Received Date:
08 July 2020 Available Online:
15 December 2020
Fund Project:
Project supported by the Major Research Plan of the National Natural Science Foundation of China (Training Program) (No. 91853118) and Youth Program of National Natural Science Foundation of China (No. 21722802)
Abstract:
Type-Ⅱ fatty acid biosynthesis pathway (FAS-Ⅱ) is the only essential biosynthesis pathway that producing saturated and unsaturated fatty acids for bacteria and plant cell assembly and cellular metabolism. It utilizes a series of individual enzymes encoded by discrete genes to stepwisely catalyze lipid chain growing carried by the substrate carrier protein-acyl carrier protein (ACP). Due to its indispensable biological role in bacteria growth, as well as the distinct biological regulation mechanisms from mammalian fatty acid biosynthesis (FAS-Ⅰ), the enzymes involved in FAS-Ⅱ have been considered as important anti-pathogenic drug targets for a long time. Hence, investigating the catalysis and dynamic regulation mechanisms of FAS-Ⅱ, developing novel anti-pathogenic drugs against the enzymes involved in FAS-Ⅱ is critical to the field. We here summarize the catalytic mechanism studies and inhibitor discovery work involved in FAS-Ⅱ so far, which may potentially facilitate further understanding of FAS-Ⅱ biological functions as well as antibacterial drug discovery for infectious diseases.
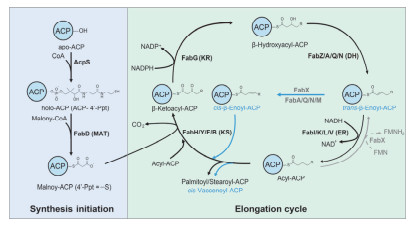
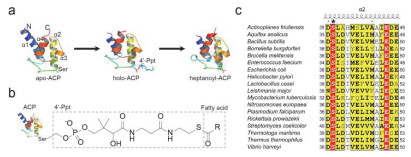
Figure 2.
Crystal structures of ACP and the primary sequence comparison of ACP conserved regions.
(a) Overall crystal structures of apo-and holo-form ACP (from Helicobacter pylori, pdb codes: 5H9G and 5H9H), and Heptanoyl-ACP (from Escherichia coli, pdb code: 2FAD). (b) Chemical structure of the 4'-Phosphopantetheine (4'-Ppt) arm attached to ACP. (c) The primary sequence comparison of ACP α2 helix among bacteria. The conserved serine that can be modified by 4'-Ppt is marked with an asterisk.
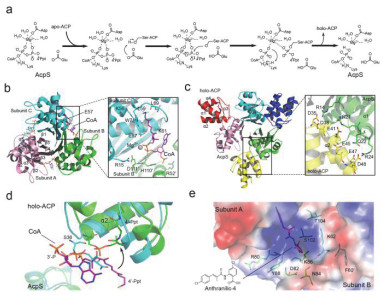
Figure 3.
Schematic diagram of AcpS catalytic mechanism and overall crystal structures
(a) The schematic diagram of AcpS catalytic mechanism. (b) Overall crystal structure of ScAcpS-CoA complex. Key residues involved in CoA (purple) binding are shown in green/cyan sticks and labeled. (c) Overall crystal structure of BsAcpS-holo-ACP complex. (d) Superposition of CoA binding pocket among EcAcpS-holo-ACP (cyan), BsAcpS-holo-ACP (green) and ScAcpS-CoA (purple) complex structures. (e) The binding mode of the inhibitor Anthranilic-4 (purple) in the active pocket of BsAcpS. The residues involved in the binding are shown in green sticks and labeled, and the chemical structure of Anthranilic-4 is also shown.
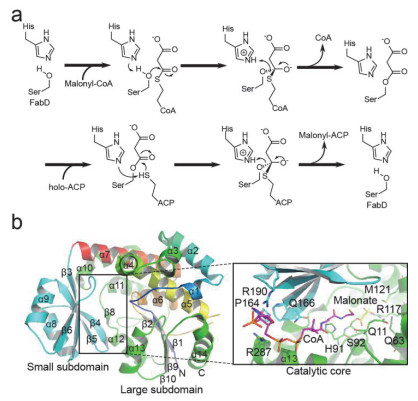
Figure 4.
Schematic diagram of FabD catalytic mechanism and overall crystal structure
(a) Schematic diagram of FabD catalytic mechanism. (b) Overall crystal structure of EcFabD in complex with malonyl-CoA (purple). Key residues involved in CoA (purple) binding are shown in green/cyan sticks and labeled.
Figure 5.
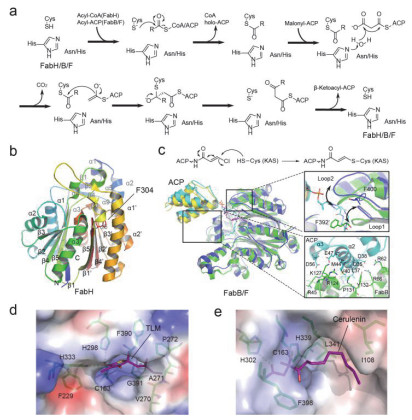
Schematic diagram of KAS family catalytic mechanisms and overall crystal structures
(a) Schematic diagram of KAS family catalytic mechanisms. (b) Overall crystal structure of EcFabH. The conserved N-and C-terminal folding regions are labeled with black fonts, while the connection and insertion regions are labeled with grey fonts. (c) Superposition of EcFabB (green)-ACP(cyan) and EcFabF(blue)-ACP(yellow) complex structures. Black arrow shows conformational changes of key residues or loop structure. (d) The binding model of inhibitor TLM (purple) in the active pocket of EcFabB. The residues involved in the binding are shown in green sticks and labeled. (e) The binding model of inhibitor Cerulenin (purple) in the active pocket of BsFabF.
Figure 6.
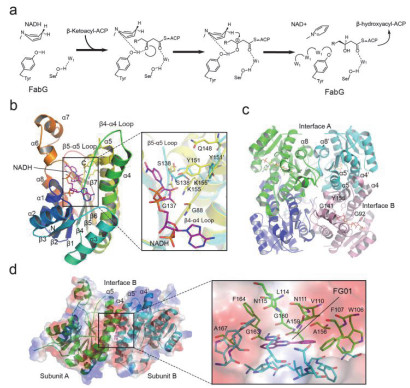
Schematic diagram of FabG catalytic mechanism and overall crystal structure
(a) Schematic diagram of FabG catalytic mechanism. (b) Overall crystal structure of EcFabG-NADH complex. The binding models of NADH (purple) in the active conformation (yellow) and inactive conformation (cyan) of EcFabG are shown. Key residues involved in the binding are shown in sticks and labeled. (c) Overall structure of VcFabG tetramer. The secondary elements on the dimer-dimer contact interface are labeled. (d) The binding model of allosteric inhibitor FG01 (purple) to PaFabG complex. Key residues involved in inhibitor binding from PaFabG A and B subunit are shown in green/cyan sticks and labeled.
Figure 7.
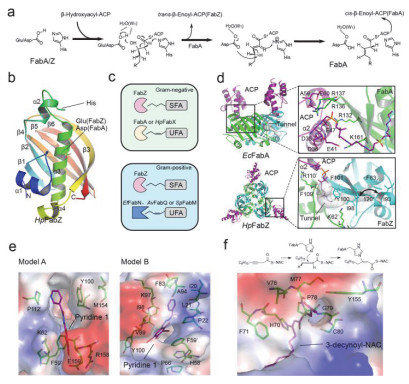
Schematic diagram of FabA/Z catalytic mechanisms and overall crystal structures
(a) Schematic diagram of FabA/Z catalytic mechanisms. (b) Overall structure of PaFabA monomer. (c) Schematic diagram of the relationship between FabZ, FabA and bacteria-specific UFA key enzymes in Gram-negative (light green) and Gram-positive bacteria (cyan). (d) Overall crystal structures of EcFabA-ACP and HpFabZ-ACP complex; Key residues involved in the binding of ACP (purple) are shown in sticks and labeled. (e) Two binding models of inhibitor Pyridine-1 (purple) to HpFabZ. Key residues involved in the binding of ACP (purple) are shown in green sticks and labeled. (f) The covalent binding model of inhibitor 3-decenyl-N-acetylcysteamine (purple) to EcFabA.
Figure 8.
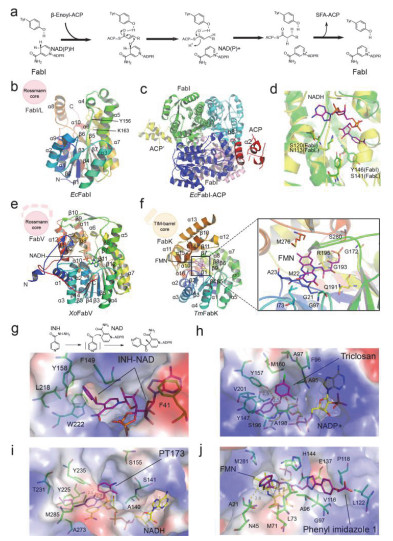
Schematic diagram of FabI/L/V/K catalytic mechanisms and overall crystal structures
(a) Schematic diagram of FabI catalytic mechanism. (b) Overall structure of EcFabI monomer. (c) Overall structure of EcFabI (green/blue/cyan/pink) in complex with ACP (yellow/red). (d) Superposition of proton transfer and NADH binding regions of EcFabI (green) and BsFabL (yellow). (e) Overall structure of XoFabV monomer. (f) Overall structure of TmFabK monomer; (g) The binding mode of inhibitor isoniazid-NAD (purple) with InhA. (h) The binding mode of inhibitor Triclosan (purple) with SaFabI. The ligand NADP+ are shown in yellow stick. (i) The binding mode of inhibitor PT173 (purple) with YpFabV. (j) The binding mode of inhibitor Phenylimidazole compound 1 (purple) with SpFabK. The ligand FMN is shown in yellow stick.
Chan, D. I.; Chu, B. C.; Lau, C. K.; Hunter, H. N.; Byers, D. M.; Vogel, H. J. J. Biol. Chem. 2010, 285, 30558.
[14]
Dall'aglio, P.; Arthur, C. J.; Williams, C.; Vasilakis, K.; Maple, H. J.; Crosby, J.; Crump, M. P.; Hadfield, A. T. Biochemistry 2011, 50, 5704.
[15]
Marcella, A. M.; Culbertson, S. J.; Shogren-Knaak, M. A.; Barb, A. W. J. Mol. Biol. 2017, 429, 3763.
[16]
Keating, D. H.; Carey, M. R.; Cronan, J. E. J. Biol. Chem. 1995, 270, 22229.
[17]
Bunkoczi, G.; Pasta, S.; Joshi, A.; Wu, X.; Kavanagh, K. L.; Smith, S.; Oppermann, U. Chem. Biol. 2007, 14, 1243.
[18]
Joseph-McCarthy, D.; Parris, K.; Huang, A.; Failli, A.; Quagliato, D.; Dushin, E. G.; Novikova, E.; Severina, E.; Tuckman, M.; Petersen, P. J.; Dean, C.; Fritz, C. C.; Meshulam, T.; DeCenzo, M.; Dick, L.; McFadyen, I. J.; Somers, W. S.; Lovering, F.; Gilbert, A. M. J. Med. Chem. 2005, 48, 7960.
[19]
Chu, M.; Mierzwa, R.; Xu, L.; Yang, S. W.; He, L.; Patel, M.; Stafford, J.; Macinga, D.; Black, T.; Chan, T. M.; Gullo, V. Bioorg. Med. Chem. Lett. 2003, 13, 3827.
[20]
Ruch, F. E.; Vagelos, P. R. J. Biol. Chem.1973, 248, 8095.
[21]
Hong, S. K.; Kim, K. H.; Park, J. K.; Jeong, K. W.; Kim, Y.; Kim, E. E. FEBS Lett. 2010, 584, 1240.
[22]
Lee, W. C.; Park, J.; Balasubramanian, P. K.; Kim, Y. Biochem. Biophys. Res. Commun. 2018, 505, 208.
[23]
Li, Z.; Huang, Y.; Ge, J.; Fan, H.; Zhou, X.; Li, S.; Bartlam, M.; Wang, H.; Rao, Z. J. Mol. Biol. 2007, 371, 1075.
[24]
Keatinge-Clay, A. T.; Shelat, A. A.; Savage, D. F.; Tsai, S.-C.; Miercke, L. J. W.; O'Connell, J. D.; Khosla, C.; Stroud, R. M. Structure 2003, 11, 147.
[25]
Liu, W.; Han, C.; Hu, L.; Chen, K.; Shen, X.; Jiang, H. FEBS Lett. 2006, 580, 697.
[26]
Kong, Y. H.; Zhang, L.; Yang, Z. Y.; Han, C.; Hu, L. H.; Jiang, H. L.; Shen, X. Acta Pharmacol. Sin. 2008, 29, 870.
Li, Y.; Florova, G.; Reynolds, K. A. J. Bacteriol. 2005, 187, 3795.
[30]
Han, L.; Lobo, S.; Reynolds, K. A. J. Bacteriol. 1998, 180, 4481.
[31]
Tsay, J. T.; Oh, W.; Larson, T. J.; Jackowski, S.; Rock, C. O. J. Biol. Chem. 1992, 267, 6807.
[32]
Gajiwala, K. S.; Margosiak, S.; Lu, J.; Cortez, J.; Su, Y.; Nie, Z.; Appelt, K. FEBS Lett. 2009, 583, 2939.
[33]
Yuan, Y.; Sachdeva, M.; Leeds, J. A.; Meredith, T. C. J. Bacteriol. 2012, 194, 5171.
[34]
Milligan, J. C.; Lee, D. J.; Jackson, D. R.; Schaub, A. J.; Beld, J.; Barajas, J. F.; Hale, J. J.; Luo, R.; Burkart, M. D.; Tsai, S. C. Nat. Chem. Biol. 2019, 15, 669.
[35]
Mindrebo, J. T.; Patel, A.; Kim, W. E.; Davis, T. D.; Chen, A.; Bartholow, T. G.; La Clair, J. J.; McCammon, J. A.; Noel, J. P.; Burkart, M. D. Nat. Commun. 2020, 11, 1727.
[36]
Nanson, J. D.; Himiari, Z.; Swarbrick, C. M.; Forwood, J. K. Sci. Rep. 2015, 5, 14797.
[37]
Price, A. C.; Choi, K. H.; Heath, R. J.; Li, Z.; White, S. W.; Rock, C. O. J. Biol. Chem. 2001, 276, 6551.
[38]
Wang, J.; Kodali, S.; Lee, S. H.; Galgoci, A.; Painter, R.; Dorso, K.; Racine, F.; Motyl, M.; Hernandez, L.; Tinney, E.; Colletti, S. L.; Herath, K.; Cummings, R.; Salazar, O.; González, I.; Basilio, A.; Vicente, F.; Genilloud, O.; Pelaez, F.; Jayasuriya, H.; Young, K.; Cully, D. F.; Singh, S. B. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 7612.
[39]
Daines, R. A.; Pendrak, I.; Sham, K.; Van Aller, G. S.; Konstantinidis, A. K.; Lonsdale, J. T.; Janson, C. A.; Qiu, X.; Brandt, M.; Khandekar, S. S.; Silverman, C.; Head, M. S. J. Med. Chem. 2003, 46, 5.
[40]
McKinney, D. C.; Eyermann, C. J.; Gu, R. F.; Hu, J.; Kazmirski, S. L.; Lahiri, S. D.; McKenzie, A. R.; Shapiro, A. B.; Breault, G. ACS Infect. Dis. 2016, 2, 456.
[41]
Wang, J.; Soisson, S. M.; Young, K.; Shoop, W.; Kodali, S.; Galgoci, A.; Painter, R.; Parthasarathy, G.; Tang, Y. S.; Cummings, R.; Ha, S.; Dorso, K.; Motyl, M.; Jayasuriya, H.; Ondeyka, J.; Herath, K.; Zhang, C.; Hernandez, L.; Allocco, J.; Basilio, A.; Tormo, J. R.; Genilloud, O.; Vicente, F.; Pelaez, F.; Colwell, L.; Lee, S. H.; Michael, B.; Felcetto, T.; Gill, C.; Silver, L. L.; Hermes, J. D.; Bartizal, K.; Barrett, J.; Schmatz, D.; Becker, J. W.; Cully, D.; Singh, S. B. Nature 2006, 441, 358.
[42]
Zheng, Z.; Parsons, J. B.; Tangallapally, R.; Zhang, W.; Rock, C. O.; Lee, R. E. Bioorg. Med. Chem. Lett. 2014, 24, 2585.
[43]
Kallberg, Y.; Oppermann, U.; Jornvall, H.; Persson, B. Eur. J. Biochem. 2002, 269, 4409.
[44]
Hou, J.; Zheng, H.; Chruszcz, M.; Zimmerman, M. D.; Shumilin, I. A.; Osinski, T.; Demas, M.; Grimshaw, S.; Minor, W. J. Bacteriol. 2016, 198, 463.
[45]
Price, A. C.; Zhang, Y.-M.; Rock, C. O.; White, S. W. Biochemistry 2001, 40, 12772.
[46]
Silva, R. G.; Rosado, L. A.; Santos, D. S.; Basso, L. A. Arch. Biochem. Biophys. 2008, 471, 1.
[47]
Price, A. C.; Zhang, Y. M.; Rock, C. O.; White, S. W. Structure 2004, 12, 417.
[48]
Cohen-Gonsaud, M.; Ducasse-Cabanot, S.; Quemard, A.; Labesse, G. Proteins 2005, 60, 392.
[49]
Cukier, C. D.; Hope, A. G.; Elamin, A. A.; Moynie, L.; Schnell, R.; Schach, S.; Kneuper, H.; Singh, M.; Naismith, J. H.; Lindqvist, Y.; Gray, D. W.; Schneider, G. ACS Chem. Biol. 2013, 8, 2518.
[50]
Lai, C. Y.; Cronan, J. E. J. Bacteriol. 2004, 186, 1869.
[51]
Sohn, M.-J.; Zheng, C.-J.; Kim, W.-G. J. Antibiot. 2008, 61, 687.
[52]
Wickramasinghe, S. R.; Inglis, K. A.; Urch, J. E.; Muller, S.; van Aalten, D. M.; Fairlamb, A. H. Biochem. J. 2006, 393, 447.
[53]
Tasdemir, D.; Lack, G.; Brun, R.; Rüedi, P.; Scapozza, L.; Perozzo, R. J. Med. Chem. 2006, 49, 3345.
[54]
Zhang, F.; Luo, S. Y.; Ye, Y. B.; Zhao, W. H.; Sun, X. G.; Wang, Z. Q.; Li, R.; Sun, Y. H.; Tian, W. X.; Zhang, Y. X. Biotechnol. Appl. Biochem. 2008, 51, 73.
[55]
Zeng, D.; Zhao, J.; Chung, H. S.; Guan, Z.; Raetz, C. R.; Zhou, P. J. Biol. Chem. 2013, 288, 5475.
[56]
Swarnamukhi, P. L.; Sharma, S. K.; Bajaj, P.; Surolia, N.; Surolia, A.; Suguna, K. FEBS Lett. 2006, 580, 2653.
Shen, S.; Hang, X.; Zhuang, J.; Zhang, L.; Bi, H.; Zhang, L. Int. J. Biol. Macromol. 2019, 128, 5.
[59]
Dodge, G. J.; Patel, A.; Jaremko, K. L.; McCammon, J. A.; Smith, J. L.; Burkart, M. D. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 6775.
[60]
Moynie, L.; Leckie, S. M.; McMahon, S. A.; Duthie, F. G.; Koehnke, A.; Taylor, J. W.; Alphey, M. S.; Brenk, R.; Smith, A. D.; Naismith, J. H. J. Mol. Biol. 2013, 425, 365.
[61]
Heath, R. J.; Rock, C. O. J. Biol. Chem. 1996, 271, 27795.
[62]
Nguyen, C.; Haushalter, R. W.; Lee, D. J.; Markwick, P. R.; Bruegger, J.; Caldara-Festin, G.; Finzel, K.; Jackson, D. R.; Ishikawa, F.; O'Dowd, B.; McCammon, J. A.; Opella, S. J.; Tsai, S. C.; Burkart, M. D. Nature 2014, 505, 427.
[63]
Bi, H.; Zhu, L.; Jia, J.; Zeng, L.; Cronan, J. E. Cell Chem. Biol. 2016, 23, 1480.
[64]
Wang, H.; Cronan, J. E. J. Biol. Chem. 2004, 279, 34489.
[65]
Bi, H.; Wang, H.; Cronan, J. E. Chem. Biol. 2013, 20, 1157.
[66]
Marrakchi, H.; Choi, K. H.; Rock, C. O. J. Biol. Chem. 2002, 277, 44809.
[67]
Aguilar, P. S.; Cronan, J. E.; de Mendoza, D. J. Bacteriol. 1998, 180, 2194.
[68]
Sharma, S. K.; Kapoor, M.; Ramya, T. N.; Kumar, S.; Kumar, G.; Modak, R.; Sharma, S.; Surolia, N.; Surolia, A. J. Biol. Chem. 2003, 278, 45661.
[69]
Zhang, L.; Liu, W.; Hu, T.; Du, L.; Luo, C.; Chen, K.; Shen, X.; Jiang, H. J. Biol. Chem. 2008, 283, 5370.
[70]
He, L.; Zhang, L.; Liu, X.; Li, X.; Zheng, M.; Li, H.; Yu, K.; Chen, K.; Shen, X.; Jiang, H.; Liu, H. J. Med. Chem. 2009, 52, 2465.
McGillick, B. E.; Kumaran, D.; Vieni, C.; Swaminathan, S. Biochemistry 2016, 55, 1091.
[74]
Leesong, M.; Henderson, B. S.; Gillig, J. R.; Schwab, J. M.; Smith, J. L. Structure 1996, 4, 253.
[75]
Moynie, L.; Hope, A. G.; Finzel, K.; Schmidberger, J.; Leckie, S. M.; Schneider, G.; Burkart, M. D.; Smith, A. D.; Gray, D. W.; Naismith, J. H. J. Mol. Biol. 2016, 428, 108.
[76]
Kim, H. T.; Kim, S.; Na, B. K.; Chung, J.; Hwang, E.; Hwang, K. Y. Biochem. Biophys. Res. Commun. 2017, 493, 28.
[77]
Rafi, S.; Novichenok, P.; Kolappan, S.; Stratton, C. F.; Rawat, R.; Kisker, C.; Simmerling, C.; Tonge, P. J. J. Biol. Chem. 2006, 281, 39285.
[78]
Kim, K. H.; Ha, B. H.; Kim, S. J.; Hong, S. K.; Hwang, K. Y.; Kim, E. E. J. Mol. Biol. 2011, 406, 403.
[79]
Neckles, C.; Pschibul, A.; Lai, C. T.; Hirschbeck, M.; Kuper, J.; Davoodi, S.; Zou, J.; Liu, N.; Pan, P.; Shah, S.; Daryaee, F.; Bommineni, G. R.; Lai, C.; Simmerling, C.; Kisker, C.; Tonge, P. J. Biochemistry 2016, 55, 2992.
[80]
Li, H.; Zhang, X.; Bi, L.; He, J.; Jiang, T. PLoS One 2011, 6, e26743.
[81]
Kim, S. H.; Khan, R.; Choi, K.; Lee, S. W.; Rhee, S. FEBS J. 2020, 281, 4710.
[82]
Saito, J.; Yamada, M.; Watanabe, T.; Iida, M.; Kitagawa, H.; Takahata, S.; Ozawa, T.; Takeuchi, Y.; Ohsawa, F. Protein Sci. 2008, 17, 691.
[83]
Qiu, X.; Abdel-Meguid, S. S.; Janson, C. A.; Court, R. I.; Smyth, M. G.; Payne, D. J. Protein Sci. 1999, 8, 2529.
[84]
Miller, W. H.; Seefeld, M. A.; Newlander, K. A.; Uzinskas, I. N.; Burgess, W. J.; Heerding, D. A.; Yuan, C. C. K.; Head, M. S.; Payne, D. J.; Rittenhouse, S. F.; Moore, T. D.; Pearson, S. C.; Berry, V.; DeWolf, W. E.; Keller, P. M.; Polizzi, B. J.; Qiu, X.; Janson, C. A.; Huffman, W. F. J. Med. Chem. 2002, 45, 3246.
[85]
Seefeld, M. A.; Miller, W. H.; Newlander, K. A.; Burgess, W. J.; DeWolf, W. E.; Elkins, P. A.; Head, M. S.; Jakas, D. R.; Janson, C. A.; Keller, P. M.; Manley, P. J.; Moore, T. D.; Payne, D. J.; Pearson, S.; Polizzi, B. J.; Qiu, X.; Rittenhouse, S. F.; Uzinskas, I. N.; Wallis, N. G.; Huffman, W. F. J. Med. Chem. 2003, 46, 1627.
[86]
Heerding, D. A.; Chan, G.; DeWolf, W. E.; Fosberry, A. P.; Janson, C. A.; Jaworski, D. D.; McManus, E.; Miller, W. H.; Moore, T. D.; Payne, D. J.; Qiu, X.; Rittenhouse, S. F.; Slater-Radosti, C.; Smith, W.; Takata, D. T.; Vaidya, K. S.; Yuan, C. C. K.; Huffman, W. F. Bioorg. Med. Chem. Lett. 2001, 11, 2061.
[87]
Seefeld, M. A.; Miller, W. H.; Newlander, K. A.; Burgess, W. J.; Payne, D. J.; Rittenhouse, S. F.; Moore, T. D.; DeWolf, W. E.; Keller, P. M.; Qiu, X.; Janson, C. A.; Vaidya, K.; Fosberry, A. P.; Smyth, M. G.; Jaworski, D. D.; Slater-Radosti, C.; Huffman, W. F. Bioorg. Med. Chem. Lett. 2001, 11, 2241.
[88]
Ramnauth, J.; Surman, M. D.; Sampson, P. B.; Forrest, B.; Wilson, J.; Freeman, E.; Manning, D. D.; Martin, F.; Toro, A.; Domagala, M.; Awrey, D. E.; Bardouniotis, E.; Kaplan, N.; Berman, J.; Pauls, H. W. Bioorg. Med. Chem. Lett. 2009, 19, 5359.
[89]
Sampson, P. B.; Picard, C.; Handerson, S.; McGrath, T. E.; Domagala, M.; Leeson, A.; Romanov, V.; Awrey, D. E.; Thambipillai, D.; Bardouniotis, E.; Kaplan, N.; Berman, J. M.; Pauls, H. W. Bioorg. Med. Chem. Lett. 2009, 19, 5355.
[90]
Fage, C. D.; Lathouwers, T.; Vanmeert, M.; Gao, L. J.; Vrancken, K.; Lammens, E. M.; Weir, A. N. M.; Degroote, R.; Cuppens, H.; Kosol, S.; Simpson, T. J.; Crump, M. P.; Willis, C. L.; Herdewijn, P.; Lescrinier, E.; Lavigne, R.; Anne, J.; Masschelein, J. Angew. Chem. Int. Ed. 2020, 59, 10549.
[91]
Karlowsky, J. A.; Laing, N. M.; Baudry, T.; Kaplan, N.; Vaughan, D.; Hoban, D. J.; Zhanel, G. G. Antimicrob. Agents Chemother. 2007, 51, 1580.
(a) Overall crystal structures of apo-and holo-form ACP (from Helicobacter pylori, pdb codes: 5H9G and 5H9H), and Heptanoyl-ACP (from Escherichia coli, pdb code: 2FAD). (b) Chemical structure of the 4'-Phosphopantetheine (4'-Ppt) arm attached to ACP. (c) The primary sequence comparison of ACP α2 helix among bacteria. The conserved serine that can be modified by 4'-Ppt is marked with an asterisk.
(a) The schematic diagram of AcpS catalytic mechanism. (b) Overall crystal structure of ScAcpS-CoA complex. Key residues involved in CoA (purple) binding are shown in green/cyan sticks and labeled. (c) Overall crystal structure of BsAcpS-holo-ACP complex. (d) Superposition of CoA binding pocket among EcAcpS-holo-ACP (cyan), BsAcpS-holo-ACP (green) and ScAcpS-CoA (purple) complex structures. (e) The binding mode of the inhibitor Anthranilic-4 (purple) in the active pocket of BsAcpS. The residues involved in the binding are shown in green sticks and labeled, and the chemical structure of Anthranilic-4 is also shown.
(a) Schematic diagram of FabD catalytic mechanism. (b) Overall crystal structure of EcFabD in complex with malonyl-CoA (purple). Key residues involved in CoA (purple) binding are shown in green/cyan sticks and labeled.
(a) Schematic diagram of KAS family catalytic mechanisms. (b) Overall crystal structure of EcFabH. The conserved N-and C-terminal folding regions are labeled with black fonts, while the connection and insertion regions are labeled with grey fonts. (c) Superposition of EcFabB (green)-ACP(cyan) and EcFabF(blue)-ACP(yellow) complex structures. Black arrow shows conformational changes of key residues or loop structure. (d) The binding model of inhibitor TLM (purple) in the active pocket of EcFabB. The residues involved in the binding are shown in green sticks and labeled. (e) The binding model of inhibitor Cerulenin (purple) in the active pocket of BsFabF.
(a) Schematic diagram of FabG catalytic mechanism. (b) Overall crystal structure of EcFabG-NADH complex. The binding models of NADH (purple) in the active conformation (yellow) and inactive conformation (cyan) of EcFabG are shown. Key residues involved in the binding are shown in sticks and labeled. (c) Overall structure of VcFabG tetramer. The secondary elements on the dimer-dimer contact interface are labeled. (d) The binding model of allosteric inhibitor FG01 (purple) to PaFabG complex. Key residues involved in inhibitor binding from PaFabG A and B subunit are shown in green/cyan sticks and labeled.
(a) Schematic diagram of FabA/Z catalytic mechanisms. (b) Overall structure of PaFabA monomer. (c) Schematic diagram of the relationship between FabZ, FabA and bacteria-specific UFA key enzymes in Gram-negative (light green) and Gram-positive bacteria (cyan). (d) Overall crystal structures of EcFabA-ACP and HpFabZ-ACP complex; Key residues involved in the binding of ACP (purple) are shown in sticks and labeled. (e) Two binding models of inhibitor Pyridine-1 (purple) to HpFabZ. Key residues involved in the binding of ACP (purple) are shown in green sticks and labeled. (f) The covalent binding model of inhibitor 3-decenyl-N-acetylcysteamine (purple) to EcFabA.
(a) Schematic diagram of FabI catalytic mechanism. (b) Overall structure of EcFabI monomer. (c) Overall structure of EcFabI (green/blue/cyan/pink) in complex with ACP (yellow/red). (d) Superposition of proton transfer and NADH binding regions of EcFabI (green) and BsFabL (yellow). (e) Overall structure of XoFabV monomer. (f) Overall structure of TmFabK monomer; (g) The binding mode of inhibitor isoniazid-NAD (purple) with InhA. (h) The binding mode of inhibitor Triclosan (purple) with SaFabI. The ligand NADP+ are shown in yellow stick. (i) The binding mode of inhibitor PT173 (purple) with YpFabV. (j) The binding mode of inhibitor Phenylimidazole compound 1 (purple) with SpFabK. The ligand FMN is shown in yellow stick.

 下载:
下载:










































 下载:
下载:









