引用本文:
张鹏程, 郭佳, 朱根, 方文玉, 唐乾元, 鲍磊, 康文斌. 无序随机多肽组分相关的结构转变的蒙特卡洛模拟:以赖氨酸、谷氨酸和异亮氨酸组成的随机多肽为例[J]. 化学学报,
2020, 78(9): 994-1000.
doi:
10.6023/A20060249 Citation:
Zhang Pengcheng, Guo Jia, Zhu Gen, Fang Wenyu, Tang Qianyuan, Bao Lei, Kang Wenbin. Monte Carlo Simulations of Composition-Related Structural Transition of Disordered Peptides: The Case Study of Random Peptides Composed of Lysine, Glutamic Acids and Isoleucine[J]. Acta Chimica Sinica,
2020, 78(9): 994-1000.
doi:
10.6023/A20060249
Monte Carlo Simulations of Composition-Related Structural Transition of Disordered Peptides: The Case Study of Random Peptides Composed of Lysine, Glutamic Acids and Isoleucine
Received Date:
18 June 2020 Available Online:
15 September 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 11947006), the Cultivating Project for Young Scholar at Hubei University of
Medicine (Nos. 2019QDJZR12, 2018QDJZR22), and the Natural Science Foundation of Hubei Provincial Department of Education (No. B2018434).
Abstract:
Intrinsically disordered proteins (IDPs) are a unique class of proteins without stable native structures. Like globular proteins, the structure and the dynamics of IDPs are also encoded in their amino acid sequences. IDPs usually contain a larger proportion of hydrophilic or charged amino acids than globular proteins. Interestingly, even with the same hydrophobicity and number of charged residues, the differences in sequence arrangement can lead to different structures of the peptides. In this work, to model such an effect, we conduct molecular simulations based on a series of peptides with randomly composed of charged residues (including glutamic acids and lysines) and isoleucine. In the simulation, we use the ABSINTH (self-Assembly of Biomolecules Studied by an Implicit, Novel, and Tunable Hamiltonian) implicit solvation model and employ the all-atom Markov Chain Monte Carlo method with replica-exchange sampling. Our simulations clearly show a transition between the extended conformations to compact structures for each peptide. The corresponding transition temperature is found to be dependent on the portion of the hydrophobic and charged residues. When the mean hydrophobicity is larger than a certain threshold, the transition temperature is higher than the room temperature, and vice versa. Such a result has outlined the borderline between intrinsically disordered proteins and the folded proteins. It is also consistent with previous analysis based on bioinformatics techniques. Furthermore, the contributions of different kinds of interactions to the structural variation of peptides are analyzed based on the contact statistics and the charge-pattern dependence of the gyration radii of the peptides. Our simulation results imply that the hydrophobicity of the sequence dominates the order-disorder transitions of IDPs, while the charge distribution can also affect such transitions. Based on these results, we achieve a comprehensive understanding of the sequence-structure relation of the natural proteins and the underlying physics. Our results may broaden our perspective of the sequence-structure relation of protein systems and shed light on the design of both ordered and disordered proteins.
Table 1.
The 31 sequences analyzed in this research. In the table, Column 1 shows the label of each sequence. Column 2 shows the sequence, with Glu (E) residues in red, Lys (K) residues in blue, and Ile (I) residues in black. Column 3 shows the κ-values. Column 4 shows the mean hydrophobicity.
Figure 1.
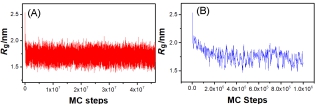
(A) The gyration radius Rg as function of the simulation steps (MC step) at T=298 K for I50 peptide, and (B) shows the relaxation process during the first 1.0×106 steps of the simulation.
Figure 2.
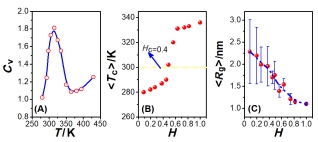
(A) The specific heat capacity Cv vs. the temperature T for the sequence E20I10K20 with κ=0.5. The transition temperature Tc corresponds to the position of the peak of the curve. (B) The averaged transition temperature <Tc> vs. the mean hydrophobicity H, and (C) the gyration radius <Rg> vs. the mean hydrophobicity H for electrically neutral EIK systems with different amino-acid compositions.
Figure 3.
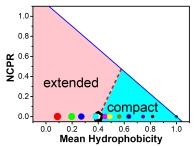
The phase diagram based on the hydrophobicity (H) and the net charge per residues (NCPR). The region with the pink (or cyan) color corresponds to the compositions of peptides with extended (or compact) conformations. As a reference, the boundary between the IDPs and folded proteins based on bioinformatical analysis is given as the red dashed line. All the simulated peptides are marked on the diagram with dots whose sizes are proportional to the average gyration radii of the peptides.
Figure 4.
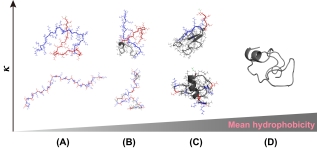
Representative conformations sampled from the simulation trajectories (T=298 K) for electrically neutral peptides with different hydrophobicity. Here, the lysine (K), glutamate acid (E) and isoleucines (I) are colored in blue, red, and gray, respectively. The conformations are rendered with PyMOL package. (A) H=0.08855, (B) H=0.38021, (C) H=0.81771, (D) H=1.0. For (A)~(C), the upper and lower subfigures denote peptide sequences with κ=0 and κ=1, respectively.
Figure 5.
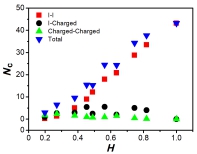
The average number of contacts Nc vs. mean hydrophobicity H. We divided all contacts into three different types: isoleucine-isoleucine (red squares), isoleucine-charged residues (black circle), and contacts of charged residue pairs (green upper triangles). The total number of residue contacts is also plotted in the graph (blue down triangles).
Figure 6.
(A) The gyration radius Rg vs. the mean hydrophobicity H for the peptides. The cases with different κ are given in various colors, as indicated by the legend. (B) The fluctuation of the gyration radius ΔRg vs. the mean hydrophobicity H for the peptides.
Apicella, A.; Marascio, M.; Colangelo, V.; Soncini, M.; Gautieri, A.; Plummer, C. J. J. Biomol. Struct. Dyn.2017, 35, 1813. doi: 10.1080/07391102.2016.1196151
[17]
Dyson, H. J.; Wright, P. E. Nat. Rev. Mol. Cell Biol.2005, 6, 197. doi: 10.1038/nrm1589
[18]
Gao, M.; Yang, F.; Zhang, L.; Su, Z.; Huang, Y. J. Biomol. Struct. Dyn.2018, 36, 1171. doi: 10.1080/07391102.2017.1316519
[19]
Jain, A.; Ashbaugh, H. S. J. Chem. Phys.2008, 129, 174505. doi: 10.1063/1.3003577
[20]
Kang, W. B.; He, C.; Liu, Z. X.; Wang, J.; Wang, W. J. Biomol. Struct. Dyn.2019, 37 1956. doi: 10.1080/07391102.2018.1472669
Figure 1
(A) The gyration radius Rg as function of the simulation steps (MC step) at T=298 K for I50 peptide, and (B) shows the relaxation process during the first 1.0×106 steps of the simulation.
Figure 2
(A) The specific heat capacity Cv vs. the temperature T for the sequence E20I10K20 with κ=0.5. The transition temperature Tc corresponds to the position of the peak of the curve. (B) The averaged transition temperature <Tc> vs. the mean hydrophobicity H, and (C) the gyration radius <Rg> vs. the mean hydrophobicity H for electrically neutral EIK systems with different amino-acid compositions.
Figure 3
The phase diagram based on the hydrophobicity (H) and the net charge per residues (NCPR). The region with the pink (or cyan) color corresponds to the compositions of peptides with extended (or compact) conformations. As a reference, the boundary between the IDPs and folded proteins based on bioinformatical analysis is given as the red dashed line. All the simulated peptides are marked on the diagram with dots whose sizes are proportional to the average gyration radii of the peptides.
Figure 4
Representative conformations sampled from the simulation trajectories (T=298 K) for electrically neutral peptides with different hydrophobicity. Here, the lysine (K), glutamate acid (E) and isoleucines (I) are colored in blue, red, and gray, respectively. The conformations are rendered with PyMOL package. (A) H=0.08855, (B) H=0.38021, (C) H=0.81771, (D) H=1.0. For (A)~(C), the upper and lower subfigures denote peptide sequences with κ=0 and κ=1, respectively.
Figure 5
The average number of contacts Nc vs. mean hydrophobicity H. We divided all contacts into three different types: isoleucine-isoleucine (red squares), isoleucine-charged residues (black circle), and contacts of charged residue pairs (green upper triangles). The total number of residue contacts is also plotted in the graph (blue down triangles).
Figure 6
(A) The gyration radius Rg vs. the mean hydrophobicity H for the peptides. The cases with different κ are given in various colors, as indicated by the legend. (B) The fluctuation of the gyration radius ΔRg vs. the mean hydrophobicity H for the peptides.
Table 1.
The 31 sequences analyzed in this research. In the table, Column 1 shows the label of each sequence. Column 2 shows the sequence, with Glu (E) residues in red, Lys (K) residues in blue, and Ile (I) residues in black. Column 3 shows the κ-values. Column 4 shows the mean hydrophobicity.

 下载:
下载:


 下载:
下载:







