Received Date:
07 July 2020 Available Online:
15 December 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21772003 and 21933001)
Abstract:
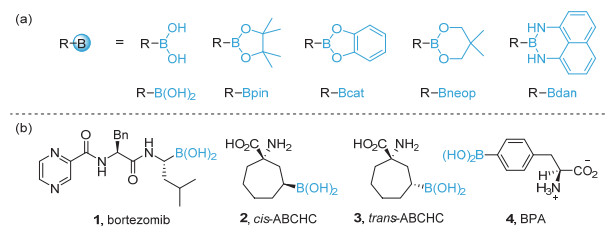
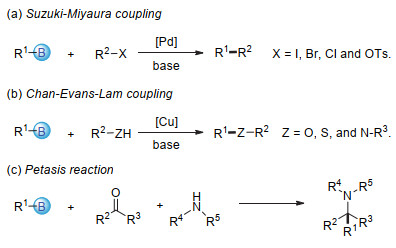
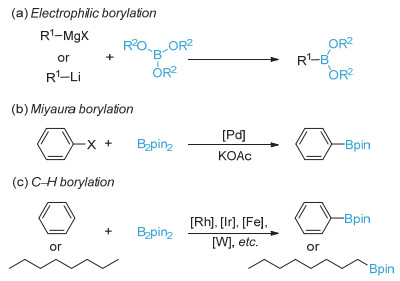
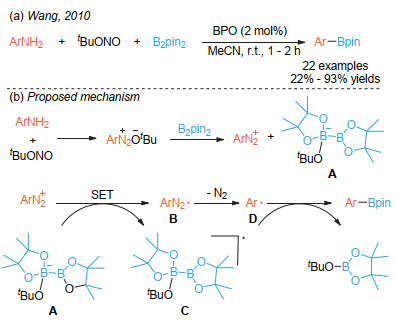
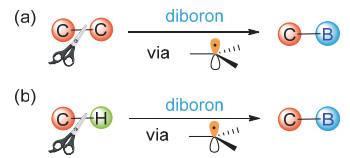
Organoboronic acids and esters are highly valuable building blocks in cross-coupling reactions and practical intermediates of various functional group transformations. Additionally, organoboronic acids can be utilized directly as small molecule drugs. Therefore, development of efficient methods to synthesize organoboronic compounds is of significant importance. Traditional pathways to synthesize organoboronic compounds mainly rely on electrophilic borylation of organometallic reagent and transition-metal-catalyzed borylation. Radical intermediates have unique chemical properties which are quite different from those of polar intermediates resulted from the heterolysis of chemical bonds and those of the organometallic compounds during transition metal catalysis. As such, borylation based on radical mechanism possesses distinctive reaction process, substrate scope, reaction selectivity, etc., and have great potential in synthesis of organoboronic compounds. In 2010, the Wang's group first reported borylation via a radical mechanism. This method realized an efficient direct conversion of anilines into aryl organoboronic esters. Inspired by this innovative work, more and more borylation methods via radical intermediates have been reported and developed as new avenues for C-B bond formation in the past decade. A series of studies show that organoboronic acids and esters could be efficiently constructed by the reaction of aryl/alkyl radicals with diboron compounds. In this paper, we summarize the recent development of borylation reactions via radical mechanisms, including aryl and alkyl radical borylation. As for aryl radical borylation, the activation of substrates containing C-N, C-O, C-S, C-X (X=halogen) bonds and carboxylic acids to C-B bond is summarized respectively. As for alkyl radical borylation, the activation of substrates containing C-N, C-O, C-X (X=halogen), C-C bonds and carboxylic acids to C-B bond is summarized respectively. Finally, we provide a perspective on the future development direction of this research area.
(a) Barth, R. F.; Kabalka, G. W.; Yang, W.; Huo, T.; Nakkula, R. J.; Shaikh, A. L.; Haider, S. A.; Chandra, S. Appl. Radiat. Isotopes 2014, 88, 38; (b) Barth, R. F.; Mi, P.; Yang, W. Cancer Commun. 2018, 38, 35.
[2]
(a) San Miguel, J. F.; Schlag, R.; Khuageva, N. K.; Dimopoulos, M. A.; Shpilberg, O.; Kropff, M.; Spicka, I.; Petrucci, M. T.; Palumbo, A.; Samoilova, O. S.; Dmoszynska, A.; Abdulkadyrov, K. M.; Schots, R.; Jiang, B.; Mateos, M. V.; Anderson, K. C.; Esseltine, D. L.; Liu, K.; Cakana, A.; Van De Velde, H.; Richardson, P. G. New Engl. J. Med. 2008, 359, 906; (b) Beenen, M. A.; An, C.; Ellman, J. A. J. Am. Chem. Soc. 2008, 130, 6910.
[3]
(a) Chemler, S. R.; Trauner, D.; Danishefsky, S. J. Angew. Chem. Int. Ed. 2001, 40, 4544; (b) Moreno-Maas, M.; Pérez, M.; Pleixats, R. J. Org. Chem. 1996, 61, 2346.
[4]
(a) Miyaura, N.; Suzuki, A., J. Chem. Soc. Chem. Commun. 1979, 866; (b) Miyaura, N.; Yamada, K.; Suzuki, A. Tetrahedron Lett. 1979, 20, 3437.
[5]
(a) Schneider, N.; Lowe, D. M.; Sayle, R. A.; Tarselli, M. A.; Landrum, G. A. J. Med. Chem. 2016, 59, 4385; (b) Bostrm, J.; Brown, D. G.; Young, R. J.; Keserü, G. M. Nat. Rev. Drug Discov. 2018, 17, 709.
[6]
Torborg, C.; Beller, M. Adv. Synth. Catal. 2009, 351, 3027.
[7]
(a) Liu, M.; Su, S.-J.; Jung, M.-C.; Qi, Y.; Zhao, W.-M.; Kido, J. Chem. Mater. 2012, 24, 3817; (b) Wong, K.-T.; Hung, T. S.; Lin, Y.; Wu, C.-C.; Lee, G.-H.; Peng, S.-M.; Chou, C. H.; Su, Y. O. Org. Lett. 2002, 4, 513.
[8]
(a) Chan, D. M. T.; Monaco, K. L.; Wang, R.-P.; Winters, M. P. Tetrahedron Lett. 1998, 39, 2933; (b) Evans, D. A.; Katz, J. L.; West, T. R. Tetrahedron Lett. 1998, 39, 2937; (c) Lam, P. Y. S.; Clark, C. G.; Saubern, S.; Adams, J.; Winters, M. P.; Chan, D. M. T.; Combs, A. Tetrahedron Lett. 1998, 39, 2941; (d) Herradura, P. S.; Pendola, K. A.; Guy, R. K. Org. Lett. 2000, 2, 2019.
[9]
Petasis, N. A.; Akritopoulou, I. Tetrahedron Lett. 1993, 34, 583.
[10]
Wu, P.; Givskov, M.; Nielsen, T. E. Chem. Rev. 2019, 119, 11245.
[11]
(a) Brown, H. C.; Cole, T. E. Organometallics. 1983, 2, 1316; (b) Brown, H. C.; Srebnik, M.; Cole, T. E. Organometallics. 1986, 5, 2300.
[12]
Ishiyama, T.; Murata, M.; Miyaura, N. J. Org. Chem. 1995, 60, 7508.
[13]
Li, Z.; Zheng, J.; Li, C.; Wu, W.; Jiang, H. Chin. J. Chem. 2019, 37, 140.
[14]
Yoshida, H. ACS Catal. 2016, 6, 1799.
[15]
Li, S.; Li, J.; Xia, T.; Zhao, W. Chin. J. Chem. 2019, 37, 462.
[16]
He, Z.; Fan, M.; Xu, J.; Hu, Y.; Wang, L.; Wu, X.; Xia, C.; Liu, C. Chin. J. Org. Chem. 2019, 39, 3438.
(a) Chen, H.; Schlecht, S.; Semple, T. C.; Hartwig, J. F. Science 2000, 287, 1995; (b) Mkhalid, I. A. I.; Barnard, J. H.; Marder, T. B.; Murphy, J. M.; Hartwig, J. F. Chem. Rev. 2010, 110, 890; (c) Jiang, Z.-T.; Wang, B.-Q.; Shi, Z.-J. Chin. J. Chem. 2018, 36, 950; (d) Zhan, M.; Song, P.; Jiao, J.; Li, P. Chin. J. Chem. 2020, 38, 665.
[19]
(a) (a) Xiao, L. Li, J.-H, Wang, T. Acta Chim. Sinica, 2019, 77, 841(in Chinese). (肖丽, 李嘉恒, 王挺, 化学学报2019, 77, 841.) (b) Ye, S.-Q, Wu, J. Acta Chim. Sinica, 2019, 77, 814(in Chinese). (叶盛青, 吴劼, 化学学报2019, 77, 814.)
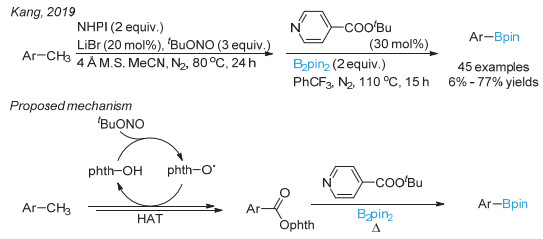
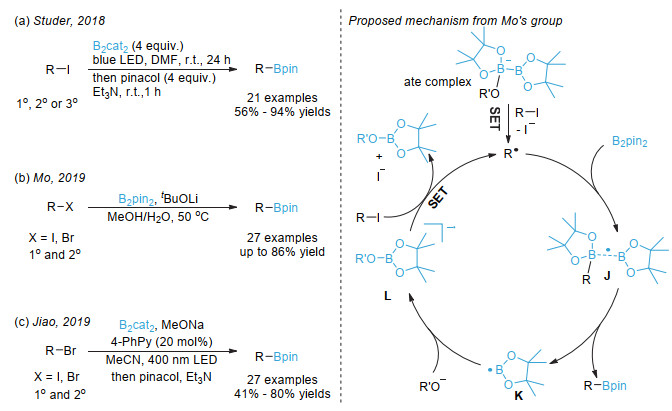
Jin, S.; Dang, H. T.; Haug, G. C.; He, R.; Nguyen, V. D.; Nguyen, V. T.; Arman, H. D.; Schanze, K. S.; Larionov, O. V. J. Am. Chem. Soc. 2020, 142, 1603.
[28]
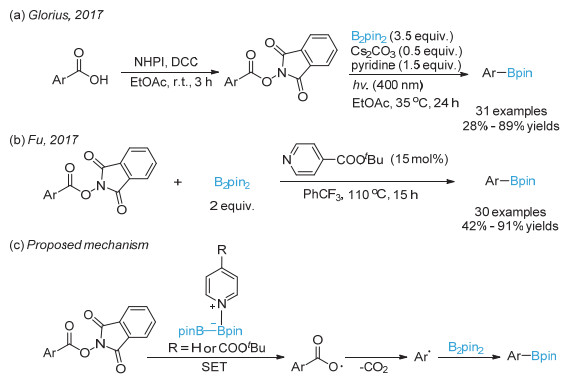
Candish, L.; Teders, M.; Glorius, F. J. Am. Chem. Soc. 2017, 139, 7440.
[29]
Cheng, W.-M.; Shang, R.; Zhao, B.; Xing, W.-L.; Fu, Y. Org. Lett. 2017, 19, 4291.
Mfuh, A. M.; Doyle, J. D.; Chhetri, B.; Arman, H. D.; Larionov, O. V. J. Am. Chem. Soc. 2016, 138, 2985.
[37]
Mukai, K.; de Sant'Ana, D. P.; Hirooka, Y.; Mercado-Marin, E. V.; Stephens, D. E.; Kou, K. G. M.; Richter, S. C.; Kelley, N.; Sarpong, R. Nat. Chem. 2018, 10, 38.
[38]
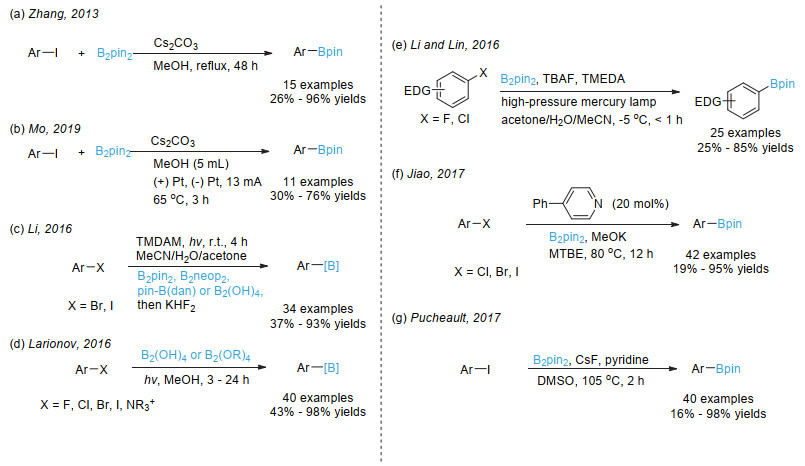
Zhang, L.; Jiao, L. J. Am. Chem. Soc. 2017, 139, 607.
[39]
Zhang, L.; Jiao, L. Chem. Sci. 2018, 9, 2711.
[40]
Pinet, S.; Liautard, V.; Debiais, M.; Pucheault, M. Synthesis 2017, 49, 4759.
[41]
Hu, D.; Wang, L.; Li, P. Org. Lett. 2017, 19, 2770.
[42]
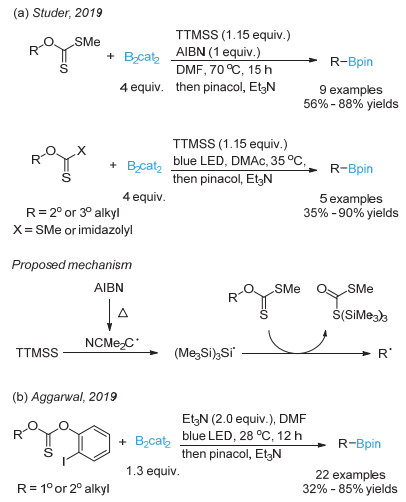
Fawcett, A.; Pradeilles, J.; Wang, Y.; Mutsuga, T.; Myers, E. L.; Aggarwal, V. K. Science 2017, 357, 283.
[43]
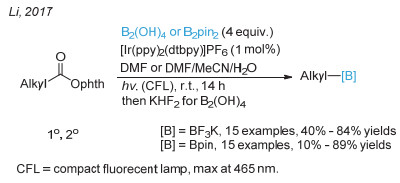
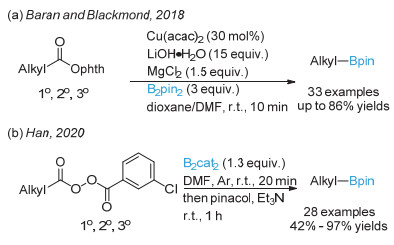
Wang, J.; Shang, M.; Lundberg, H.; Feu, K. S.; Hecker, S. J.; Qin, T.; Blackmond, D. G.; Baran, P. S. ACS Catal. 2018, 8, 9537.
[44]
Wei, D.; Liu, T.-M.; Zhou, B.; Han, B. Org. Lett. 2020, 22, 234.
[45]
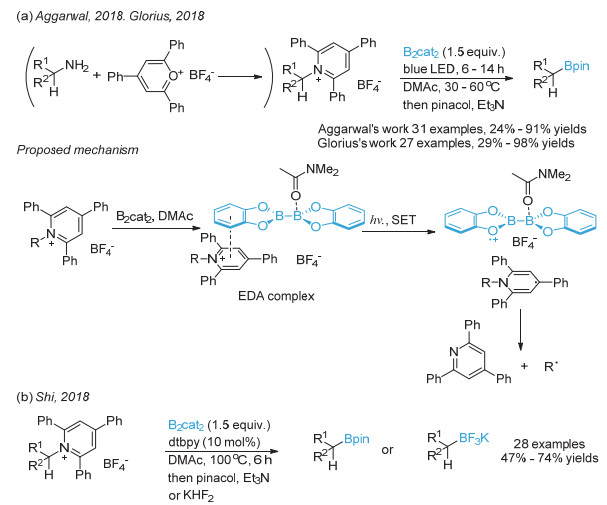
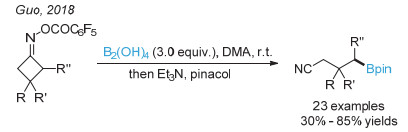
(a) Wu, J.; He, L.; Noble, A.; Aggarwal, V. K. J. Am. Chem. Soc. 2018, 140, 10700; (b) Sandfort, F.; Strieth-Kalthoff, F.; Klauck, F. J. R.; James, M. J.; Glorius, F. Chem. Eur. J. 2018, 24, 17210.

 下载:
下载:
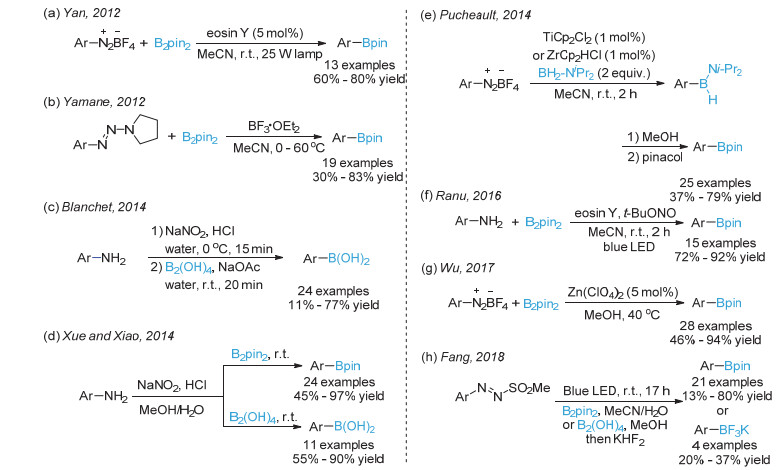
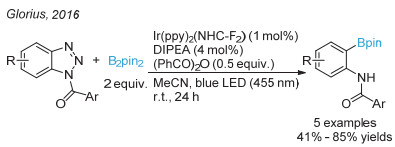
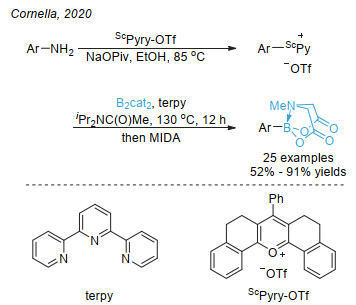
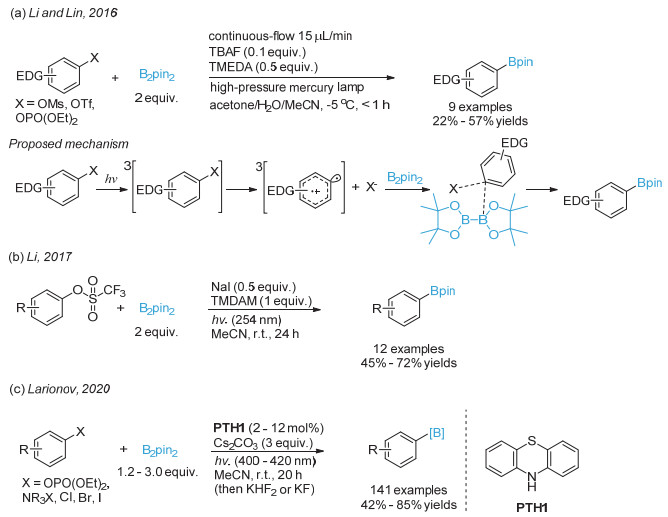


 下载:
下载:





















