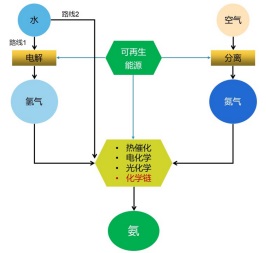
图 1.
可再生能源驱动的“绿色”合成氨过程
Figure 1.
Green ammonia synthesis processes driven by renewable energy

氨是氮肥的主要原料, 同时, 亦是人工合成几乎所有重要含氮化学品的原料[1]. 在合成氨工业建立一个世纪后, 氨又因其能量密度高(3 kWh•kg-1)、氢含量高(质量分数17.7%)、易储运、相关基础设施成熟等特点而被认为是重要的能源载体(Energy carrier)[2-5]. 目前, 可再生能源的利用面临间歇性、波动性、并网发电难等问题, 寻找安全高效的能量存储方式是当前可再生能源利用所面临的巨大挑战. 而将可再生能源通过合成氨反应储存于氨分子中, 利用氨的易储运特性, 在终端用户处通过氨分解制氢、氨燃烧、氨燃料电池等方式释放能量, 即是氨作为能源载体的含义[3].
目前工业合成氨采用的是Haber-Bosch (H-B)工艺, 它需要在高温高压条件(350~550 ℃, 压力10~30 MPa)下实施, 消耗全球能源供应总量的1%~2%[6]; 同时, 该过程需要使用大量化石资源用于产氢和供能, 每年排放大约5亿吨二氧化碳, 约占全球二氧化碳排放的1.8%. 在节能减排和可持续发展为首要任务的今天, 开发清洁高效的“绿色”合成氨工艺尤为重要和迫切[7].
目前, 由可再生能源驱动, 并通过多相催化、电催化、光催化和化学链等方式进行的“绿色”合成氨的路线大致有两条[8]. 一是利用可再生能源制取氢气, 氢气再和氮气进行反应生成氨, 如图 1路线1所示; 事实上, 在20世纪60年代以前, 挪威、埃及等国曾利用本国丰富的水电, 制取氢气, 氢气再和氮气反应合成氨. 另一途径则是直接以氮气和水反应生成氨, 如图 1路线2所示, 该路线可跳过制氢过程, 减少中间环节, 但目前效率还较低, 离实际应用还有较长一段距离.
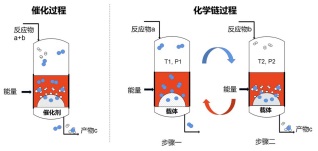
化学链过程(Chemical looping)是指将目标反应拆分为两个或多个分步反应, 各分步反应可在不同的空间、时间或反应条件下分别进行, 分步反应可以逐一优化以达到整个目标反应的最优化. 在化学链过程中, 常常需要借助某种“反应载体(Carrier)”的消耗和再生完成整个化学循环. 根据目标反应的不同, 反应载体可用于氧、氮、碳或氢等物种的传递, 分别称之为载氧体[9]、载氮体[10]、载碳体[11]或载氢体[12]等.
化学链过程与多相催化过程有着密切的关联. 二者本质上均涉及气固或液固表界面化学反应, 有些多相催化剂亦可用作化学链过程中的反应载体. 但是, 化学链不同于多相催化过程, 载体亦不同于多相催化剂. 二者之间存在诸多不同, 现以催化与化学链过程合成氨反应(N2+H2→NH3)为例来具体说明二者之间的差别, 如图 2所示.
目前, 化学链技术已在化石能源燃烧与二氧化碳捕获、水分解制氢、化学品及液体燃料制备等重要领域得到了广泛的关注与研究. 如化学链燃烧(Chemical looping combustion, CLC)具有CO2内分离、能量利用率高、能耗低、污染低等优点, 是一种很有前景、经济环保、效益高的燃烧方式. 迄今为止, 国内外研究者进行了大量卓有成效的工作[13].
此外, 化学链技术在烷烃氧化脱氢制烯烃[14, 15]、甲烷氧化制取甲醇[16, 17]、甲烷氧化偶联[18]、甲烷制合成气[19, 20]、水分解制氢[21, 22]、合成氨[10]等领域也取得了许多新的研究进展. 感兴趣的读者可参阅近期发表的相关综述文章[23, 24], 本综述仅详细介绍化学链合成氨的研究进展.
将合成氨催化过程解耦为固氮及产氨等两步或两步以上分立的化学反应, 即为化学链合成氨过程. 早在19世纪时, Tessie du Motay就曾提出利用Ti3N2和TiN之间的循环转换合成氨[25], 即Ti3N2固氮生成富氮的TiN, TiN与H2高温反应放氨并再生贫氮的Ti3N2, 如方程式(1), (2)所示.
|
$ 2{\rm{T{i}}_3}{{\rm{N}}_2} + {{\rm{N}}_2} \to 6{\rm{TiN}} $ |
(1) |
|
$ 6{\rm{TiN}} + 3{{\rm{H}}_2} \to 2{\rm{T{i}}_3}{{\rm{H}}_2} + 2{\rm{N{H}}_3} $ |
(2) |
1908年, Frank[26]开发了一种通过碳化钙(CaC2)固氮生成氰氨化钙(CaCN2), 继而氰氨化钙水解产氨的分步合成氨方法, 水解产物碳酸钙与焦炭混合加热反应可再生CaC2, 其反应方程式如(3)~(6)所示. 此方法在Haber-Bosch工艺开发前是工业上主要的固氮方法. 电石再生的条件十分苛刻, 需要消耗大量的能量, 据估计该工艺的能耗为210 GJ•t-1 NH3, 这使得该工艺在经济上缺乏竞争力[27].
|
$ {\rm{Ca{C}}_2} + {{\rm{N}}_2} \to {\rm{CaC{N}}_2} + {\rm{C}} \quad\Delta H = - 288.4 \; {\rm{kJ \bullet mo{l}}^{ - 1}} $ |
(3) |
|
$ {\rm{CaC{N}}_2} + 3{{\rm{H}}_2}{\rm{O}} \to 2{\rm{N{H}}_3} + {\rm{CaC{O}}_3} \quad \Delta H = - 94.2 \; {\rm{kJ \bullet mo{l}}^{ - 1}} $ |
(4) |
|
$ {\rm{CaC{O}}_3} + 5/2{\rm{C}} \to {\rm{Ca{C}}_2} + 3/2{\rm{C{O}}_2} \quad \Delta H = 557.8 \; {\rm{kJ \bullet mo{l}}^{ - 1}} $ |
(5) |
|
$ \text{总包反应:}{{\rm{N}}_2} + 3{{\rm{H}}_2}{\rm{O}} + 3/2{\rm{C}} \to 2{\rm{N{H}}_3} + 3/2{\rm{C{O}}_2} $ |
(6) |
Haber等[28]曾经使用Mn-Mn3N2和CaH2-Ca3N2进行了化学链合成氨的尝试, 其反应方程式为(7)~(10). 但该过程的合成氨产率极低, 因而未得到进一步研究.
|
$ 3{\rm{Ca{H}}_2} + {{\rm{N}}_2} \to {\rm{C{a}}_3}{{\rm{N}}_2} + 3{{\rm{H}}_2} $ |
(7) |
|
$ {\rm{C{a}}_3}{{\rm{N}}_2} + 6{{\rm{H}}_2} \to 3{\rm{Ca{H}}_2} + 2{\rm{N{H}}_3} $ |
(8) |
|
$ 3{\rm{Mn}} + {{\rm{N}}_2} \to {\rm{M{n}}_3}{{\rm{N}}_2} $ |
(9) |
|
$ {\rm{M{n}}_3}{{\rm{N}}_2} + 3{{\rm{H}}_2} \to 3{\rm{Mn}} + 2{\rm{N{H}}_3} $ |
(10) |
自20世纪初Haber-Bosch合成氨工业建立以来, 关于化学链合成氨的研究便逐渐淡出历史舞台. 近年来, 随着可再生能源合成氨的需求日益旺盛, 将太阳能、风能等可再生能源与化学链过程相耦合, 使得化学链作为一种替代合成氨方式得到了复兴[29]. 与合成氨催化过程相比, 化学链过程具有以下几个特点: 可在常压条件下操作, 有助于简化工艺流程, 可用于分布化、小型化产氨; 由于形成稳定的氮化物或氧化物, 易于启停, 易于与可再生能源耦合; 可分别对固氮、产氨等各步骤中的反应物、温度、压力等进行优化; 可规避N2与H2或H2O的竞争吸附问题.
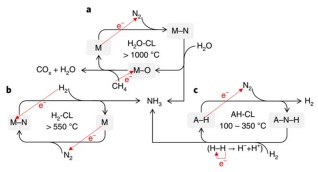
根据载氮体(水解或加氢)放氨形式的不同, 化学链合成氨过程可分为以水为氢源的化学链(表示为H2O-CL)和以氢气为氢源的化学链过程(表示为H2-CL), 如图 3所示. 人们对于H2O-CL过程的兴趣源于其避免了昂贵的、高能耗的天然气或煤重整制氢过程, 是目前受到关注较多的体系. 而对于H2-CL过程, 氢气则可来源于可再生能源, 通过电解水等制氢方式得到, 再通过化学链方式制氨.
在化学链合成氨过程中, 载氮体材料的选择是决定过程效率的关键因素. 近年来, 随着化学链合成氨技术逐渐引起学者的关注, 一系列载氮体得到了广泛而深入的研究, 从而大大丰富了化学链合成氨的研究内涵. 接下来, 就近期化学链合成氨载氮体材料的发展做一介绍.
目前研究的载氮体材料大多是无机含氮化合物, 包括金属氮化物(nitride)、氮氧化物(oxynitride)、氮氢化物(nitride hydride)、(亚)氨基化合物(amide or imide)等, 分别构成氮化物-氧化物对、贫氮氮化物-富氮氮化物对或氢化物-亚氨基化合物对等实现固氮及产氨反应. 根据化学键性质的不同, 氮化物一般可分为共价型氮化物、过渡金属氮化物和离子型氮化物. 据此, 本综述将载氮体材料分为以下三种.
共价氮化物的主要代表是AlN. 2007年, Steinfeld 等[30, 31]设计了一种太阳能集热驱动的两步化学链合成氨过程(图 4). 在固氮过程中, Al2O3在氮气气氛中与甲烷或者碳等还原性原料共同反应生成AlN和CO; AlN与水发生高温水解反应, 可以生成氨同时AlN重新回到Al2O3. 其反应方程式及反应焓变如(11)~(13)所示. 从热力学上看, 第一步反应为强吸热过程, 反应需要高温才能发生. 第二步则是放热反应.
|
$ \text{固氮:}{\rm{A{l}}_2}{{\rm{O}}_3} + 3{\rm{C}} + {{\rm{N}}_2} \to 2{\rm{AlN}} + 3{\rm{CO}} \\ \ \ \ \qquad \qquad \quad \Delta H = 708.1 \; {\rm{kJ \bullet mo{l}}^{ - 1}} $ |
(11) |
|
$ \begin{array}{l} {\rm{A{l}}_2}{{\rm{O}}_3} + 3{\rm{C{H}}_4} + {{\rm{N}}_2} \to 2{\rm{AlN}} + 6{{\rm{H}}_2} + 3{\rm{CO}}\\ \ \ \ \qquad \quad \Delta H = 931.9 \; {\rm{kJ \bullet mo{l}}^{ - 1}} \end{array} $ |
(12) |
|
$ \begin{array}{l} \text{产氨:} 2{\rm{AlN}} + 3{{\rm{H}}_2}{\rm{O}} \to 2{\rm{A{l}}_2}{{\rm{O}}_3} + 2{\rm{N{H}}_3}\\ \ \ \ \qquad \qquad \quad \Delta H = - 274.1 \; {\rm{kJ \bullet mo{l}}^{ - 1}} \end{array} $ |
(13) |
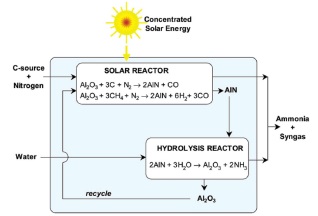
这一AlN-Al2O3化学链过程的特点在于原料Al2O3的价格低廉、常压操作工艺、利用太阳能集热器供能、无CO2排放、副产CO等; 缺点为两步反应都需要高温条件, 第一步需要1500~1700 ℃, 第二步需要900~1200 ℃, 能耗较大, 对反应器设备的要求较高. 且高温时, 氨分解为氮气和氢气的反应是热力学自发进行的, 从而降低了氨的产率.
研究者对于AlN-Al2O3化学链合成氨过程的影响因素, 如Al2O3结构、催化剂、反应条件等进行了研究. Al2O3的结构和存在形式影响固氮反应速率, 反应性的顺序为γ-Al2O3>Al(OH)3>α-Al2O3[32]. 为改善反应动力学性能, 研究者尝试了多种添加剂或催化剂. 针对固氮反应, 研究发现一些含钙化合物, 如CaF2、CaCO3、Ca(OH)2和CaC2等在1350 ℃下与Al2O3形成铝酸盐, 而铝酸盐在高温下气化离解形成铝蒸气, 可以快速氮化. 此外, Y2O3、Yb2O3、Cr2O3、Mg、Na2CO3、Li2CO3、NiCO3等添加剂也可以提高反应(11)中原料的转化率[32].
对于水解产氨步骤, Wu等[33]发现Fe2O3和CuO的加入对AlN的水解反应有明显的催化作用, 其中Fe2O3的催化作用优于CuO. Fe2O3的加入, 使得AlN水解反应的表观活化能从186.3 kJ•mol-1降低到119.2 kJ•mol-1, 降低了近70 kJ•mol-1. Liu等[34]报道TiO2对AlN的水解产氨反应亦表现出良好的催化作用, 这是由于TiO2表面具有良好的H2O解离吸附性能, 所产生的羟基在水解反应中起着重要作用. 随着TiO2的负载量、反应温度和水蒸汽浓度的增加, AlN的转化率和NH3的产率都有所增加. 近期, 他们[35]又发现ZrO2能促进NH3的形成, 因为NH3分子吸附在ZrO2表面, 从而阻止NH3的分解. 温度和水蒸气浓度的升高促进了AlN的水解和氨的生成, 但随着温度的升高, 氨的生成效率明显降低. 另外, 添加过量的γ-Al2O3可以增加AlN载氮体的孔隙率, 提高固氮效率, 但是随着温度的升高, 载氮体的孔隙率明显下降, 抑制放氨过程[36]. 对于水解产氨反应, 如何抑制高温下氨的分解, 是提高氨收率必须要考虑的因素.
与AlN-Al2O3不同, 过渡金属元素具有丰富的可变化学价态, 可形成种类繁多的氮化物/氧化物, 且氮/氧化物的稳定性较弱, 是目前受到关注较多的载氮体材料. 尤其是Cr、Mo、Mn等载氮体材料得到了较为系统的研究. 除了H2O直接作为反应物实施水解产氨反应外, 过渡金属氮化物还可以与H2反应构成化学链合成氨过程. 下面分别进行介绍.
(1) H2O-CL
2011年, Michalsky等[37]提出了氮化铬作为载氮体, 利用Cr-Cr2N-Cr2O3间的循环, 通过3步化学反应合成氨的化学链过程. 其反应方程式如(14)~(17)所示. 首先, Cr2O3被CO或者H2还原生成Cr, 第二步Cr被氮化生成Cr2N, 最后Cr2N水解放氨.
|
$ {\rm{C{r}}_2}{{\rm{O}}_3} + 3{\rm{H}} \to 2{\rm{Cr}} + 3{{\rm{H}}_2}{\rm{O}} \quad \Delta H = 360.0 \; {\rm{kJ \bullet mo{l}}^{ - 1}} $ |
(14) |
|
$ {\rm{C{r}}_2}{{\rm{O}}_3} + 3{\rm{CO}} \to 2{\rm{Cr}} + 3{\rm{C{O}}_2} \quad \Delta H = 278.6 \; {\rm{kJ \bullet mo{l}}^{ - 1}} $ |
(15) |
|
$ 2{\rm{Cr}} + 1/2{{\rm{N}}_2} \quad \Delta H = - 122.6\; {\rm{kJ \bullet mo{l}}^{ - 1}} $ |
(16) |
|
$ \begin{array}{l} {\rm{C{r}}_2}{\rm{N}} + 3{{\rm{H}}_2}{\rm{O}} \to {\rm{C{r}}_2}{{\rm{O}}_3} + {\rm{N{H}}_3} + 3/2{{\rm{H}}_2} \\ \ \ \ \qquad \qquad \quad \; \Delta H = - 321.1 \; {\rm{kJ \bullet mo{l}}^{ - 1}} \end{array} $ |
(17) |
氧化铬的还原为强吸热反应, 从热力学上看, 需要较高的温度. 在实验中, 由于避免了使用固态还原剂, 因而还原温度较AlN-Al2O3体系有所降低, 但仍需要1200~1600 ℃的高温条件, 在1600 ℃, Cr还原的速率为2.7×10-3 molCr•molCr2O3-1•min-1; 第二步和第三步是放热反应, 但是由于存在动力学阻力, 实际反应中需要500~1000 ℃, 在1000 ℃, Cr氮化的速度为4.13×10-2 molN2•molCr-1•min-1, 首先生成Cr2N, 继续氮化生成CrN, 氮化铬水解生成氨的速率为1.07×10-4 molNH3• molCr-1•min-1.
该工艺除了使用Cr基化合物作为载氮体, 还可以将其他过渡金属设计成类似的循环, 更一般的反应方程式如(18)~(20)所示[27]. Michalsky等[38]对不同氮化物中晶格氮的扩散动力学对合成氨性能的影响进行了详细研究, 发现氮化物的离子性(Ionicity)与晶格氮的扩散系数有重要的关联, 即晶格氮的扩散系数随氮化物离子性的增加而增大(增大的离子性可增加氮化物中活性晶格氮的体积浓度), 从而促进氮化物水解产氨. 这一认识有助于指导研究人员通过理性的材料设计寻找更为合适的化学链载氮体材料. 其中Mn基和Mo基氮化物被认为是潜在的载氮体而得到了较多的研究. Michalsky等对Mo2N-MoO2化学链合成氨的过程进行了热力学和经济分析, 结果表明, 当氨的产量为900 t•day-1, 每吨氨的成本为534±28美元时, 经济上将具有可行性[39].
|
$ a/bc{{\rm{M}}_c}{{\rm{O}}_d} + ad/bc{\rm{R}} \to a/b{\rm{M}} + ad/bc{\rm{RO}} $ |
(18) |
|
$ a/b{\rm{M}} + 1/2{{\rm{N}}_2} \to 1/b{{\rm{M}}_a}{{\rm{N}}_b} $ |
(19) |
|
$ 1/b{{\rm{M}}_a}{{\rm{N}}_b} + ad/bc{{\rm{N}}_2}{\rm{O}} \to a/bc{{\rm{M}}_c}{{\rm{O}}_d} + {\rm{N{H}}_3} + (ad/bc - 3/2){{\rm{H}}_2} $ |
(20) |
2017年, Pfromm等[40]报道了一种用氮气、水蒸汽和CH4在大气压下制备NH3和合成气(CO-H2)的新型化学链过程: 首先, 金属锰与氮气反应生成Mn5N2, 接着氮化锰在500 ℃水解生成NH3和MnO, 第三步是MnO在1150 ℃, 4%CH4-96%N2混合蒸汽中还原, 并再生Mn5N2, 构成化学链循环. 研究还发现, 在氮化物中加入NaOH后, 氨的产量有显著的提升, 其中, Na被认为是电子助剂, 削弱了Mn–N键, 产生更多活性氮变成氨气.
(2) H2-CL
过渡金属氮化物通过加氢产氨的化学链产氨过程可以表示为:
|
$ {\rm{T{M}}_a}{{\rm{N}}_b} + 3x/2{{\rm{H}}_2} \to {\rm{T{M}}_a}{{\rm{N}}_{b - x}} + x{\rm{N{H}}_3} $ |
(21) |
|
$ {\rm{T{M}}_a}{{\rm{N}}_{b - x}} + x/2{{\rm{N}}_2} \to {\rm{T{M}}_a}{{\rm{N}}_b} $ |
(22) |
在此过程中, 氮化物通过过渡金属的变价特性, 富氮的氮化物与贫氮氮化物物相之间的转变实现产氨过程. 过渡金属氮化物的稳定性是制约其固氮及加氢产氨反应热力学和动力学性能的关键因素. 在过渡金属催化的合成氨反应中, 我们已认识到过渡金属的催化性能符合Sabatier原理, 即N物种在过渡金属上的吸附能应具有适中的吸附强度[41]. 这一原则可推广于载氮体的筛选, 事实上, 早期人们就发现合成氨催化活性与过渡金属氮化物的生成焓之间存在着火山型曲线的关系[42].
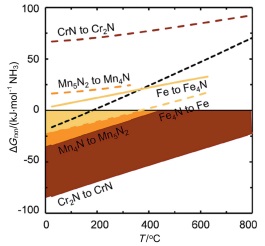
Michalsky等[43]对部分过渡金属氮化物载氮体固氮及加氢产氨的热力学进行了计算. 从图 5中可以看出, Mn4N及Cr2N氮化为富氮的氮化物(Mn5N2及CrN)均为热力学有利的反应, 而富氮氮化物的加氢产氨反应则是热力学爬坡的反应; 与此不同的是, Fe的氮化(生成Fe4N)则为热力学爬坡反应, Fe4N的加氢产氨则是热力学可行的反应. 氮化锰具有丰富的价态和氮化物结构, 且较为适中的热力学, 因而以氮化锰为载氮体的研究较多.
但是氮化锰的动力学性能较差, 如Mn6N2.58在550 ℃、0.1 MPa氢气流的条件下加氢, 最终产物为Mn4N, 反应速率为55 μmolNH3•g-1•h-1. Laassiri等[44]报道Mn3N2反应性很低, 只有3.1%的晶格氮可与氢反应生成氨. 因此, 不少研究着眼于对氮化锰的修饰改性. 为了降低氮化锰的相对稳定性, 锰基载氮体的设计关键就在于降低晶格氮的稳定性. 晶格氮空位的生成能可作为一个描述子(Descriptor), 用来衡量晶格氮转化为氨的难易程度. 通过掺杂或合金化等方式对氮化锰的组成进行调变, 可改变其局部或整体的电子结构, 从而调变过渡金属氮化物晶格氮的稳定性, 最终改变产氨性能. Michalsky等[45]计算了掺杂不同过渡金属对ζ-Mn2N相晶格氮空位的生成自由能的影响, 发现3d过渡金属(从Sc到Zn)的加入都可以降低晶格氮的稳定性. 掺杂过渡金属的d电子占据数越大, 晶格氮空位的生成自由能越低. 实验证实, 与未掺杂相比, Fe掺杂的氮化锰(Fe0.25Mn1.75N)可以降低晶格氮的稳定性, 增加氮空位的浓度. 最近, Liu等[46, 47]利用密度泛函理论(DFT)计算了Mn4N在分子水平上的加氢过程. 金属–氮(N–M, M=Mn, Fe, Ni)和金属–氢(H–M)键的强度都决定了整个加氢过程的热力学. 计算结果证实了Fe和Ni掺杂在氮化锰的表层和次表层可以干扰其局域电子结构, 从而影响产氨活性. 基于DFT的动力学模拟进一步表明, Mn4N亚层中掺杂低浓度的Fe或Ni可降低晶格氮的扩散势垒. 此外, 这些杂原子掺杂物种, 特别是Ni, 由于具有良好的氢吸附作用, 表层和次表层的Ni都可以降低还原反应活化能.
金属Co的存在对氮化锰中氮转移亦会产生影响. 除过渡金属掺杂外, 在氮化锰中掺杂低含量的Li, 可提高低温下的合成氨性能. 在Li-Mn-N体系中, 400 ℃时, 有15%的晶格氮与氢反应生成氨. 但是, Li-Mn-N经氢还原后具有较高的热化学稳定性, 导致在N2气氛中的再生步骤难以进行, 无法构成化学链过程[44].
Co-Mo-N催化剂是继Fe、Ru后发展的一类氮化物合成氨催化剂[48-50]. Hargreaves等[51-54]在研究Co3Mo3N催化剂时, 发现H2气氛中处理时, 部分晶格氮以氨的形式释放出来, 生成了贫氮的Co6Mo6N相, 而Co6Mo6N相在N2中焙烧后可再生Co3Mo3N相, 从而构成了合成氨循环. N2同位素交换实验也证实晶格氮是活性氮, 在合成氨反应条件下直接参与了氨的催化合成[53]. 而且, 对Co6Mo6N进行实验时, 发现除了存在氮交换, 还可以转变成Co3Mo3N. 这项研究证实了Co-Mo-N体系中氮的消耗和补充是可逆的, 同时也表明Co-Mo-N也是一种潜在的载氮体材料.
其它过渡金属氮化物作为载氮体的例子并不多. Hargreaves等[55, 56]考察了一系列氮化物, 如Ni3N、Cu3N、Zn3N2、Ta3N5等在氢气气氛下的脱氮性能, 其中Ni3N和Cu3N由于稳定性较低, 因此在相对较低温度(250 ℃)可以进行脱氮反应, 脱氮率分别是25%和30%, 但是产物进行氨化再生氮化物却比较困难. 稳定性相对较高的Zn3N2在400 ℃时有约23%的氮转化成氨, 但是产物氨化再生的量也很有限. 不同的是, Ta3N5虽然传递到氨的氮量很低(13%), 但是产物可以氨化再生. 氮化铁在400 ℃能脱掉70%的氮, 氮化钴在250 ℃可以完全脱氮, 氮化铼在350 ℃下也能脱去接近90%的氮. 由于这些氮化物的循环再生很困难, 难以形成化学链合成氨过程, 但是这些研究对合成氨载氮体的设计与选择具有借鉴意义.
Li、Mg、Ca等与N2生成相应的离子型氮化物(Li3N、Mg3N2和Ca3N2等)的反应不仅是热力学可行的, 且动力学性能良好. 一个典型的例子是, 碱金属Li在室温就能和N2反应生成Li3N[57]. 这类离子型氮化物水解产氨的速率也往往较快, 但由生成的氧化物或氢氧化物再生金属或氮化物则是非常困难的. 比如MgO的碳热反应温度高于1500 ℃. 此外, LiOH的熔点较低(471 ℃), 再生Li3N时可能会因为熔化和气化从而带来产物分离的问题.
2019年, Halas等[58]提出了一种利用等离激元(Plasmonic)材料TiN的光热转化的三步法合成氨过程: TiN纳米颗粒在光照射作用下产生热, 驱动CH4将MgO还原为金属Mg, 并生成物质的量比1∶2的CO和H2, 可以作为费托合成的原料. 第二步是金属镁在氮气气氛中被氮化, 生成Mg3N2. 最后, Mg3N2水解生成氨气, 再生氧化镁, 完成循环. 由于材料只有有限的表面能够暴露在光照下, 所以产氨速率很低, 约为1.67 μmolNH3• gTiN/MgO-1•h-1. 为提高光热耦合化学链产氨速率, 需要增大其光热转化效率或尝试其它等离激元材料.
|
$ {\rm{MgO}} + {\rm{C{H}}_4} \to {\rm{Mg}} + {\rm{CO}} + 2{{\rm{H}}_2} $ |
(23) |
|
$ 3{\rm{Mg}} + {{\rm{N}}_2} \to {\rm{M{g}}_3}{{\rm{N}}_2} $ |
(24) |
|
$ {\rm{M{g}}_3}{{\rm{N}}_2} + {{\rm{H}}_2}{\rm{O}} \to 3{\rm{MgO}} + {\rm{N{H}}_3} $ |
(25) |
除了光热耦合化学链合成氨外, 热电耦合是另一种可再生能源利用的方式. 近期, Jaramillo等[59].提出了一种基于Li循环的化学链过程, 该过程的化学反应如(26)~(28)所示. 第一步是LiOH电解生成金属锂; 第二步是将金属Li暴露在流动态的氮气中, 温度维持在22~100 ℃, 形成Li3N; 第三步是Li3N水解产生氨气. 作者通过电解的方式, 使该反应可以在300 ℃条件下进行, 电流效率达到了88.5%. 该过程中由LiOH生成Li的反应热力学不利, 电解过程需要消耗大量的电能. 该工作表明通过其他形式能量的注入, 可以调变反应的热力学, 使反应在更温和条件下进行.
|
$ 6{\rm{LiOH}} \to 6{\rm{Li}} + 3{{\rm{H}}_2}{\rm{O}} + 3/2{{\rm{O}}_2}({\rm{g}}) $ |
(26) |
|
$ 6{\rm{Li}} + {{\rm{N}}_2}({\rm{g}}) \to 2{\rm{L{i}}_3}{\rm{N(s)}} $ |
(27) |
|
$ 2{\rm{L{i}}_3}{\rm{N(s)}} + 6{{\rm{H}}_2}{\rm{O}} \to 6{\rm{LiOH}} + 2{\rm{N{H}}_3} $ |
(28) |
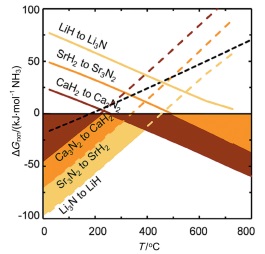
碱(土)金属氮化物作为载氮体, 也可以利用加氢反应产氨. Michalsky等[43]计算了碱(土)金属氮化物与氢化物相互转化反应的热力学(图 6), 可以看出, 碱(土)金属氢化物(Hydride)如LiH、CaH2和SrH2转化成Li3N、Ca3N2和Sr3N2的反应高温有利, 碱(土)金属氮化物生成氢化物则是低温有利的反应; 选择合适的温度及压力有可能实现化学链合成氨. 作者考察了Ca3N2和Sr2N与H2的反应, 在550 ℃、0.1 MPa条件下, 二者加氢后的最终产物分别为Ca2NH和SrH2, 产氨速率分别为98和81 μmolNH3•g-1•h-1. 但对于固氮反应, 作者并未进行考察.
Ichikawa等[60]报道Li3N在300 ℃和0.5 MPa H2流中可生成LiH和NH3. 最近, 他们发现金属Li可与14族金属元素如Sn等形成Li-Sn合金(Li17Sn4), Li-Sn合金在400 ℃可以和氮气反应生成纳米Li3N和Li7Sn2合金(式29), Li3N和H2反应生成LiH和NH3, 350 ℃后再生Li-Sn合金(式30)[61, 62].
|
$ 2{\rm{L{i}}_{17}}{\rm{S{n}}_4} + {{\rm{N}}_2} \to 2{\rm{L{i}}_3}{\rm{N}} + 4{\rm{L{i}}_7}{\rm{S{n}}_2} $ |
(29) |
|
$ 2{\rm{L{i}}_3}{\rm{N}} + 4{\rm{L{i}}_7}{\rm{S{n}}_2} + 3{{\rm{H}}_2} \to 2{\rm{L{i}}_{17}}{\rm{S{n}}_4} + 2{\rm{N{H}}_3} $ |
(30) |
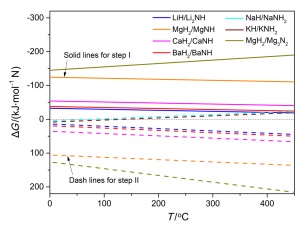
近期, Gao等[10, 63]提出了一种以碱(土)金属亚氨基化合物为载氮体的低温化学链合成氨过程. 该过程利用碱(土)金属氢化物与亚氨基化合物之间的相互转化实现固氮及加氢产氨过程, 具体反应方程式如(31), (32)所示. 在固氮步骤, 碱(土)金属氢化物与N2反应生成相应的亚氨基化合物, 随后亚氨基化合物再与H2反应放氨, 同时再生氢化物. 图 7示出了0~450 ℃温度区间内, 部分二元氢化物, 如LiH、MgH2、CaH2、BaH2的固氮及相应(亚)氨基化合物或氮化物, 如Li2NH、CaNH、MgNH、BaNH、NaNH2、KNH2及Mg3N2加氢产氨反应的热力学. 可以看出, LiH、CaH2、MgH2、BaH2等碱(土)金属氢化物固氮生成相应亚氨基化合物或氮化物均是热力学有利的反应, NaH和KH由于难以形成稳定的亚氨基化合物而无法实现固氮过程. 而亚氨基化合物或氮化物加氢产氨则为热力学不利的反应, 如Mg3N2和MgNH, 在500 ℃以下难以实现加氢放氨过程. 氨基化合物NaNH2、KNH2在温度高于150 ℃时则是热力学可行的反应. 综合考虑氢化物及亚氨基化合物的固氮及加氢产氨的热力学, BaH2-BaNH和LiH-Li2NH体系具有较为合适的反应热力学, 有可能在较低温度和压力条件下进行, 实施氨的化学链合成[64].
固氮:
|
$ {\rm{A}}{{\rm{H}}_x} + x{{\rm{N}}_2} \to 2x{{\rm{A}}_{{\rm{2/x}}}}{\rm{NH}} + x{{\rm{H}}_2} $ |
(31) |
加氢放氨:
|
$ x{{\rm{A}}_{2/x}}{\rm{NH}} + 2x{{\rm{H}}_2} \to 2{\rm{A}}{{\rm{H}}_x} + x{\rm{N}}{{\rm{H}}_{\rm{3}}} $ |
(32) |
其中, A=Li、Mg、Ca、Ba 等; x 是 A 的表观价态.
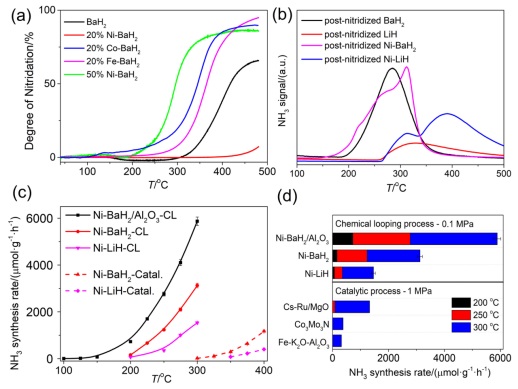
实验结果证实了LiH和BaH2分别在400和250 ℃即可实施固氮反应, 而Li2NH、BaNH在300和200 ℃以上时可实现加氢产氨, 如图(8a和8b)所示. 特别需要指出的是, 氢化物的固氮过程是通过氢化物与N2的氧化还原反应完成的, 其中N2被还原, 氢化物中的负氢物种(H‾)失去电子生成质子和H2. 这与过渡金属或碱(土)金属作为电子给体还原N2的反应是有显著区别的. 氢化物的固氮及加氢产氨温度较AlN和过渡金属氮化物均有大幅度降低. 研究还发现, 3d过渡金属如Fe、Co和Ni可作为催化剂显著加速氢化物固氮及亚氨基化合物加氢产氨速率, 其催化效果顺序为Ni>Co>Fe. 在传统的合成氨催化过程中, Ni长期以来一直被认为是活性很差的过渡金属[65-67], 这是由于N2在金属Ni表面解离能垒较高[67]且H2与N2存在竞争吸附的缘故. 然而, Ni加速了氢化物的固氮反应速率这一事实表明了Ni与氢化物之间存在着某种协同作用, 从而加速了N2的解离. 研究者在固定床反应器中, 通过交替通入N2和H2的操作方式考察了氢化物-亚氨基化合物的化学链产氨速率. 如图 8c和8d所示, 与合成氨催化过程相比, 亚氨基化合物作为载氮体的化学链产氨速率有了显著提高. 比如Al2O3负载的Ni-BaH2在100 ℃时即实现了氨的合成(2.3 μmol•g-1•h-1), 在常压、250 ℃的产氨速率较催化过程(使用高活性Cs-Ru/MgO为催化剂, 250 ℃, 1 MPa)高一个数量级(图 8d), 这一对比结果充分彰显了化学链过程的优势. 研究者认为, 在化学链过程中N2和H2分步进料的模式很大程度上避免了反应物在催化剂上的竞争吸附, 因而展现了明显的优势. 但是此过程中亚氨基化合物的加氢放氨为吸热反应(图 7), 从热力学上看, 平衡氨浓度较低. 通过调变氢化物–亚氨基化合物的组成可调变其热力学性能, 从而提高产氨浓度. 此外, 该类材料的稳定性也是制约其实际应用的重要因素.
2017年, Steinfeld等[68] 提出了一种由钙钛矿型氮氧化物作为载氮体的化学链合成氨过程, 反应方程式如(33), (34)所示. 第一步, 在高温和低氧分压下, 利用太阳能集热部分还原钙钛矿, 产生氧空位用来活化和固定氮气分子. 第二步是氮氧化物水解产氨并再生钙钛矿氧化物. 在这个过程中, 氧空位的产生被认为是速率控制步骤. 钙钛矿化合物具有晶相稳定、组成可调等特点, 是燃料电池、二氧化碳和水的热化学裂解等研究领域受到较多关注的材料, 其化学链合成氨性能还有待进一步实验研究.
|
$ {\rm{AB}}{{\rm{O}}_3} + \varepsilon /2{{\rm{N}}_2} \to {\rm{AB}}{{\rm{O}}_{{\rm{3}} - \delta }}{{\rm{N}}_\varepsilon } + \delta /2{{\rm{O}}_2} $ |
(33) |
|
$ {\rm{AB}}{{\rm{O}}_{3 - \delta }}{{\rm{N}}_{2\delta /3}} + \delta {{\rm{H}}_2}{\rm{O}} \to {\rm{AB}}{{\rm{O}}_3} + 2\delta /3{\rm{N}}{{\rm{H}}_3} $ |
(34) |
以上总结了近年来化学链合成氨的研究进展, 表 1对几种主要化学链合成氨载氮体的操作条件及产氨速率进行了总结. 可以看到目前大部分载氮体材料的操作温度较高, 产氨速率较低, 距实际应用还有较大的距离. 高效载氮体材料的开发是目前该领域所面临的主要挑战.
 下载:
导出CSV
下载:
导出CSV
| No. | 载氮体对 | 反应 | 反应条件 | 产氨速率/(μmol•g-1•h-1) | 特点 | Ref. |
| 1 | AlN-Al2O3 | Al2O3+3C+N2→ 2AlN+3CO 2AlN+3H2O→ Al2O3+2NH3 |
固氮: 0.1 MPa, 1500~1700 ℃ 水解: 0.1 MPa, 900~1200 ℃ |
无数据 | 高温反应, 动力学性能差, 碳基 化合物作为还原剂, 太阳能聚 热 |
[30, 31] |
| 2 | Cr2N-Cr2O3 | Cr2O3+3H2→ 2Cr+3H2O 2Cr+1/2N2→Cr2N Cr2N+3H2O→ Cr2O3+NH3+3/2H2 |
还原: 0.1 MPa, 1200~1500 ℃ 固氮: 0.1 MPa, 500~1000 ℃ 水解: 0.1 MPa, 500~1000 ℃ |
1000 ℃时, Cr2N水解产氨速 率为108 μmol•g-1•h-1 |
高温反应, 动力学性能差, H2为 还原剂, 过渡金属作为电子给 体还原N2 |
[37] |
| 3 | MaNb-MaNb-d (Mn) |
MaNb-d+d/2N2→ MaNb MaNb+3d/2H2→ MaNb-d+dNH3 |
固氮: 200~ 1000 ℃ 加氢: 0.1 MPa, >300 ℃ |
Mn6N2.58在550 ℃时加氢产 氨速率为55.3 μmol•g-1•h-1 |
动力学性能差, 过渡金属作为 电子给体还原N2 |
[43] |
| 4 | MH2-MaNb
(Ca, Sr) |
MH2+b/2N2→ MaNb+aH2 MaNb+(a+3/2b)H2→ aMH2+bNH3 |
固氮: 200~ 1000 ℃ 加氢: 0.1 MPa, >300 ℃ |
Ca3N2和Sr2N在550 ℃时的加氢产氨速率分别为98和81 μmol•g-1•h-1 | H-作为电子和质子给体还原N2 | [43] |
| 5 | Mg-Mg3N2-MgO | MgO+CH4→ Mg+CO+2H2 3Mg+N2→Mg3N2 Mg3N2+3H2O→ 3MgO+2NH3 |
光源加热 还原&水解: 压力<13.3 Pa 固氮: 0.1 MPa |
1.67 μmol•g-1•h-1 | 光致等离子体加热 | [58] |
| 6 | BaH2-BaNH LiH-Li2NH |
以Ba为例: BaH2+N2→ BaNH+1/2H2 BaNH+2H2→ BaH2+NH3 |
固氮: 0.1 MPa, 100~500 ℃ 加氢: 0.1 MPa, 100~500 ℃ Ni催化剂 |
300 ℃时BaH2-BaNH 的总包反应速率为 5870 μmol•g-1•h-1 |
H‾作为电子和质子给体还 原N2, H2异裂产生H+和H‾ |
[10] |
| 7 | Li-Li3N-LiOH | 6LiOH→ 6Li+3H2O+3/2O2 6Li+N2→2Li3N 2Li3N+6H2O→ 6LiOH+2NH3 |
电解: 0.1 MPa, 450 ℃, 3 V 固氮&水解: 0.1 MPa, 22~100 ℃ |
无数据 | 电热耦合 | [59] |
| 8 | Mn5N2-MnO | Mn5N2+5H2O→ 5MnO+2NH3+2H2 5MnO+5CH4+N2→ Mn5N2+10H2+5CO |
水解: 0.1 MPa, 500 ℃ 固氮: 0.1 MPa, 1150 ℃ |
无数据 | 过渡金属价态发生变化 | [40] |
在化学链合成氨过程中, 载氮体通过固氮和加氢(或水解)产氨的循环交替反应来实现合成氨循环. 载氮体在此循环中既传递晶格氮, 同时伴随着能量的传递过程, 是整个化学链循环的核心. 为了实现高效化学链合成氨过程, 载氮体材料的开发是其关键核心技术. 总体而言, 理想的载氮体应该具备以下几个条件: 高的载氮容量; 适中的固氮及加氢或水解产氨反应热力学; 良好的动力学性能; 高的机械强度以及稳定性; 原料易得、成本低; 制备能耗低且环境友好.
化学链过程中, 载氮体参与的各分步反应的吉布斯自由能变化(∆G)是决定其性能优劣的关键因素. 因而, 选择载氮体材料时, 首先应对所有相关反应的热力学进行分析[27]. 理想的情形是, 载氮体在一定的温度、压力区间内, 各反应均满足∆G≤0的条件. 遗憾的是, 这样的载氮体目前尚未发现, 如前文所述, 载氮体材料的筛选应符合Sabatier原理, 即载氮体中晶格氮的稳定性应适中. 以此为理论依据, 热力学计算可高通量筛选载氮体材料, 可大大缩小材料选择的范围, 为载氮体材料的开发提供了强有力的工具.
Bartel等[69]对1148对二元金属氮化物–金属氧化物构成的化学链合成氨过程进行了高通量的热力学计算. 对于包括氮化物水解产氨并生成氧化物、H2还原氧化物以及固氮再生金属氮化物的三步化学链产氨过程, 计算结果揭示了循环中反应极限吉布斯自由能与氮化物和氧化物的生成能之间呈现一火山型曲线(图 9), 其中Mn、Fe、W、Tc和Yb的氮化物–氧化物对可能是良好的载氮体材料. 事实上, Mn基氮化物正是目前研究较多的载氮体材料之一. 平衡产物分布分析表明除了CrN-Cr2O3、MoN-MoO2、WN2-WO3、MnN-MnO和Mn2N-MnO外, B、V、Fe和Ce的氮化物是潜在的新型载氮体, 值得进一步实验验证.
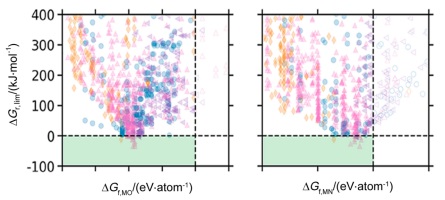
需要说明的是, 这项研究工作仅考察了二元氮化物材料, 但这些化合物只占已知材料的10%. 因此, 载氮体材料开发的空间还很大. 一方面, 可以通过高通量热力学计算快速筛选混合阳离子的三元或多元氮化物材料. 在此过程中, 除了对已知的氮化物进行反应热力学分析外, 还需要人工设计新型的氮化物结构, 从而帮助实验研究人员缩小材料筛选范围, 选择性的合成氮化物材料并验证其固氮、产氨反应的动力学性能. 另一方面, 除了对氮化物的金属阳离子组成进行调变外, 也可将O、C、H、F、Cl、Br等阴离子引入氮化物晶格, 形成混合阴离子化合物, 如氮氧化物、氮碳化物、氮氢化物等, 来调变氮化物的热力学性能. 相较于前者, 阴离子调变更具有挑战性. 目前, 这类混合阴离子化合物还很少, 人们对于这类材料的合成、性质、反应等还知之甚少. 仅有几个作为载氮体的例子, 因此, 混合阴离子载氮体材料的设计与开发是一个非常值得探索的崭新领域.
对于具有良好热力学性能的载氮体材料, 考察其固氮及产氨动力学性能是接下来需要做的工作. 由于化学链合成氨过程为非平衡态过程, 涉及到表面反应、晶格氮/氧/氢等物种在固相中的传递以及相变过程等, 动力学阻力往往较大. 在固氮反应中, N2分子的解离吸附通常被认为是动力学上的慢步骤. 在这种情况下, 可加入合适的催化剂, 比如过渡金属Ni的加入使得BaH2固氮反应的表观活化能从109 kJ•mol-1大幅降低至46 kJ• mol-1, 从而加快了固氮速率[10]. 在产氨反应中, 已有研究表明, 对于过渡金属氮化物载氮体, 高温时的速控步可能是固相扩散[38]; 在这种情况下, 可通过掺杂、合金化等方式削弱金属-氮键的强度, 降低晶格氮的稳定性.
总的来说, 一种性能良好的载氮体不仅需要适中的热力学性能, 更要结合动力学性能统一考量. 所以, 从热力学和动力学两方面对载氮体的性能进行优化仍然是化学链合成氨研究领域的重要课题.
可再生能源驱动的化学链合成氨过程是实现“绿色”合成氨的重要途径之一. 在过去的十多年里, 研究人员对化学链合成氨进行了积极的探索, 特别是对于不同载氮体作为媒介的固氮及(水解或加氢)产氨反应的热力学进行了大量的分析, 这对于载氮体材料的设计与开发具有重要的指导意义. 尽管近年来化学链合成氨研究取得了诸多进展, 总体来看, 产氨速率低仍是制约该过程走向应用的首要障碍. 高性能载氮体材料的开发应是近期化学链合成氨研究的核心内容.
对于H2O-CL, 金属氧化物还原为金属或直接氮化为氮化物的反应常常是热力学不利的, 且动力学阻力较大, 需要高温的苛刻条件, 从而对反应器提出了很高的要求. 开发三元或多元氧化物, 以降低二元金属氧化物的热力学稳定性, 是值得研究和验证的可行策略之一. 相比于H2O-CL过程, 过渡金属氮化物如氮化锰的固氮和加氢产氨反应(即H2-CL)的热力学较为适中, 是非常具有前景的载氮体材料. 但是, 其动力学阻力较大, 产氨速率低. 如何改善其固氮及加氢产氨的动力学性能是目前研究的重点. 碱(土)金属亚氨基化合物, 作为一类新型的载氮体材料, 具有适中的固氮及加氢产氨热力学; 在过渡金属的催化作用下, 其展示出了良好的低温动力学性能, 是目前文献报道的具有最高化学链产氨速率的载氮体材料. 除对已有载氮体材料进行热力学和动力学调变外, 新型载氮体的探索与开发仍将持续, 也是这一领域长期的研究课题.
载氮体材料在化学链过程中的形貌、组成及结构变化是影响其性能的重要因素, 但目前对于这方面的认识还不够深入. 随着先进材料表征技术及理论计算的发展, 将极大促进人们对于化学链过程微观机制的理解与认识. 比如原位电镜、同步辐射表征手段等可以在原位条件下观察载氮体的氮传递过程, 理论计算则可以从原子水平上深入理解载氮体与N2、H2的相互作用及催化剂的作用机制等, 从而为载氮体材料的设计提供新的研究思路.
除太阳能集热外, 将光、电、等离子体等不同能量输入形式与化学链合成氨过程相耦合是目前该领域研究的新动向. 光、电等外界能量的输入可以打破载氮体的固氮及产氨反应的热力学平衡限制; 同时, 对反应动力学、中间物种的种类及浓度、反应路径等亦会产生不同程度的影响. 考察这些影响因素与化学链产氨性能的关联是该领域一个新的研究课题.
反应器的设计也是影响化学链过程效率的重要因素. 化学链反应器包括固定床和流化床反应器等. 目前化学链合成氨的研究主要集中在载氮体的设计与优化, 反应器大多以固定床反应器为主, 关于反应器的设计与优化方面的研究还比较少, 有待进一步研究. 此外, 化学链合成氨过程的生命周期分析、能量效率、成本核算等也应随着载氮体材料的开发逐渐提上研究日程.
化学链合成氨技术作为实现“绿色”合成氨的可能方案之一, 方兴未艾. 化学链合成氨技术走向应用有赖于催化化学、材料科学、化学工程、计算化学等多学科交叉. 随着化学链合成氨日益受到研究者的关注, 这一技术在氨的绿色合成及可再生能源的储存与转化过程中或将发挥重要的作用.
刘化章, 化工进展, 2013, 32, 1995. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498Liu, H. Z. Chem. Ind. Eng. Prog. 2013, 32, 1995(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Klerke, A.; Christensen, C. H.; Norskov, J. K.; Vegge, T. J. Mater. Chem. 2008, 18, 2304. doi: 10.1039/b720020j
Guo, J. P.; Chen, P. Chem 2017, 3, 709. doi: 10.1016/j.chempr.2017.10.004
Valera-Medina, A.; Xiao, H.; Owen-Jones, M.; David, W. I. F.; Bowen, P. J. Prog. Energy Combust. Sci. 2018, 69, 63. doi: 10.1016/j.pecs.2018.07.001
Smith, C.; Hill, A. K.; Torrente-Murciano, L. Energy Environ. Sci. 2020, 13, 331. doi: 10.1039/C9EE02873K
Erisman, J. W.; Sutton, M. A.; Galloway, J.; Klimont, Z.; Winiwarter, W. Nat. Geosci. 2008, 1, 636. doi: 10.1038/ngeo325
王倩茹, 郭建平, 陈萍, 能源化学, 2019, 36, 25. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498Wang, Q. R.; Guo, J. P.; Chen, P. J. Energy Chem. 2019, 36, 25(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Norskov, J. K.; Chen, J. G. Sustainable Ammonia Synthesis, US Department of Energy, 2016. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Zeng, L.; Cheng, Z.; Fan, J. A.; Fan, L. S.; Gong, J. L. Nat. Rev. Chem. 2018, 2, 349. doi: 10.1038/s41570-018-0046-2
Gao, W. B.; Guo, J. P.; Wang, P. K.; Wang, Q. R.; Chang, F.; Pei, Q. J.; Zhang, W. J.; Liu, L.; Chen, P. Nat. Energy 2018, 3, 1067. doi: 10.1038/s41560-018-0268-z
Koerts, T.; Vansanten, R. A. J. C. S. Chem. Commun. 1991, 1281. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Wang, Q. R.; Guo, J. P.; Chen, P. Joule 2020, 4, 705. doi: 10.1016/j.joule.2020.02.008
曾亮, 罗四维, 李繁星, 范良士, 中国科学:化学, 2012, 42, 260. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498Zeng, L.; Luo, S. W.; Li, F. X.; Fan, L. S. Sci. China Chem. 2012, 42, 260(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Chen, S.; Zeng, L.; Mu, R. T.; Xiong, C. Y.; Zhao, Z. J.; Zhao, C. J.; Pei, C. L.; Peng, L. M.; Luo, J.; Fan, L. S.; Gong, J. L. J. Am. Chem. Soc. 2019, 141, 18653. doi: 10.1021/jacs.9b09235
Gao, Y. F.; Wang, X. J.; Liu, J. C.; Huang, C. D.; Zhao, K.; Zhao, Z. L.; Wang, X. D.; Li, F. X. Sci. Adv. 2020, 6, eaaz9339.
Tomkins, P.; Ranocchiari, M.; van Bokhoven, J. A. Acc. Chem. Res. 2017, 50, 418. doi: 10.1021/acs.accounts.6b00534
Groothaert, M. H.; Smeets, P. J.; Sels, B. F.; Jacobs, P. A.; Schoonheydt, R. A. J. Am. Chem. Soc. 2005, 127, 1394. doi: 10.1021/ja047158u
Cheng, Z.; Baser, D. S.; Nadgouda, S. G.; Qin, L.; Fan, J. A.; Fan, L. S. ACS Energy Lett. 2018, 3, 1730. doi: 10.1021/acsenergylett.8b00851
Huang, C. D.; Wu, J.; Chen, Y. T.; Tian, M.; Rykov, A. I.; Hou, B. L.; Lin, J.; Chang, C. R.; Pan, X. L.; Wang, J. H.; Wang, A. Q.; Wang, X. D. Commun. Chem. 2018, 1, 55. doi: 10.1038/s42004-018-0050-y
Liu, Y.; Qin, L.; Cheng, Z.; Goetze, J. W.; Kong, F. H.; Fan, J. A.; Fan, L. S. Nat. Commun. 2019, 10, 6. doi: 10.1038/s41467-018-07858-8
Xu, B. J.; Bhawe, Y.; Davis, M. E. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 9260. doi: 10.1073/pnas.1206407109
Abanades, S.; Flamant, G. Solar Energy 2006, 80, 1611. doi: 10.1016/j.solener.2005.12.005
Zhu, X.; Imtiaz, Q.; Donat, F.; Muller, C. R.; Li, F. X. Energy Environ. Sci. 2020, 13, 772. doi: 10.1039/C9EE03793D
段一菲, 陈存壮, 张军社, 王新赫, 魏进家, 中国科学:化学, 2020, 50, 337. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498Duan, Y. F.; Chen, C. Z.; Zhang, J. S.; Wang, X. H.; Wei, J. J. Sci. China Chem. 2020, 50, 337(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Jennings, J. R. Catalytic ammonia synthesis:Fundamentals and practice, Plenum Press, New York, 1991. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Frank, A. R. Trans. Faraday Soc. 1908, 4, 099. doi: 10.1039/tf9080400099
Michalsky, R.; Pfromm, P. H. AlChE J. 2012, 58, 3203. doi: 10.1002/aic.13717
Haber, F.; van Oordt, G. Z. Anorg. Chem. 1905, 44, 341. doi: 10.1002/zaac.19050440122
Chen, J. G.; Crooks, R. M.; Seefeldt, L. C.; Bren, K. L.; Bullock, R. M.; Darensbourg, M. Y.; Holland, P. L.; Hoffman, B.; Janik, M. J.; Jones, A. K.; Kanatzidis, M. G.; King, P.; Lancaster, K. M.; Lymar, S. V.; Pfromm, P.; Schneider, W. F.; Schrock, R. R. Science 2018, 360, eaar6611.
Galvez, M. E.; Halmann, M.; Steinfeld, A. Ind. Eng. Chem. Res. 2007, 46, 2042. doi: 10.1021/ie061550u
Galvez, M. E.; Frei, A.; Halmann, M.; Steinfeld, A. Ind. Eng. Chem. Res. 2007, 46, 2047. doi: 10.1021/ie061551m
Molisani, A. L.; Yoshimura, H. N. Mater. Res. Bull. 2010, 45, 733. doi: 10.1016/j.materresbull.2010.02.012
Wu, Y.; Jiang, G. D.; Zhang, H. B.; Sun, Z.; Gao, Y.; Chen, X. P.; Liu, H. Z.; Tian, H. J.; Lai, Q. H.; Fan, M. H.; Liu, D. Chem. Commun. 2017, 53, 10664. doi: 10.1039/C7CC04742H
Gao, Y.; Wu, Y.; Zhang, Q.; Chen, X. P.; Jiang, G. D.; Liu, D. Int. J. Hydrogen Energy 2018, 43, 16589. doi: 10.1016/j.ijhydene.2018.07.042
Wu, Y.; Gao, Y.; Zhang, Q.; Cai, T.; Chen, X.; Liu, D.; Fan, M. Fuel 2020, 264, 116821. doi: 10.1016/j.fuel.2019.116821
Zhang, Q.; Wu, Y.; Gao, Y.; Chen, X.; Liu, D.; Fan, M. Int. J. Hydrogen Energy 2020, 45, 9903. doi: 10.1016/j.ijhydene.2020.01.172
Michalsky, R.; Pfromm, P. H. Solar Energy 2011, 85, 2642. doi: 10.1016/j.solener.2011.08.005
Michalsky, R.; Pfromm, P. H. J. Phys. Chem. C 2012, 116, 23243. doi: 10.1021/jp307382r
Michalsky, R.; Parman, B. J.; Amanor-Boadu, V.; Pfromm, P. H. Energy 2012, 42, 251. doi: 10.1016/j.energy.2012.03.062
Heidlage, M. G.; Kezar, E. A.; Snow, K. C.; Pfromm, P. H. Ind. Eng. Chem. Res. 2017, 56, 14014. doi: 10.1021/acs.iecr.7b03173
Medford, A. J.; Vojvodic, A.; Hummelshoj, J. S.; Voss, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Norskov, J. K. J. Catal. 2015, 328, 36. doi: 10.1016/j.jcat.2014.12.033
Appl, M. Ammonia:Principles and industrial practice, Wiley-VCH, Weinheim, 1999. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Michalsky, R.; Avram, A. M.; Peterson, B. A.; Pfromm, P. H.; Peterson, A. A. Chem. Sci. 2015, 6, 3965. doi: 10.1039/C5SC00789E
Laassiri, S.; Zeinalipour-Yazdi, C. D.; Catlow, C. R. A.; Hargreaves, J. S. J. Appl. Catal. B 2018, 223, 60. doi: 10.1016/j.apcatb.2017.04.073
Michalsky, R.; Pfromm, P. H.; Steinfeld, A. Interface Focus 2015, 5, 20140084. doi: 10.1098/rsfs.2014.0084
Shan, N.; Chikan, V.; Pfromm, P.; Liu, B. J. Phys. Chem. C 2018, 122, 6109. doi: 10.1021/acs.jpcc.7b12569
Shan, N. N.; Huang, C. R.; Lee, R. T.; Manavi, N.; Xu, L. B.; Chikan, V.; Pfromm, P. H.; Liu, B. ChemCatChem 2020, 12, 2233. doi: 10.1002/cctc.201902383
Jacobsen, C. J. H. Chem. Commun. 2000, 1057. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Jacobsen, C. J. H.; Dahl, S.; Clausen, B. S.; Bahn, S.; Logadottir, A.; Norskov, J. K. J. Am. Chem. Soc. 2001, 123, 8404. doi: 10.1021/ja010963d
Kojima, R.; Aika, K. Chem. Lett. 2000, 514.
McKay, D.; Gregory, D. H.; Hargreaves, J. S. J.; Hunter, S. M.; Sun, X. Chem. Commun. 2007, 3051. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Hunter, S. M.; McKay, D.; Smith, R. J.; Hargreaves, J. S. J.; Gregory, D. H. Chem. Mater. 2010, 22, 2898. doi: 10.1021/cm100208a
Hunter, S. M.; Gregory, D. H.; Hargreaves, J. S. J.; Richard, M.; Duprez, D.; Bion, N. ACS Catal. 2013, 3, 1719. doi: 10.1021/cs400336z
Zeinalipour-Yazdi, C. D.; Hargreaves, J. S. J.; Catlow, C. R. A. J. Phys. Chem. C 2015, 119, 28368. doi: 10.1021/acs.jpcc.5b06811
Alexander, A. M.; Hargreaves, J. S. J.; Mitchell, C. Top. Catal. 2012, 55, 1046. doi: 10.1007/s11244-012-9890-3
Alexander, A. M.; Hargreaves, J. S. J.; Mitchell, C. Top. Catal. 2013, 56, 1963. doi: 10.1007/s11244-013-0133-z
Roy, D.; Navarro-Vazquez, A.; Schleyer, P. V. R. J. Am. Chem. Soc. 2009, 131, 13045. doi: 10.1021/ja902980j
Swearer, D. F.; Knowles, N. R.; Everitt, H. O.; Halas, N. J. ACS Energy Lett. 2019, 4, 1505. doi: 10.1021/acsenergylett.9b00860
McEnaney, J. M.; Singh, A. R.; Schwalbe, J. A.; Kibsgaard, J.; Lin, J. C.; Cargnello, M.; Jaramillo, T. F.; Nrskov, J. K. Energy Environ. Sci. 2017, 10, 1621. doi: 10.1039/C7EE01126A
Goshome, K.; Miyaoka, H.; Yamamoto, H.; Ichikawa, T.; Ichikawa, T.; Kojima, Y. Mater. Trans. 2015, 56, 410. doi: 10.2320/matertrans.M2014382
Yamaguchi, S.; Ichikawa, T.; Wang, Y. M.; Nakagawa, Y.; Isobe, S.; Kojima, Y.; Miyaoka, H. ACS Omega 2017, 2, 1081. doi: 10.1021/acsomega.6b00498
Yamaguchi, T.; Shinzato, K.; Yamamoto, K.; Wang, Y.; Nakagawa, Y.; Isobe, S.; Ichikawa, T.; Miyaoka, H.; Ichikawa, T. Int. J. Hydrogen Energy 2020, 45, 6806. doi: 10.1016/j.ijhydene.2019.12.190
Gao, W. B.; Guo, J. P.; Chen, P. Chin. J. Chem. 2019, 37, 442. doi: 10.1002/cjoc.201800586
Veser, G. Nat. Energy 2018, 3, 1025. doi: 10.1038/s41560-018-0293-y
Hagen, S.; Barfod, R.; Fehrmann, R.; Jacobsen, C. J. H.; Teunissen, H. T.; Chorkendorff, I. J. Catal. 2003, 214, 327. doi: 10.1016/S0021-9517(02)00182-3
Liu, T.; Temprano, I.; Jenkins, S. J.; King, D. A. J. Chem. Phys. 2013, 139, 184708 http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Vojvodic, A.; Medford, A. J.; Studt, F.; Abild-Pedersen, F.; Khan, T. S.; Bligaard, T.; Norskov, J. K. Chem. Phys. Lett. 2014, 598, 108. doi: 10.1016/j.cplett.2014.03.003
Michalsky, R.; Steinfeld, A. Catal. Today 2017, 286, 124. doi: 10.1016/j.cattod.2016.09.023
Bartel, C. J.; Rumptz, J. R.; Weimer, A. W.; Holder, A. M.; Musgrave, C. B. ACS Appl. Mater. Interfaces 2019, 11, 24850. doi: 10.1021/acsami.9b01242

图 1 可再生能源驱动的“绿色”合成氨过程
Figure 1 Green ammonia synthesis processes driven by renewable energy

图 2 催化(a)与化学链(b)过程之对比
Figure 2 Comparison of catalytic process (a) with chemical looping process (b)






图 8 金属亚氨基化合物作为载氮体的化学链合成氨. (a) 氢化物的氮化过程. (b) 氮化后样品的加氢放氨. (c) Ni-氢化物化学链产氨速率与热催化速率对比. (d) 氢化物化学链产氨速率与高活性合成氨催化剂热催化产氨速率的对比[10]
Figure 8 Metal imides as nitrogen carriers for chemical looping ammonia synthesis. (a) Nitridation of hydride samples. (b) Hydrogenation of post-nitridized samples. (c) Temperature-dependent ammonia production rates of Ni–AH samples in chemical looping and thermo-catalytic processes, respectively. (d) Comparison of NH3 production rates of Ni catalyzed AH-CL (0.1 MPa) and the conventional thermo-catalytic process (1 MPa). Reprinted from Ref. [10] with permission from Nature Energy, Springer Nature

表 1 常见 CLAS 过程总结
Table 1. Summary of nitrogen carriers for CLAS
| No. | 载氮体对 | 反应 | 反应条件 | 产氨速率/(μmol•g-1•h-1) | 特点 | Ref. |
| 1 | AlN-Al2O3 | Al2O3+3C+N2→ 2AlN+3CO 2AlN+3H2O→ Al2O3+2NH3 |
固氮: 0.1 MPa, 1500~1700 ℃ 水解: 0.1 MPa, 900~1200 ℃ |
无数据 | 高温反应, 动力学性能差, 碳基 化合物作为还原剂, 太阳能聚 热 |
[30, 31] |
| 2 | Cr2N-Cr2O3 | Cr2O3+3H2→ 2Cr+3H2O 2Cr+1/2N2→Cr2N Cr2N+3H2O→ Cr2O3+NH3+3/2H2 |
还原: 0.1 MPa, 1200~1500 ℃ 固氮: 0.1 MPa, 500~1000 ℃ 水解: 0.1 MPa, 500~1000 ℃ |
1000 ℃时, Cr2N水解产氨速 率为108 μmol•g-1•h-1 |
高温反应, 动力学性能差, H2为 还原剂, 过渡金属作为电子给 体还原N2 |
[37] |
| 3 | MaNb-MaNb-d (Mn) |
MaNb-d+d/2N2→ MaNb MaNb+3d/2H2→ MaNb-d+dNH3 |
固氮: 200~ 1000 ℃ 加氢: 0.1 MPa, >300 ℃ |
Mn6N2.58在550 ℃时加氢产 氨速率为55.3 μmol•g-1•h-1 |
动力学性能差, 过渡金属作为 电子给体还原N2 |
[43] |
| 4 | MH2-MaNb
(Ca, Sr) |
MH2+b/2N2→ MaNb+aH2 MaNb+(a+3/2b)H2→ aMH2+bNH3 |
固氮: 200~ 1000 ℃ 加氢: 0.1 MPa, >300 ℃ |
Ca3N2和Sr2N在550 ℃时的加氢产氨速率分别为98和81 μmol•g-1•h-1 | H-作为电子和质子给体还原N2 | [43] |
| 5 | Mg-Mg3N2-MgO | MgO+CH4→ Mg+CO+2H2 3Mg+N2→Mg3N2 Mg3N2+3H2O→ 3MgO+2NH3 |
光源加热 还原&水解: 压力<13.3 Pa 固氮: 0.1 MPa |
1.67 μmol•g-1•h-1 | 光致等离子体加热 | [58] |
| 6 | BaH2-BaNH LiH-Li2NH |
以Ba为例: BaH2+N2→ BaNH+1/2H2 BaNH+2H2→ BaH2+NH3 |
固氮: 0.1 MPa, 100~500 ℃ 加氢: 0.1 MPa, 100~500 ℃ Ni催化剂 |
300 ℃时BaH2-BaNH 的总包反应速率为 5870 μmol•g-1•h-1 |
H‾作为电子和质子给体还 原N2, H2异裂产生H+和H‾ |
[10] |
| 7 | Li-Li3N-LiOH | 6LiOH→ 6Li+3H2O+3/2O2 6Li+N2→2Li3N 2Li3N+6H2O→ 6LiOH+2NH3 |
电解: 0.1 MPa, 450 ℃, 3 V 固氮&水解: 0.1 MPa, 22~100 ℃ |
无数据 | 电热耦合 | [59] |
| 8 | Mn5N2-MnO | Mn5N2+5H2O→ 5MnO+2NH3+2H2 5MnO+5CH4+N2→ Mn5N2+10H2+5CO |
水解: 0.1 MPa, 500 ℃ 固氮: 0.1 MPa, 1150 ℃ |
无数据 | 过渡金属价态发生变化 | [40] |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们