School of Chemistry and Chemical Engineering, Advanced Research Institute of Multidisciplinary Science, Beijing Institute of Technology, Beijing 100081, China
Received Date:
22 June 2020 Available Online:
15 October 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21922502, 21674012)
Abstract:
Metal-organic frameworks (MOFs) and covalent organic frameworks (COFs) are representative crystalline porous polymers. Due to their high surface area, high porosity, open channels, abundant functional groups and easy functionalization, they show great applications in gas storage and separation, catalysis, energy storage, photovoltaic devices, etc. Amino acids are the basic structural units that constitute peptides and proteins, which not only have important biological functions, but also play an important role in industrial applications such as pharmaceutical production, biodegradable plastics, and chiral catalysts. The introduction of amino acids into MOFs and COFs could endow them with diverse and flexible frameworks, special pore environment, and chiral sites, improving their biocompatibility and degradability to some extent and enriching their functions and applications. This review focuses on the progress of the amino acid functionalized MOFs and COFs, including their synthetic strategies, such as employing amino acids and their derivatives as building unit, covalent modification of amino acids onto the framework, and utilizing amino acids as modulators. The advantages and disadvantages of these strategies are compared and their challenges are discussed. In addition, we also introduce their applications in chiral separation, catalysis, adsorption and proton conduction. Finally, we summarize the current challenges in the preparation of amino acid functionalized crystalline porous polymers and outlook the future research direction in this field.
Figure 3.
(a) Schematic representation of linkers L1, L2, and L3 that react with Cd(II) to produce corresponding MOF architectures (MOF 1a/1b, 2a/2b, and 3a/3b). (b) MOFs 1a, 2a, and 3a with their SBUs and topological simplification model[17]
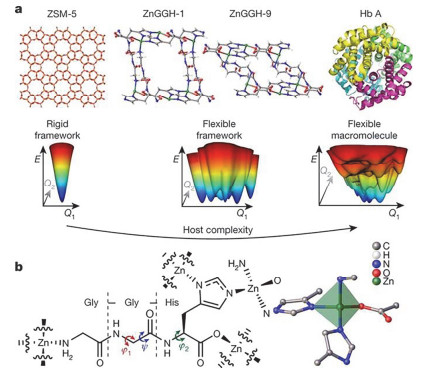
Figure 5.
The structural flexibility of ZnGGH in the context of a conformational energy landscape. (a) ZSM-5 is a rigid porous framework, the new ZnGGH compound reported here is a flexible porous framework. In contrast to the macromolecular state of proteins, ZnGGH is a crystalline solid framework, which restricts the available conformational space of the linker. All energy landscapes drawn in this figure are hypothetical illustrations that reflect the structural properties of the corresponding materials. (b) The doubly deprotonated tripeptide Gly-Gly-L-His acts as a tetratopic linker to form the ZnGGH framework. Adapted from the literature [25]
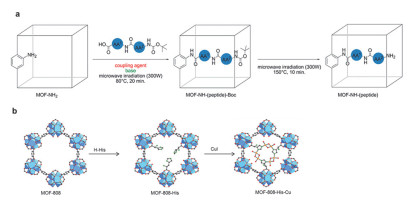
Figure 6.
(a) The two-step peptide grafting process into various MOFs. (b) Synthesis of the catalysts comprising the replacement of formate with imidazole-containing ligands and metalation with Cu(I). Adapted from the literature [29], [31]
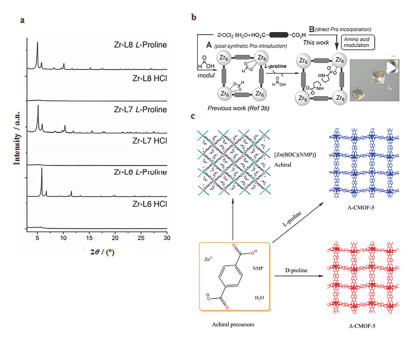
Figure 7.
(a) PXRD patterns showing the effects of incorporation of 5 equivalents of L-proline and one of HCl into solvothermal syntheses of Zr MOFs of L6~L8 compared to addition of HCl alone. (b) Anchoring of L-proline onto a Zr MOF and amino acid-modulated MOF synthesis. (c) Synthesis of CMOF-5. The presence of L- or D-proline additives induces chirality and enables the growth and isolation of CMOF-5 crystals. An achiral square grid net, [Zn(BDC)(NMP)], forms in the absence of proline. Adapted from the literature [32], [33], [36]
Figure 9.
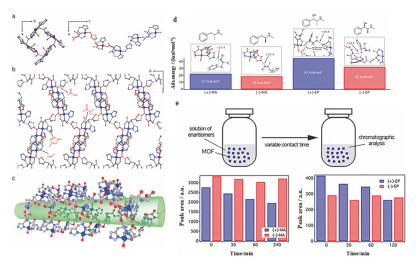
(a) Structure of helicoidal chains in Cu(GHG) MOF. (b) Functional groups in the peptidic backbone decorating the surface of the pores. (c) 1D channels in Cu(GHG) are surrounded by functional sites prone to establish supramolecular interactions, well suited for chiral recognition and discrimination. (d) Representative MC binding geometries of (+, -) MA and EP enantiomers within the structure of Cu(GHG) and corresponding adsorption energies as absolute values calculated with respect to gas phase. (e) Evolution of the enantioselective recognition of Cu(GHG) for chiral drugs MA and EP. Adapted from the literature [45]
Figure 10.
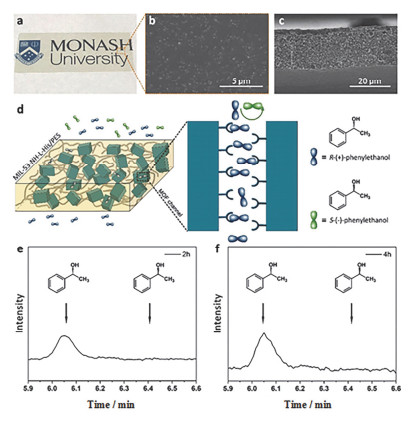
Fabrication and characterization of homochiral MOF-based MMMs for chiral separation of 1-phenylethanol enantiomers. Adapted from the literature [48]
Figure 11.
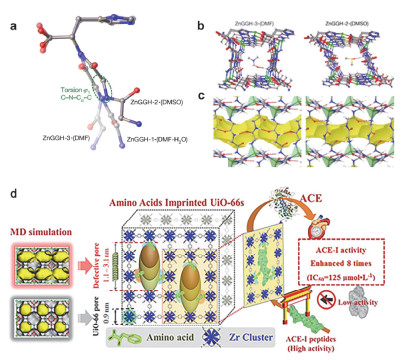
(a) Conformation of the GGH linker in the refined experimental crystal structures ZnGGH-1•(DMF-H2O), ZnGGH-2•(DMSO) and ZnGGH-3•(DMF).
(b) The view down the one-dimensional pores of ZnGGH-3•(DMF) and ZnGGH-2•(DMSO). (c) The side view reveals the intraframework hydrogen-bond pattern for each structure. (d) Amino acid imprinted UiO-66s for highly recognized adsorption of small angiotensin- converting-enzyme-inhibitory peptides. Adapted from the literature [25], [34]
Figure 12.
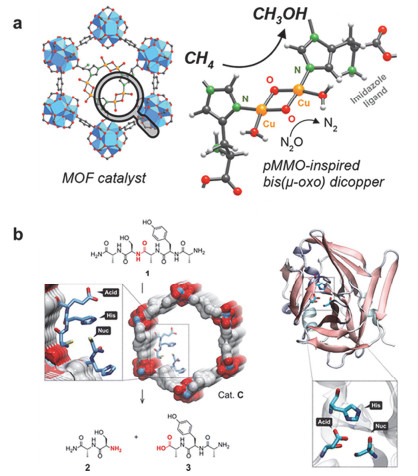
(a) Bioinspired metal-organic framework catalysts for selective methane oxidation to methanol. (b) Catalytic cleavage of pentapeptide 1 by Cat. C [MTVIRMOF-74-III-(CH3)0.6(CH2NH-Asp-His-Cys-NH2)0.1] in the specific sequence containing serine, and cartoon representation of the enzyme TEV endoprotease, highlighting the three amino acids that participate in the catalysis. Adapted from the literature [30], [31]
Figure 13.
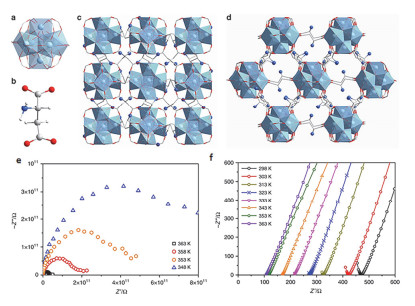
(a) The 12-connected Zr6(µ3-O)4(µ3-OH)4(COO-)12 cluster SBU. (b) An aspartic acid linker. (c) The crystal structure of MIP-202(Zr) viewed along the a-axis. (d) The crystal structure of MIP-202(Zr) viewed along the (101) plane. (e) and (f) Nyquist plots from AC impedance data for MIP-202(Zr) at 0% and 95% RH. Adapted from the literature [16]
(a) Ploetz, E.; Engelke, H.; Lchelt, U.; Wuttke, S. Adv. Funct. Mater. 2020, DOI: 10.1002/adfm.201909062. (b) Ferey, G. Chem. Soc. Rev. 2008, 37, 191. (c) Ferey, G.; Mellot-Draznieks, C.; Serre, C.; Millange, F. Acc. Chem. Res. 2005, 38, 217. (d) Horike, S.; Shimomura, S.; Kitagawa, S. Nat. Chem. 2009, 1, 695. (e) Kitagawa, S.; Kitaura, R.; Noro, S. Angew. Chem. Int. Ed. 2004, 43, 2334. (f) Yaghi, O. M. Mol. Front. J. 2019, 03, 66.
[2]
(a) Wang, Q.; Astruc, D. Chem. Rev.2019, 120, 1438. (b) Stock, N.; Biswas, S. Chem. Rev.2012, 112, 933. (c) Rowsell, J. L. C.; Yaghi, O. M. Micropor. Mesopor. Mat.2004, 73, 3. (d) Furukawa, H.; Ko, N.; Go, Y. B.; Aratani, N.; Choi, S. B.; Choi, E.; Yazaydin, A. O.; Snurr, R. Q.; O'Keeffe, M.; Kim, J.; Yaghi, O. M. Science2010, 329, 424.
(a) Zhao, X.; Wang, Y.; Li, D. S.; Bu, X.; Feng, P. Adv. Mater.2018, 30, 1705189. (b) Zeng, Y.; Zou, R.; Zhao, Y. Adv. Mater.2016, 28, 2855. (c) Wang, Y.; Huang, N. Y.; Shen, J. Q.; Liao, P. Q.; Chen, X. M.; Zhang, J. P. J. Am. Chem. Soc.2018, 140, 38. (d) Rodenas, T.; Luz, I.; Prieto, G.; Seoane, B.; Miro, H.; Corma, A.; Kapteijn, F.; Llabrés i Xamena, F. X.; Gascon, J. Nat. Mater.2014, 14, 48. (e) Furukawa, H.; Cordova, K. E.; O'Keeffe, M.; Yaghi, O. M. Science2013, 341, 974. (f) Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O'Keeffe, M.; Yaghi, O. M. Science2002, 295, 469. (g) Guo, Z.; Zhang, Y.; Feng, X. Acta Chim. Sinica2020, 78, 397 (in Chinese). (郭振彬, 张媛媛, 冯霄, 化学学报, 2020, 78, 397.) (h) Peng, Z.; Ding, H.; Chen, R.; Gao, C.; Wang, C. Acta Chim. Sinica2019, 77, 681. (in Chinese). (彭正康, 丁慧敏, 陈如凡, 高超, 汪成, 化学学报, 2019, 77, 681.)
[5]
(a) Pascanu, V.; Gonzalez Miera, G.; Inge, A. K.; Martin-Matute, B. J. Am. Chem. Soc.2019, 141, 7223. (b) Liu, Y.; Xuan, W.; Cui, Y. Adv. Mater.2010, 22, 4112. (c) Kang, Y.-S.; Lu, Y.; Chen, K.; Zhao, Y.; Wang, P.; Sun, W.-Y. Coordin. Chem. Rev.2019, 378, 262. (d) Ding, S. Y.; Gao, J.; Wang, Q.; Zhang, Y.; Song, W. G.; Su, C. Y.; Wang, W. J. Am. Chem. Soc.2011, 133, 19816. (e) Liu, J.; Zhang, M.; Wang, N.; Wang, C.; Ma, L. Acta Chim. Sinica2020, 78, 311 (in Chinese). (刘建国, 张明月, 王楠, 王晨光, 马隆龙, 化学学报, 2020, 78, 311.) (h) Hang, G.; Chen, Z.; Jiang, H. Acta Chim. Sinica2016, 74, 113. (in Chinese). (黄刚, 陈玉贞, 江海龙, 化学学报, 2016, 74, 113.) (i) Xiao, J.; Li, D.; Jiang, H. Sci. Sin. Chim.2018, 48, 1058.(in Chinese). (肖娟定, 李丹丹, 江海龙, 化学学报, 2018, 48, 1058.) (j) Cai, G.; Ding, M.; Wu, Q.; Jiang, H. Natl. Sci. Rev. 2020, 7, 37.
[6]
(a) Wales, D. J.; Grand, J.; Ting, V. P.; Burke, R. D.; Edler, K. J.; Bowen, C. R.; Mintova, S.; Burrows, A. D. Chem. Soc. Rev.2015, 44, 4290. (b) Hussain, M.; Wackerlig, J.; Lieberzeit, P. A. Biosensors2013, 3, 89. (c) Kong, B.; Selomulya, C.; Zheng, G. F.; Zhao, D. Y. Chem. Soc. Rev.2015, 44, 7997.
[7]
(a) Horcajada, P.; Serre, C.; Vallet-Regi, M.; Sebban, M.; Taulelle, F.; Ferey, G. Angew. Chem. Int. Ed.2006, 45, 5974. (b) Horcajada, P.; Gref, R.; Baati, T.; Allan, P. K.; Maurin, G.; Couvreur, P.; Ferey, G.; Morris, R. E.; Serre, C. Chem. Rev.2012, 112, 1232. (c) Zeng, J.; Wang, X.; Zhang, X.; Zhou, R. Acta Chim. Sinica2019, 77, 1156. (in Chinese). (曾锦跃, 王小双, 张先正, 卓仁禧, 化学学报, 2019, 77, 1156.)
[8]
(a) Mu, J.; He, L.; Huang, P.; Chen, X. Coordin. Chem. Rev.2019, 399, 213039. (b) Cai, H.; Huang, Y.-L.; Li, D. Coordin. Chem. Rev.2019, 378, 207. (c) Anderson, S. L.; Stylianou, K. C. Coordin. Chem. Rev.2017, 349, 102.
Kathalikkattil, A. C.; Roshan, R.; Tharun, J.; Babu, R.; Jeong, G. S.; Kim, D. W.; Cho, S. J.; Park, D. W. Chem. Commun.2016, 52, 280. doi: 10.1039/C5CC07781H
Wang, S. H.; Zheng, F. K.; Zhang, M. J.; Liu, Z. F.; Chen, J.; Xiao, Y.; Wu, A. Q.; Guo, G. C.; Huang, J. S. Inorg. Chem.2013, 52, 10096. doi: 10.1021/ic401409b
Kutzscher, C.; Hoffmann, H. C.; Krause, S.; Stoeck, U.; Senkovska, I.; Brunner, E.; Kaskel, S. Inorg. Chem.2014, 54, 1003.
[25]
Katsoulidis, A. P.; Antypov, D.; Whitehead, G. F. S.; Carrington, E. J.; Adams, D. J.; Berry, N. G.; Darling, G. R.; Dyer, M. S.; Rosseinsky, M. J. Nature2019, 565, 213. doi: 10.1038/s41586-018-0820-9
[26]
(a) Deria, P.; Mondloch, J. E.; Karagiaridi, O.; Bury, W.; Hupp, J. T.; Farha, O. K. Chem. Soc. Rev.2014, 43, 5896. (b) Cohen, S. M. J. Am. Chem. Soc.2017, 139, 2855. (c) Li, B.; Zhang, Y.; Ma, D.; Li, L.; Li, G.; Li, G.; Shi, Z.; Feng, S. Chem. Commun.2012, 48, 6151. (d) Li, F.; Wang, D.; Xing, Q.-J.; Zhou, G.; Liu, S.-S.; Li, Y.; Zheng, L.-L.; Ye, P.; Zou, J.-P. Appl. Catal., B2019, 243, 621.
Bonnefoy, J.; Legrand, A.; Quadrelli, E. A.; Canivet, J.; Farrusseng, D. J. Am. Chem. Soc.2015, 137, 9409. doi: 10.1021/jacs.5b05327
[30]
Fracaroli, A. M.; Siman, P.; Nagib, D. A.; Suzuki, M.; Furukawa, H.; Toste, F. D.; Yaghi, O. M. J. Am. Chem. Soc.2016, 138, 8352. doi: 10.1021/jacs.6b04204
[31]
Baek, J.; Rungtaweevoranit, B.; Pei, X.; Park, M.; Fakra, S. C.; Liu, Y.-S.; Matheu, R.; Alshmimri, S. A.; Alshehri, S.; Trickett, C. A.; Somorjai, G. A.; Yaghi, O. M. J. Am. Chem. Soc.2018, 140, 18208. doi: 10.1021/jacs.8b11525
[32]
Marshall, R. J.; Hobday, C. L.; Murphie, C. F.; Griffin, S. L.; Morrison, C. A.; Moggach, S. A.; Forgan, R. S. J. Mater. Chem. A2016, 4, 6955. doi: 10.1039/C5TA10401G
[33]
Gutov, O. V.; Molina, S.; Escudero-Adan, E. C.; Shafir, A. Chemistry2016, 22, 13582. doi: 10.1002/chem.201600898
Chan, J. Y.; Zhang, H.; Nolvachai, Y.; Hu, Y.; Zhu, H.; Forsyth, M.; Gu, Q.; Hoke, D. E.; Zhang, X.; Marriot, P. J.; Wang, H. Angew. Chem., Int. Ed.2018, 130, 17376. doi: 10.1002/ange.201810925
[48]
Lu, Y.; Zhang, H.; Chan, J. Y.; Ou, R.; Zhu, H.; Forsyth, M.; Marijanovic, E. M.; Doherty, C. M.; Marriott, P. J.; Holl Banaszak, M. M.; Wang, H. Angew. Chem., Int. Ed.2019, 131, 17084. doi: 10.1002/ange.201910408
[49]
Mon, M.; Ferrando-Soria, J.; Grancha, T.; Fortea-Perez, F. R.; Gascon, J.; Leyva-Perez, A.; Armentano, D.; Pardo, E. J. Am. Chem. Soc.2016, 138, 7864. doi: 10.1021/jacs.6b04635
[50]
Perez-Cejuela, H. M.; Mon, M.; Ferrando-Soria, J.; Pardo, E.; Armentano, D.; Simo-Alfonso, E. F.; Herrero-Martinez, J. M. Mikrochim. Acta2020, 187, 201. doi: 10.1007/s00604-020-4185-z
[51]
Feng, X.; Jena, H. S.; Leus, K.; Wang, G.; Ouwehand, J.; Van Der Voort, P. J. Catal.2018, 365, 36. doi: 10.1016/j.jcat.2018.06.013
[52]
Jeong, G. S.; Kathalikkattil, A. C.; Babu, R.; Chung, Y. G.; Won Park, D. Chin. J. Catal.2018, 39, 63. doi: 10.1016/S1872-2067(17)62916-4
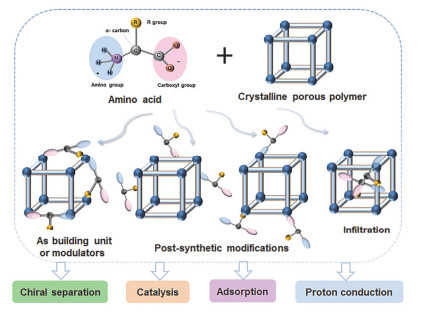
图 1
氨基酸功能化晶态多孔聚合物的制备策略.
Figure 1
Schematic illustration of synthetic strategies for amino acid functionalized crystalline porous polymer.
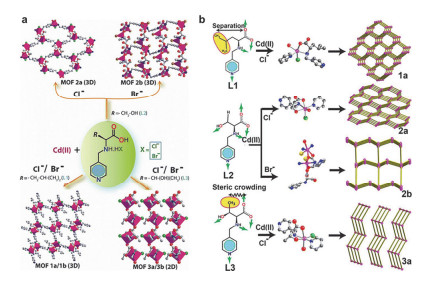
Figure 3
(a) Schematic representation of linkers L1, L2, and L3 that react with Cd(II) to produce corresponding MOF architectures (MOF 1a/1b, 2a/2b, and 3a/3b). (b) MOFs 1a, 2a, and 3a with their SBUs and topological simplification model[17]
Figure 5
The structural flexibility of ZnGGH in the context of a conformational energy landscape. (a) ZSM-5 is a rigid porous framework, the new ZnGGH compound reported here is a flexible porous framework. In contrast to the macromolecular state of proteins, ZnGGH is a crystalline solid framework, which restricts the available conformational space of the linker. All energy landscapes drawn in this figure are hypothetical illustrations that reflect the structural properties of the corresponding materials. (b) The doubly deprotonated tripeptide Gly-Gly-L-His acts as a tetratopic linker to form the ZnGGH framework. Adapted from the literature [25]
Figure 6
(a) The two-step peptide grafting process into various MOFs. (b) Synthesis of the catalysts comprising the replacement of formate with imidazole-containing ligands and metalation with Cu(I). Adapted from the literature [29], [31]
Figure 7
(a) PXRD patterns showing the effects of incorporation of 5 equivalents of L-proline and one of HCl into solvothermal syntheses of Zr MOFs of L6~L8 compared to addition of HCl alone. (b) Anchoring of L-proline onto a Zr MOF and amino acid-modulated MOF synthesis. (c) Synthesis of CMOF-5. The presence of L- or D-proline additives induces chirality and enables the growth and isolation of CMOF-5 crystals. An achiral square grid net, [Zn(BDC)(NMP)], forms in the absence of proline. Adapted from the literature [32], [33], [36]
Figure 9
(a) Structure of helicoidal chains in Cu(GHG) MOF. (b) Functional groups in the peptidic backbone decorating the surface of the pores. (c) 1D channels in Cu(GHG) are surrounded by functional sites prone to establish supramolecular interactions, well suited for chiral recognition and discrimination. (d) Representative MC binding geometries of (+, -) MA and EP enantiomers within the structure of Cu(GHG) and corresponding adsorption energies as absolute values calculated with respect to gas phase. (e) Evolution of the enantioselective recognition of Cu(GHG) for chiral drugs MA and EP. Adapted from the literature [45]
Figure 10
Fabrication and characterization of homochiral MOF-based MMMs for chiral separation of 1-phenylethanol enantiomers. Adapted from the literature [48]
Figure 11
(a) Conformation of the GGH linker in the refined experimental crystal structures ZnGGH-1•(DMF-H2O), ZnGGH-2•(DMSO) and ZnGGH-3•(DMF).
(b) The view down the one-dimensional pores of ZnGGH-3•(DMF) and ZnGGH-2•(DMSO). (c) The side view reveals the intraframework hydrogen-bond pattern for each structure. (d) Amino acid imprinted UiO-66s for highly recognized adsorption of small angiotensin- converting-enzyme-inhibitory peptides. Adapted from the literature [25], [34]
Figure 12
(a) Bioinspired metal-organic framework catalysts for selective methane oxidation to methanol. (b) Catalytic cleavage of pentapeptide 1 by Cat. C [MTVIRMOF-74-III-(CH3)0.6(CH2NH-Asp-His-Cys-NH2)0.1] in the specific sequence containing serine, and cartoon representation of the enzyme TEV endoprotease, highlighting the three amino acids that participate in the catalysis. Adapted from the literature [30], [31]
Figure 13
(a) The 12-connected Zr6(µ3-O)4(µ3-OH)4(COO-)12 cluster SBU. (b) An aspartic acid linker. (c) The crystal structure of MIP-202(Zr) viewed along the a-axis. (d) The crystal structure of MIP-202(Zr) viewed along the (101) plane. (e) and (f) Nyquist plots from AC impedance data for MIP-202(Zr) at 0% and 95% RH. Adapted from the literature [16]

 下载:
下载:

 下载:
下载:














