Key Laboratory of Functional Inorganic Material Chemistry of Education Ministry, School of Chemistry and Materials Science, Heilongjiang University, Harbin 150080, China
Received Date:
02 July 2020 Available Online:
15 October 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21671060), the Natural Science Foundation of Heilongjiang Province (No. LH2019B029) and the Heilongjiang Touyan Innovation Team Program
Abstract:
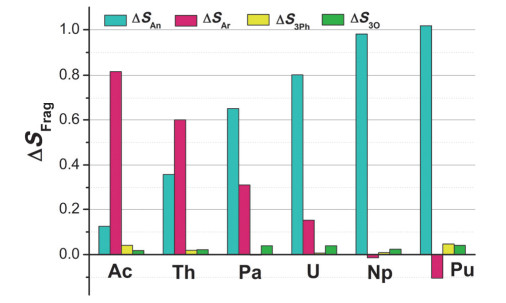
It is of great significance to identify new oxidation state of actinide, which will enrich actinide coordination chemistry and advance its exploration of chemical bond and reactivity. So far, uranium with +3~+6 oxidation states has been widely recognized in complexes. Comparatively, isolated, crystallographically identified U(Ⅱ) complexes remain rare. Inspired by the pioneering work of Evans and co-workers that Y·[UⅡ(Cp')3] (Y=[K(2.2.2-cryptand)]+, Cp'=[C5H4SiMe3]-) was structurally characterized, several uranium(Ⅱ) complexes such as Y·[ULE] (LE=[(Ad, MeArO)3 mesitylene]3-, Ad=adamantyl), [U(NHAriPr6)2] (AriPr6=2, 6-(2, 4, 6-iPr3C6H2)2C6H3), Y·[U{N(SiMe3)2}3] and[U(η5-C5iPr5)2] were synthetically accessible. Inspection finds that all these U(Ⅱ) complexes were prepared in the same route, i.e., utilizing potassium graphite or potassium sphere to reduce respective U(Ⅲ) parent at low temperature. Cyclopentadiene (Cp) and arene (Ar)-based ligands are involved. They are key to determine U(Ⅱ) electron configuration, leading to 5f36d1 and 5f4, respectively. Moreover, δ(U-Ar) bonds play a significant role in stabilizing arene-ligated complexes. With the supporting of Cp-derived ligands, actinide(Ⅱ) complexes were extended to Th, Np and Pu. Unfortunately, it is not the case for the arene ligands, even with massive efforts. Given the prevailing route that actinide(Ⅱ) complex was synthesized by reducing its trivalent parent, the exploration of redox property will help to guide the synthesis of more novel U(Ⅱ) and even other actinide(Ⅱ) complexes. In this respect, theoretical computation based on accurate methodology is greatly appealing. Herein, relativistic density functional theory was exploited to investigate structural and redox properties of[AnL]z (An=Ac~Pu; L=[(Me, MeArOH)3Ar]3-; z=0 and -1), where analogues of uranium complexes were experimentally known. It is found that the central arene moiety is redox-active for Ac and Th complexes in the reduction reaction, while the metal center is reduced for other complexes. So Ac and Th in reduced products still remain +3 oxidation states, whereas metals in others turn +2. The 5fn electronic configuration is unraveled for actinide of[AnL]- (An=Pa~Pu), having 3~6 electrons, respectively. Calculated redox potential (E0) increases from Ac to Pu in general, where U and Np show lower values than adjacent elements. A good correlation has been built between E0 and Δ(An-CAr/An-Arcent)/electron affinity. In brief, the study is expected to provide theoretical support for the synthesis of novel arene-based actinide(Ⅱ) complexes.
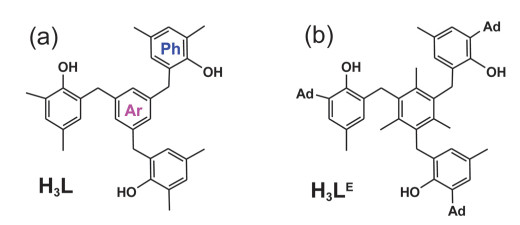
Figure 1.
Ligands of theoretically computed H3L (a) and experimentally synthesized H3LE (b), where the central arene group is labeled as Ar and the three arm ones are named as 3Ph
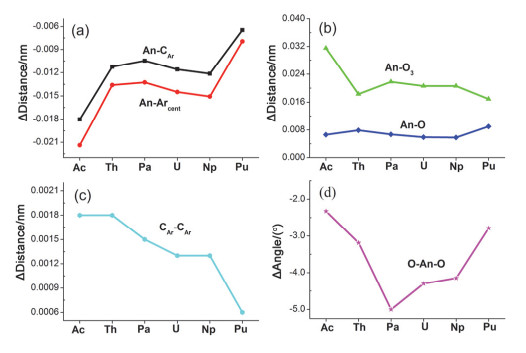
aAverage value; bThe distance between An and the centroid of arene; cAn-O3 denotes the normal distance from the An ion to the plane defined by three oxygen donors. Noting that a positive value means that An is situated between the O3 and Ar planes. d Experimental values from Ref. [8].
Table 3.
Energies (eV) of the single-electron reduction reactions of complexes from [AnL] to [AnL]–, along with reduction potential E0 (V, versus Fc+/Fc) in THF
aThe total energy (ΔrE), total energy including zero-point vibrational energy (ΔrE0) and free energy ΔrG(gas) in the gas phase for the single-electron reduction reactions of actinide complexes. bThe solvation energy is included in ΔrG(sol), and both solvation and spin-orbit energies are added in ΔrG(sol-so). These energies were calculated by the ADF code. cThe free energies, both ΔrG(sol) and ΔrG(sol-so), for the reference electrode of Fc+/Fc were calculated to be -4.99 eV. Thus, reduction potentials, E0(sol) and E0(sol-so), were obtained according to Eq. (2).
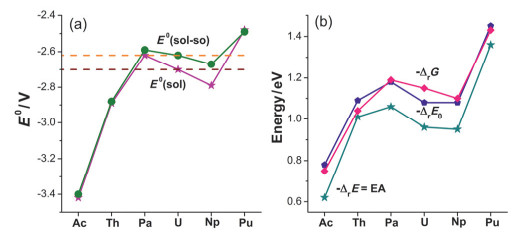
Figure 5.
Calculated reduction potential (E0 in V on the left side) of [AnL]/[AnL]– relative to the reference electrode Fc+/Fc in THF, along with various reaction energies (eV on the right side) where the minus value of ΔrE is equal to the electron affinity (EA)
岳国宗, 高瑞, 赵鹏翔, 褚明福, 帅茂兵, 化学学报, 2016, 74, 657. doi: 10.6023/A16050260Yue, G.; Gao, R.; Zhao, P.; Chu, M.; Shuai, M. Acta Chim. Sinica2016, 74, 657 (in Chinese). doi: 10.6023/A16050260
[7]
MacDonald, M. R.; Fieser, M. E.; Bates, J. E.; Ziller, J. W.; Furche, F.; Evans, W. J. J. Am. Chem. Soc.2013, 135, 13310. doi: 10.1021/ja406791t
[8]
La Pierre, H. S.; Scheurer, A.; Heinemann, F. W.; Hieringer, W.; Meyer, K. Angew. Chem. Int. Ed.2014, 53, 7158. doi: 10.1002/anie.201402050
[9]
Billow, B. S.; Livesay, B. N.; Mokhtarzadeh, C. C.; McCracken, J.; Shores, M. P.; Boncella, J. M.; Odom, A. L. J. Am. Chem. Soc.2018, 140, 17369. doi: 10.1021/jacs.8b10888
[10]
Ryan, A. J.; Angadol, M. A.; Ziller, J. W.; Evans, W. J. Chem. Commun.2019, 55, 2325. doi: 10.1039/C8CC08767A
Langeslay, R. R.; Fieser, M. E.; Ziller, J. W.; Furche, F.; Evans, W. J. Chem. Sci.2015, 6, 517. doi: 10.1039/C4SC03033H
[16]
Su, J.; Windorff, C. J.; Batista, E. R.; Evans, W. J.; Gaunt, A. J.; Janicke, M. T.; Kozimor, S. A.; Scott, B. L.; Woen, D. H.; Yang, P. J. Am. Chem. Soc.2018, 140, 7425. doi: 10.1021/jacs.8b03907
[17]
Dutkiewicz, M. S.; Apostolidis, C.; Walter, O.; Arnold, P. L. Chem. Sci.2017, 8, 2553. doi: 10.1039/C7SC00034K
[18]
Windorff, C. J.; Chen, G. P.; Cross, J. N.; Evans, W. J.; Furche, F.; Gaunt, A. J.; Janicke, M. T.; Kozimor, S. A.; Scott, B. L. J. Am. Chem. Soc.2017, 139, 3970. doi: 10.1021/jacs.7b00706
[19]
Wu, Q. Y.; Lan, J. H.; Wang, C. Z.; Cheng, Z. P.; Chai, Z. F.; Gibson, J. K.; Shi, W. Q. DaltonTrans. 2016, 45, 3102. doi: 10.1039/C5DT04540A
Fieser, M. E.; Palumbo, C. T.; La Pierre, H. S.; Halter, D. P.; Voora, V. K.; Ziller, J. W.; Furche, F.; Meyer, K.; Evans, W. J. Chem. Sci.2017, 8, 7424. doi: 10.1039/C7SC02337E
[28]
Lewis, A. J.; Carroll, P. J.; Schelter, E. J. J. Am. Chem. Soc.2013, 135, 13185. doi: 10.1021/ja406610r
Baerends, E. J.; Ziegler, T.; Autschbach, J.; Bashford, D.; Bérces, A.; Bickelhaupt, F. M.; Bo, C.; Boerrigter, P. M.; Cavallo, L.; Chong, D. P.; Deng, L.; Dickson, R. M.; Ellis, D. E.; van Faassen, M.; Fan, L.; Fischer, T. H.; Fonseca Guerra, C.; Franchini, M.; Ghysels, A.; Giammona, A.; van Gisbergen, S. J. A.; Gö tz, A. W.; Groeneveld, J. A.; Gritsenko, O. V.; Grüning, M.; Gusarov, S.; Harris, F. E.; van den Hoek, P.; Jacob, C. R.; Jacobsen, H.; Jensen, L.; Kaminski, J. W.; van Kesse, G.; Kootstra, F.; Kovalenko, A.; Krykunov, M. V.; van Lenthe, E.; McCormack, D. A.; Michalak, A.; Mitoraj, M.; Morton, S. M.; Neugebauer, J.; Nicu, V. P.; Noodleman, L.; Osinga, V. P.; Patchkovskii, S.; Pavanello, M.; Philipsen, P. H. T.; Post, D.; Pye, C. C.; Ravenek, W.; Rodríguez, J. I.; Ros, P.; Schipper, P. R. T.; van Schoot, H.; Schreckenbach, G.; Seldenthuis, J. S.; Seth, M.; Snijders, J. G.; Solà, M.; Swart, M.; Swerhone, D.; te Velde, G.; Vernooijs, P.; Versluis, L.; Visscher, L.; Visser, O.; Wang, F.; Wesolowski, T. A.; van Wezenbeek, E. M.; Wiesenekker, G.; Wolff, S. K.; Woo, T. K.; Yakovlev, A. L., ADF (2014.06 version), SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2014.
[34]
Klamt, A.; Schuurmann, G. J. Chem. Soc., Perkin Trans.1993, 799.
Figure 1
Ligands of theoretically computed H3L (a) and experimentally synthesized H3LE (b), where the central arene group is labeled as Ar and the three arm ones are named as 3Ph
Figure 5
Calculated reduction potential (E0 in V on the left side) of [AnL]/[AnL]– relative to the reference electrode Fc+/Fc in THF, along with various reaction energies (eV on the right side) where the minus value of ΔrE is equal to the electron affinity (EA)
Table 1.
Optimized geometry parameters for [AnL]– and [AnL] (Distance in nm and angle in (°))
Complexes.
Approaches
An-CAra
An-Arcentb
An-Oa
An-O3c
CAr-CAra
O-An-Oa
[AcL]–
Calc.
0.2897
0.2523
0.2400
0.0380
0.1426
117.6
[ThL]–
Calc.
0.2654
0.2237
0.2286
0.0622
0.1443
113.0
[PaL]–
Calc.
0.2554
0.2111
0.2230
0.0772
0.1440
108.7
[UL]–
Calc.
0.2576
0.2143
0.2213
0.0685
0.1431
110.9
Expt. d
0.2615
0.2180
0.2236
0.0668
0.1432
111.5
[NpL]–
Calc.
0.2595
0.2168
0.2214
0.0655
0.1428
111.6
[PuL]–
Calc.
0.2700
0.2298
0.2262
0.0580
0.1418
113.8
[AcL]
Calc.
0.3078
0.2737
0.2334
0.0065
0.1408
119.9
[ThL]
Calc.
0.2767
0.2373
0.2207
0.0439
0.1425
116.2
[PaL]
Calc.
0.2658
0.2244
0.2163
0.0554
0.1425
113.7
[UL]
Calc.
0.2692
0.2288
0.2154
0.0479
0.1418
115.2
Expt. d
0.2749
0.2350
0.2168
0.0480
0.1433
115.3
[NpL]
Calc.
0.2716
0.2319
0.2156
0.0449
0.1415
115.8
[PuL]
Calc.
0.2765
0.2378
0.2172
0.0411
0.1412
116.5
aAverage value; bThe distance between An and the centroid of arene; cAn-O3 denotes the normal distance from the An ion to the plane defined by three oxygen donors. Noting that a positive value means that An is situated between the O3 and Ar planes. d Experimental values from Ref. [8].
Table 3.
Energies (eV) of the single-electron reduction reactions of complexes from [AnL] to [AnL]–, along with reduction potential E0 (V, versus Fc+/Fc) in THF
An
ΔrEa
ΔrE0a
ΔrG(gas)a
ΔrG(sol)b
ΔrG(sol-so)b
E0(sol)c
E0(sol-so)c
Ac
-0.62
-0.78
-0.75
-1.57
-1.59
-3.42
-3.40
Th
-1.01
-1.09
-1.04
-2.10
-2.10
-2.89
-2.88
Pa
-1.06
-1.18
-1.19
-2.37
-2.39
-2.62
-2.59
U
-0.96
-1.08
-1.15
-2.29
-2.37
-2.70
-2.62
Np
-0.95
-1.08
-1.10
-2.20
-2.32
-2.79
-2.67
Pu
-1.36
-1.45
-1.43
-2.51
-2.50
-2.48
-2.49
aThe total energy (ΔrE), total energy including zero-point vibrational energy (ΔrE0) and free energy ΔrG(gas) in the gas phase for the single-electron reduction reactions of actinide complexes. bThe solvation energy is included in ΔrG(sol), and both solvation and spin-orbit energies are added in ΔrG(sol-so). These energies were calculated by the ADF code. cThe free energies, both ΔrG(sol) and ΔrG(sol-so), for the reference electrode of Fc+/Fc were calculated to be -4.99 eV. Thus, reduction potentials, E0(sol) and E0(sol-so), were obtained according to Eq. (2).

 下载:
下载:

 下载:
下载:






