H. In International Conference on Neural Information Processing Systems, MIT Press, Siem Reap, 2014, p. 32.
de Pablo, J. J.; Jackson, N. E.; Webb, M. A.; Chen, L. Q.; Moore, J. E.; Morgan, D.; Jacobs, R.; Pollock, T.; Schlom, D. G.; Toberer, E. S.; Analytis, J.; Dabo, I.; DeLongchamp, D. M.; Fiete, G. A.; Grason, G. M.; Hautier, G.; Mo, Y.; Rajan, K.; Reed, E. J.; Zhao, J. C. npj Comput. Mater.2019, 5, 41.
Figure 1
The development of artificial intelligence concept. The biggest difference between artificial intelligence and automation is the source of the program: the automation program is a fixed model manually set by human, which responds to specific inputs; the artificial intelligence program is a model that can mine laws from existing data and conduct self-learning to improve itself, generate reliable model automatically, this model can also correctly respond to unexpected inputs, and has the ability to predict and analyze new situations.
Figure 2
The machine learning process. (1) Preprocessing: Divide the raw data into a training set and a test set, the training set serves as the model's "learning material", and the test set verifies the model's ability to respond to new situations. (2) Training: Build the structure of training set to model to test set. Adjust the optimization parameters of the algorithm inside the model and iterate multiple times until the model output meets the expected value. (3) Evaluation: Use the test set to evaluate the expressive ability of the model.
Figure 3
(a) Values of 3D molecular descriptors. (b) Molecular descriptors calculated with MOE[4, 19]. Reprinted with permission from ref. [19]. Copyright [MDPI, Basel, Switzerland].
Figure 4
Rough classification of molecular descriptors. The qualitative descriptor is also called molecular fingerprint, by using a certain code to represent the molecular structure, property, fragment or substructure information[21].
Figure 14
Synthesis planning with 3N-MCTS. (a) MCTS searches by iterating over four phases. In the selection phase (1), the most urgent node for analysis is chosen on the basis of the current position values. In phase (2) this node may be expanded by processing the molecules of the position A with the expansion procedure (b), which leads to new positions B and C, which are added to the tree. Then, the most promising new position is chosen, and a rollout phase (3) is performed by randomly sampling transformations from the rollout policy until all molecules are solved or a certain depth is exceeded. In the update phase (4), the position values are updated in the current branch to reflect the result of the rollout. (b) Expansion procedure. First, the molecule A to retroanalyse is converted to a fingerprint and fed into the policy network, which returns a probability distribution over all possible transformations (T1 to Tn). Then, only the k most probable transformations are applied to molecule A. This yields the reactants necessary to make A, and thus complete reactions R1 to Rk. For each reaction, the reaction prediction is performed using the in-scope filter, returning a probablity score. Improbable reactions are then filtered out, which leads to the list of admissible actions and corresponding precursor positions B and C[40].
Figure 16
Closed research route designed by Dezhen Xue et al. Describe materials with data from previous experiments and physical models and related features. This part of the information is input into the machine learning framework for prediction, and the global optimization method is used to optimize the machine learning model. Carrying out new synthetic characterization experiments based on the model prediction results has the dual purpose of improving the model and discovering new materials. The above results are used to update the database. The updated database provides input for the next cycle[49].
Figure 17
Experimental process of combining liquid handling robot and artificial intelligence to explore chemical space. The liquid-handling robot performs reactions by choosing reactants from the pool of starting materials. Online analytics is used for real-time interpretation of reaction outcomes as reactive or non-reactive, and the reaction database stores reaction outcomes. Machine learning is used to build a model of the chemical space, recommend the next experiments and control the robot[49].
Figure 18
Schematic diagram of data classification by SVM when the given data is linearly separable. The minimum distance of hyperplane A0 from two groups of points > the minimum distance of hyperplane A1 from two groups of points, so hyperplane P0 is a hyperplane with better classification function.
Figure 23
Schematic diagram of convolutional neural network (CNN). CNN requires the input to be in the form of image data. Each convolutional layer extracts and retains spatial information, and learns a representation, which is then passed to the traditional fully connected feedforward neural network before the output layer[76].

 下载:
下载:

 下载:
下载:
























