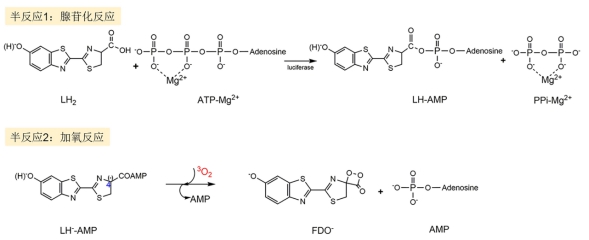
图 1.
荧光素酶催化氧化反应阶段的两个半反应
Figure 1.
Two half reactions in enzymatic oxygenation of luciferin (LH2)

生物发光是一种有趣的现象, 给自然界增加了闪亮的色彩[1, 2]. 大多数的发光生物(80%)生存于海洋之中[3], 而人们最熟知的发光生物是萤火虫(firefly)[4]. 萤火虫生物发光的光量子产率达0.41[5], 已在基因表达、体内成像、药物筛选等生命科学和医学技术领域得到广泛的实际应用. 萤火虫属于鞘翅目萤科(Lampyrinae)[6], 目前发现有超过2000种萤火虫广泛分布于世界各地. 经过多年的实验[7-9]和理论研究[10-13], 人们认识到萤火虫生物发光的过程包含荧光素(LH2)催化氧化生成高能中间体、高能中间体解离生成发光体、发光体发出可见光和荧光素分子的再生四个阶段. 后三个阶段的机理得到了详细的理论研究和机理阐释[14-16], 但第一阶段的反应机理至今仍未有全面可靠的理论研究. 这一氧化反应是萤火虫生物发光, 也是所有氧气依赖型的生物发光体系的启动反应. 对该反应的正确认知对所有氧气依赖型的生物发光的机理理解都具有重要意义. 这正是本工作的研究目的.
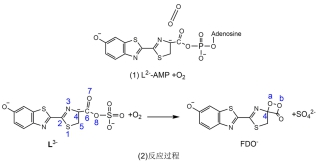
荧光素氧化生成高能中间体的反应可以细化为两个半反应[17](图 1): 腺苷化反应和加氧反应. 腺苷化反应在众多生物体系中普遍存在, 已有成熟的实验[18, 19]和理论研究[20-22], 研究结果一致认为该反应过程为: LH2在荧光素酶的催化下与腺苷三磷酸-镁离子复合物(ATP-Mg2+)发生SN2亲核取代反应, 由亲核试剂进攻ATP的α位点, 脱去焦磷酸-镁离子复合物(PPi-Mg2+), 生成荧光素-腺苷一磷酸复合物(LH-AMP). 其中ATP为萤火虫闪烁发光提供能量[23], Mg2+用于稳定构象和屏蔽ATP中的负电荷[23]. 加氧反应的大致过程为: LH-AMP复合物与O2发生加氧反应, 伴随AMP的离去, 生成四元环结构的高能中间体即萤火虫1, 2-二氧环丁酮(FDO)[24]. 2015年, Branchini等[25]在萤火虫生物发光的化学模型反应中检测到了超氧负离子(O2•−), 由此提出了萤火虫生物发光由LH2到O2的单电子转移(SET)诱发. 首先荧光素分子的C4位(见图 1)的质子被附近的氨基酸残基脱去而形成LH2−离子, 这一过程也得到了理论计算的验证[26]. 随后从LH--AMP到O2的SET产生LH-AMP和O2•−, 从而诱发之后的加氧反应. 尽管SET机理产生LH-AMP和O2•−得到很强的实验证据支持, 但随后从LH-AMP+O2•−到FDO−的反应过程和路径并没有明确完整的描述. LH-AMP中苯并噻唑环羟基上的质子(见图 1), 在荧光素酶环境中容易失去[7, 27]. 另外, 实验[28]和理论计算[14, 15]工作都指认了FDO−(苯并噻唑环羟基去质子化)是LH2催化氧化的产物和下一阶段反应-高能中间体解离的反应物. 因此, LH2催化氧化反应的实际反应物为L2−-AMP+O2 (见图 2). AMP侧链柔性很大且距离反应中心很远, 为了计算的方便, 我们使用去质子化的磺酸基(-OSO3−)代替AMP. 本工作最终采用的计算模型为L3−+O2→FDO−+SO42−(见图 2). 目前在真实蛋白环境计算整个反应过程不现实. 计算考虑了荧光素酶活性口袋的极性, 详见计算方法部分.
三重态的O2分子与单重态的底物分子L3−反应, 必须经过自旋翻转, 才会最终得到单重态的产物FDO−. 三重态转换为单重态的过程在没过渡金属离子等磁性物质催化的条件下, 只能通过两个二重态的自由基重排的方式生成自旋对称性允许的单重态双自由基. 首先L3−的C4位负电荷转移到的3O2(SET)形成L•2−和O2•−自由基对. 这一步已得到证实, 无需进一步计算. 我们要研究的是从L•2−和O2•−形成的反应复合物RC到FDO−的过程. RC自由基对存在两种形式3[L•2−…O2•−]和 1[L•2−…O2•−].
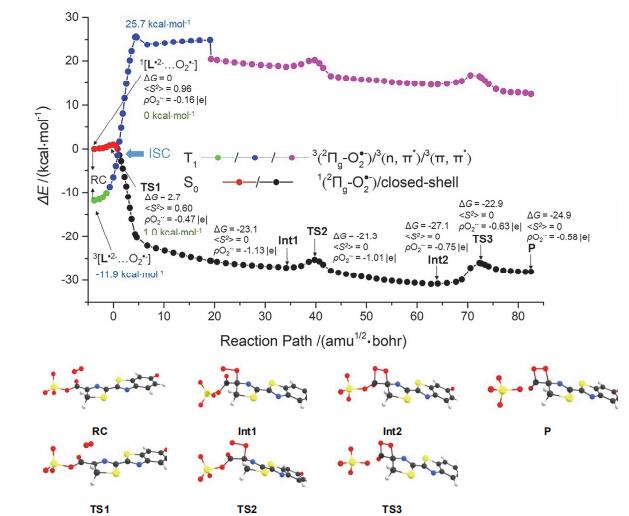
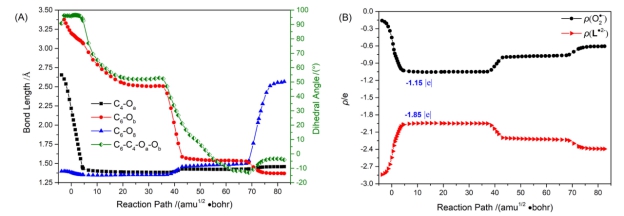
首先以1[L•2−…O2•−]作为初始反应物. 在1[L•2−…O2•−]→FDO−的反应过程中, 从RC到产物(P)共优化得到两个中间体(Int1和Int2)和三个过渡态(TS1、TS2和TS3), 见图 3. 频率分析显示TS1 (−266.6cm−1)、TS2 (−194.2cm−1)和TS3 (−173.5cm−1)的唯一虚频分别对应C4—Oa、C6—Ob和C6—O8键的伸缩振动. 各稳定点的关键几何结构参数见附录数据(SI)中的表S1. 关键的几何结构参数的变化及O2•−和L•2−两部分电荷变化分别总结在图 4中. 1[L•2−…O2•−]中Oa—Ob键长为1.218 Å, 长于正常的三重态氧气分子3O2的键长(约为1.205 Å). O2•−由L3−到3O2 的SET刚刚生成时, 其电荷密度 ρ(O2•−)应为−1.00 |e|. 而在RC复合物形成过程中O2•−的电荷部分转移给L•3−, O2•−部分的电荷变为−0.16 |e|. 结合图 3和图 4A可见, 随着反应的进行, C4—Oa的键长由RC中的2.655 Å缩短为2.219 Å, 并伴随C6-C4-Oa-Ob二面角向四元环闭合方向的调整, 形成TS1. RC和TS1的<S2>分别为0.96和0.60, 说明两者都具有双自由基性质. TS1之后, 双自由基性质消失. 从RC到TS1克服2.7 kcal•mol−1的能垒之后, C4—Oa键长继续缩短, C6-C4-Oa-Ob二面角扭转, 达到Int1. Int1之后C4—Oa键长几乎不再变化. 如图 4B所示, 在此区域内O2•−的电荷 ρ(O2•−)约为−1.15 |e|, 可以说明Int1就是C4−过氧键加成离子. 随后C6—Ob键长曲线和C6-C4-Oa-Ob二面角曲线先快速下降, 之后C6—Ob键长曲线趋于平坦, 翻越1.8 kcal•mol−1的TS2能垒. C6-C4-Oa-Ob二面角继续扭转形成四元环结构, 得到Int2. C6—O8键长从加氧反应开始就基本保持不变, Int2之后C6—O8键开始断裂, 导致去质子化的磺酸基离去, 经过能垒为4.2 kcal•mol−1的TS3, 伴随C6-C4-Oa-Ob二面角的轻微调整, 最终得到P. 如图 3所示, 在反应过程中, L•2−电子态的性质也在变化. 不同性质的代表性电子态自然轨道见表S2. 据自然轨道分析, 反应开始L•2−为2Πg组态, 随着C4—Oa键长的缩短, O2分子中单占的n轨道与底物分子L•2−上的轨道接近, 经过TS1变为闭壳层组态(closed-shell state), 此时所有成键轨道都是双占的. 我们简写为1(2Πg-O2•−)/ closed-shell. 如图 4B, TS1之前, L•2−的电荷向O2•−迁移, 随后L•2−的电荷降为−1.85 |e|, O2•−电荷升为−1.15 |e|. 在4.292到37.126 amu1/2•bohr区间内, O2•−电荷一直保持−1.15 |e|. TS2生成后O2•−电荷部分回迁至L•2−部分, 最终得到产物时L•2−和O2•−两部分的电荷分别为−2.42 |e|和−0.58 |e|. 当3[L•2−…O2•−]作为初始反应物时, 反应过程中各稳定点的第一三重态(T1)能量基于相应的S0构型进行评估, 见图 3. 对于T1态, 在RC处L•2−同样为2Πg组态. 随后3[L•2−…O2•−]在TS1附近出现(n, π*)组态的性质. 其中n指Oa和Ob上的p轨道, π*主要分布在L3−中N=C—C=N共轭结构的反键π轨道. 随着二面角向四元环闭合方向扭转, O2•−的π*轨道与分布在L3−上的π轨道耦合, 在Int1形成前表现出(π, π*)的性质. 随后C6—Ob逐渐成键, O2•−部分电荷回迁至L•2−上, T1态仍然表现出(π, π*)的性质, 这里π和π*轨道都分布在L•2−上. 我们将T1态简写为3(2Πg-O2•−)/3(n, π*)/3(π, π*). 3[L•2−…O2•−]比1[L•2−…O2•−]的能量低11.9 kcal•mol−1. 在TS1附近S0态能量下降T1态能量上升, 两条能量曲线交叉后T1态能量远远高于S0态能量. 由于TS1附近S0态与T1态能量近简并(在TS1处相差约7.6 kcal•mol−1, 见表S3)而且过TS1后能量更接近, 这意味着在TS1附近存在一个S/T交叉点(intersystem crossing, ISC). 不幸的是, 目前所使用的计算方法不能优化S0/T1交叉点. 该体系合理的活化空间大小已超出目前多组态方法的处理能力. 我们在萤火鱿生物发光的SET加氧过程中[29]也遇到类似情况. 但在研究细菌生物发光时, 对于较小体系的类似反应采用多组态计算确实得到了S0/T1交叉点[30]. 从图 4可见, 反应从3[L•2−…O2•−]出发, 先在T1位能面上进行, 随后经过S0/T1交叉点, 沿S0位能面生成1FDO−.
本工作采用密度泛函理论, 首次使用模型分子详细研究了萤火虫生物发光的起始反应-荧光素催化氧化生成高能中间体的步骤. 反应由单重态L3−到三重态O2的SET诱发, 先生成RC 3[L•2−…O2•−]. 随后O2•−亲核进攻L•2−的C4位, 并在TS1附近经过S0/T1的ISC. 之后, 反应沿S0位能面进行, 经过过渡态TS2和TS3生成 FDO−. 电子密度和自然轨道结果表明, 在ISC附近发生双自由基湮灭的快速反应. 反应从三重态位能面开始, 到达ISC约需11.9 kcal•mol−1能量, 随后反应在S0位能面进行. 在S0位能面进行的反应最高的能垒仅为4.2 kcal•mol−1. ISC附近的双自由基湮灭和S0位能面的超低能垒充分说明这个自旋禁阻的反应可以高效率发生. 研究结论对理解萤火虫高效生物发光具有重要的理论意义, 也对理解其它氧气依赖型生物发光体系的起始反应有帮助.
采用M06-2X[31]/6-31G**计算. 该级别的密度泛函理论(DFT)[32, 33]计算适合现在的研究体系[34, 35]. 因为所研究的反应涉及到三重态和开壳层单重态的体系, 因此整个反应路径上的稳定点和过渡态的优化和频率分析均使用非限制性开壳层方法结合对称性破损(broken-symmetry, BS)技术. 萤火虫荧光素酶的活性口袋是一个疏水的空腔, Gilson[36]和Pitera[37]等分别在1986年和2001年对空腔的静态介电常数(ε)进行了估算, 约为2到4. 在此我们选用二甲基亚砜(DMSO)(ε= 2.25)用类导体极化连续介质模型(conductor-like polarizable continuum model, CPCM)[38]来模拟环境的极性. DMSO也是生物发光的实验研究中常用的极性溶剂[39]. 通过内禀反应坐标(intrinsic reaction coordinate, IRC)计算确定所优化得到的各过渡态正确连接对应的反应物和产物. 反应过程的电荷变化采用了自然布居分析(natural population analysis, NPA). 所有稳定态的能量都进行了零点能校正. 计算使用了Gaussian 09程序包[40].
陈浮, 刘树深, 段欣甜, 化学学报, 2013, 71, 1035. http://www.cnki.com.cn/Article/CJFDTotal-HXXB201307010.htmChen, F.; Liu, S.; Duan, X. Acta Chim. Sinica 2013, 71, 1035(in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-HXXB201307010.htm
Wilson, T.; Hastings, J. W. Annu. Rev. Cell. Dev. Bi. 1998, 14, 197. doi: 10.1146/annurev.cellbio.14.1.197
Haddock, S. H. D.; Moline, M. A.; Case, J. F. Annu. Rev. Mar. Sci. 2010, 2, 443. doi: 10.1146/annurev-marine-120308-081028
Lee, J. Bioluminescence, the Nature of the Light, University of Georgia, 2020, pp. 102~114. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Ando, Y.; Niwa, K.; Yamada, N.; Enomot, T.; Irie, T.; Kubota, H.; Ohmiya, Y.; Akiyama, H. Nat. Photonics 2008, 2, 44. doi: 10.1038/nphoton.2007.251
Wood, K. V. Photochem. Photobiol. 1995, 62, 662. doi: 10.1111/j.1751-1097.1995.tb08714.x
Ugarova, N. N.; Brovko, L. Y. Luminescence 2002, 17, 321. doi: 10.1002/bio.688
Schmidt, S. P.; Schuster, G. B. J. Am. Chem. Soc. 1978, 100, 1966. doi: 10.1021/ja00474a074
Day, J. C.; Tisi, L. C.; Bailey, M. J. Luminescence 2004, 19, 8. doi: 10.1002/bio.749
Navizet, I.; Liu, Y.-J.; Ferre, N.; Xiao, H.-Y.; Fang, W.-H.; Lindh, R. J. Am. Chem. Soc. 2010, 132, 706. doi: 10.1021/ja908051h
Orlova, G.; Goddard, J. D.; Brovko, L. Y. J. Am. Chem. Soc. 2003, 125, 6962. doi: 10.1021/ja021255a
Wilsey, S.; Bernardi, F.; Olivucci, M.; Robb, M. A.; Murphy, S.; Adam, W. J. Phys. Chem. A 1999, 103, 1669. doi: 10.1021/jp9848086
闵春刚, 李作盛, 崔小英, 杨喜昆, 黄绍军, 王绍华, 任爱民, 有机化学, 2015, 35, 432. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498Min, C.; Li, Z.; Cui, X.; Yang, X.; Huang, S.; Wang, S.; Ren, A. Chin. J. Org. Chem. 2015, 35, 432(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Yue, L.; Liu, Y.-J.; Fang, W.-H. J. Am. Chem. Soc. 2012, 134, 11632. doi: 10.1021/ja302979t
Yue, L.; Lan, Z.; Liu, Y.-J. J. Phys. Chem. Lett. 2015, 6, 540. doi: 10.1021/jz502305g
Cheng, Y.-Y.; Liu, Y.-J. ChemPhysChem 2019, 20, 1720. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Branching, B. R.; Rosenberg, J. C.; Fontaine, D. M.; Southworth, T. L.; Behney, C. E.; Uzasci, L. J. Am. Chem. Soc. 2011, 133, 11088. doi: 10.1021/ja2041496
Marahiel, M. A.; Stachelhaus, T.; Mootz, H. D. Chem. Rev. 1997, 97, 2651. doi: 10.1021/cr960029e
Westheimer, F. H. Science 1987, 235, 1173. doi: 10.1126/science.2434996
Barrozo, A.; Blaha-Nelson, D.; Williams, N. H.; Kamerlin, S. C. L. Pure Appl. Chem. 2017, 89, 715. doi: 10.1515/pac-2016-1125
Duarte, F.; qvist, J.; Williams, N. H.; Kamerlin, S. C. L. J. Am. Chem. Soc. 2015, 137, 1081. doi: 10.1021/ja5082712
Duarte, F.; Barrozo, A.; qvist, J.; Williams, N. H.; Kamerlin, S. C. L. J. Am. Chem. Soc. 2016, 138, 10664. doi: 10.1021/jacs.6b06277
Nelson, D. L.; Cox, M. M. Lehninger Principles of Biochemistry, 7th ed., W. H. Freeman and Company, New York, 2013, pp. 1370~1380. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Koo, J.-Y.; Schuster, G. B. J. Am. Chem. Soc. 1977, 99, 6107. doi: 10.1021/ja00460a050
Branchini, B. R.; Behney, C. E.; Southworth, T. L.; Fontaine, D. M.; Gulick, A. M.; Vinyard, D. J.; Brudvig, G. W. J. Am. Chem. Soc. 2015, 137, 7592. doi: 10.1021/jacs.5b03820
Berraud-Pache, R.; Lindh, R.; Navizet, I. J. Phys. Chem. B 2018, 122, 5173. doi: 10.1021/acs.jpcb.8b00642
Hirano, T.; Hasumi, Y.; Ohtsuka, K.; Maki, S.; Niwa, H.; Yamaji, M.; Hashizume, D. J. Am. Chem. Soc. 2009, 131, 2385. doi: 10.1021/ja808836b
Shimomura, O. In Bioluminescence:Chemical Principles and Methods, World Scientific Publishing Co. Pte. Ltd., Singapore, 2006. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Ding, B. W.; Liu, Y. J. J. Am. Chem. Soc. 2017, 139, 1106. doi: 10.1021/jacs.6b09119
Luo, Y.; Liu, Y.-J. J. Phys. Chem. A 2019, 123, 4354. doi: 10.1021/acs.jpca.9b02084
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2007, 120, 215. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Perdew, J. P.; Ruzsinszky, A.; Tao, J. M.; Staroverov, V. N.; Scuseria, G. E.; Csonka, G. I. J. Chem. Phys. 2005, 123, A1133. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Dreuw, A.; Head-Gordon, M. Chem. Rev. 2005, 105, 4009. doi: 10.1021/cr0505627
Boggio-Pasqua, M.; Heully, J.-L. Theor. Chem. Acc. 2015, 135, 9. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Ess, D. H.; Cook, T. C. J. Phys. Chem. A 2012, 116, 4922. doi: 10.1021/jp300633j
Gilson, M. K.; Honig, B. H. Biopolymers 1986, 25, 2097. doi: 10.1002/bip.360251106
Pitera, J. W.; Falta, M.; van Gunsteren, W. F. Biophys. J. 2001, 80, 2546. doi: 10.1016/S0006-3495(01)76226-1
Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. J. Comput. Chem. 2003, 24, 669. doi: 10.1002/jcc.10189
Nakatani, N.; Hasegawa, J.-y.; Nakatsuji, H. J. Am. Chem. Soc. 2007, 129, 8756. doi: 10.1021/ja0611691
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision D. Gaussian Inc., Wallingford, 2009. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498

图 1 荧光素酶催化氧化反应阶段的两个半反应
Figure 1 Two half reactions in enzymatic oxygenation of luciferin (LH2)

图 3 UM06-2X/6-31G**计算的从3[L•2−…O2•−]/1[L•2−…O2•−]到FDO−的反应位能曲线及稳定点的结构. 其中红色、黄色、黑色、蓝色和白色小球分别代表O、S、C、N和H原子. (ΔG单位为kcal•mol−1)
Figure 3 UM06-2X/6-31G** calculated potential energy curve of reaction from 3[L•2−…O2•−]/1[L•2−…O2•−]. Here, red, yellow, black, blue and white balls represent oxygen, sulfur, carbon, nitrogen and hydrogen atoms. (The units of ΔGis kcal•mol−1)

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:

