图 1.
肝素酶底物四糖A的结构
Figure 1.
Structure of tetrasaccharide A relevant to the substrate of heparinase

肝素(Heparin)和硫酸肝素(Heparan Sulfate)是一类高度硫酸化的天然直链聚糖, 由重复的二糖单元: 1→4连接的己糖醛酸(α-L-艾杜糖醛酸或者β-D-葡萄糖醛酸)与己糖胺(α-D-氨基葡萄糖)交替连接而成, 其分子量分布在5000~30000之间.肝素和硫酸肝素的硫酸化位点主要在氨基葡萄糖基的3位、6位和糖醛酸的2位, 另外, 氨基葡萄糖基的2位氨基可以有硫酸化、乙酰化和裸露三种形式.
肝素最早由约翰霍普金斯大学的McLean和Howell于1916年从动物的肝脏中分离得到, 并发现了它具有强抗凝血作用[1].近年来, 受益于生物学技术的发展, 肝素类分子越来越多的生物学功能被揭示[2].特别是其独特的抗凝血作用机理得到阐明[3]:肝素主要通过与抗凝血酶Ⅲ(ATⅢ)的结合, 抑制凝血酶原激活物和凝血酶的形成和活性, 从而促进纤维蛋白(血栓)的溶解; 并且通过抑制血小板的粘附和聚集, 间接影响血小板内凝血因子的释放.除了具有抗凝血和抗血栓的功能外, 肝素还具有降血脂、抗动脉粥样硬化、抗中膜平滑肌细胞(SMC)增生、抗炎、抗过敏、抗病毒、抗癌等多种生物学功能[4].
由于肝素结构的复杂性和微观不均一性, 从天然肝素中分离得到结构单一的片断非常困难.同时, 肝素结构中众多的O-硫酸基、N-硫酸基以及羧酸基对其生物活性的影响非常大.通过合成手段得到精确结构的肝素寡糖, 对于其构效关系和生物学研究是极其必要的[3].确实, 肝素寡糖的合成一直是备受化学家关注的课题[2, 5].
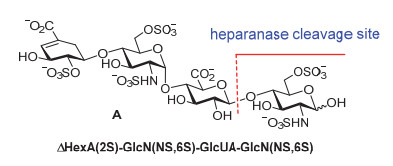
乙酰肝素酶(Heparanase, Hpa)是哺乳动物体内的内切葡萄糖醛酸水解酶, 它能通过选择性水解葡萄糖醛酸(GlcA)与己胺糖(GlcN)之间的β-糖苷键来降解肝素和硫酸肝素(图 1).乙酰肝素酶通过释放肝素寡糖参与多种重要的生理过程, 如肿瘤细胞的浸润和转移[6]、血管的生成[7]、炎症[8]、和平滑肌细胞的增生[9].发展乙酰肝素酶抑制剂用于抑制肿瘤细胞的增殖和转移、新血管的生成和炎症的发生等, 是近来药物研究的一个热点课题.
2002年, Okada和Yamada等采用酸性条件裂解肝素, 分离得到一系列结构确定的肝素寡糖片段; 随后, 测试肝素酶对这些肝素片段的水解活性, 较系统、明确地阐明了人源肝素酶所能识别的底物结构[10].他们发现四糖A和结构相关的六糖是肝素酶能较好识别的底物.四糖A结构为[ΔHexUA(2S)-GlcN(NS, 6S)-GlcUA-GlcN(NS, 6S)](图 1), 由于其中只含一个酶切割位点, 适合用作底物对肝素酶的水解活性进行定量分析.他们的研究还发现, 肝素酶底物的结构中必须具有-GlcN(NS, 6S)-GlcUA-GlcN(NS, 6S)-三糖片段.其中葡萄糖醛酸上的羧基、非还原端己胺糖6位羟基上的硫酸根和还原端己胺糖氨基上的硫酸根至关重要.非还原末端己胺糖上的N-硫酸根和还原端己胺糖6位上的O-硫酸根对于肝素酶的识别有促进作用, 而还原端己胺糖上3位羟基的硫酸根则对肝素酶的活性有抑制作用.
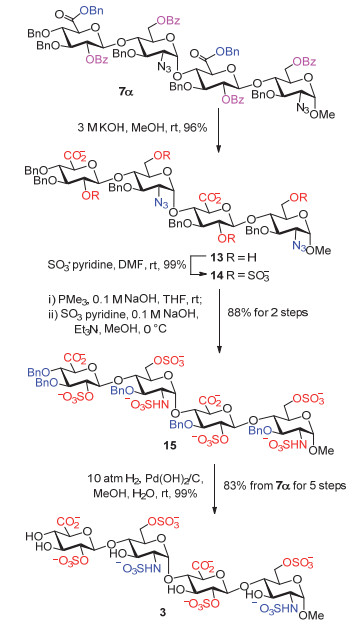
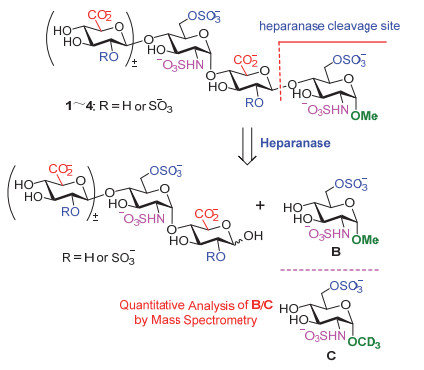
基于上述研究成果, 我们根据人源肝素酶所能较好识别的肝素四糖片段A和最小的三糖片段(-GlcN(NS, 6S)-GlcUA-GlcN(NS, 6S)-)的结构, 拟合成肝素寡糖1~4(图 2), 以用于肝素酶底物选择性的进一步研究, 并发展可用于肝素酶活测试的底物.为了更好地模拟天然肝素分子和方便地表征合成中间体和目标产物, 我们将四糖A中非还原端的不饱和葡萄糖醛酸还原成天然的葡萄糖醛酸, 并把还原端用α-甲苷锁定.我们预期, 合成寡糖1~4与肝素酶作用后, 将被水解成二糖或三糖片段和单糖B; 只要加入内标, 如氘代α-甲苷C, 即可通过定量质谱测定B和C的比例来定量肝素酶的活性.
这里, 我们报道对肝素三糖和四糖1~4的化学合成.
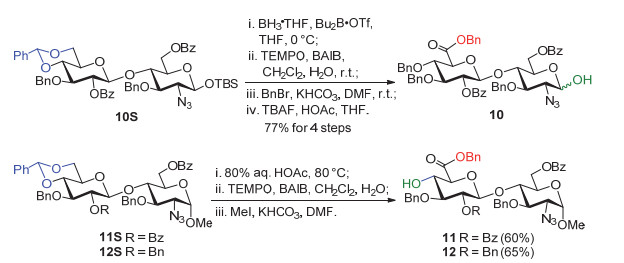
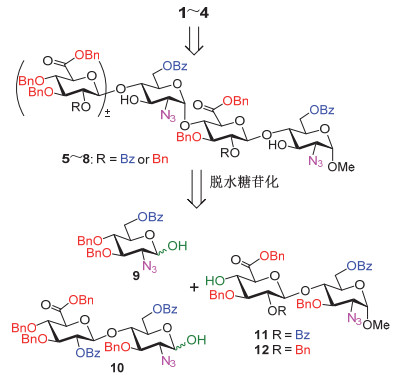
如图 3所示, 我们拟采取精简的保护基策略, 将需要硫酸化的羟基用苯甲酰基(Bz)保护, 需要硫酸化的氨基采用叠氮基(N3)取代, 裸露的羟基和糖醛酸6位的羧酸根采用苄基(Bn)保护.采用汇聚式的[1+2]和[2+2]糖苷化策略, 寡糖5~8可以从单糖砌块9和二糖砌块10~12拼接制备; 我们拟采用直接脱水糖苷化方法来立体选择性地构建其中关键的α-(1→4)糖苷键[11].
如图式 1所示, 从已知二糖砌块10S[12b]出发, 采用硼烷四氢呋喃(BH3•THF)/二丁基硼三氟甲磺酸(Bu2B•OTf)区域选择性的还原苄叉保护基, 裸露出6位羟基[13].对于6位羟基进行2, 2, 6, 6-四甲基哌啶-氮-氧化物(TEMPO)/醋酸碘苯(BAIB)氧化[14], 随后, 苄酯化; 接着, 采用四丁基氟化铵(TBAF)/醋酸(HOAc)条件, 脱除异头位的叔丁基二甲基硅基(TBS), 以四步77%的收率, 制备得到二糖半缩醛10.从已知二糖砌块11S和12S[12b]出发, 用醋酸水溶(φ醋酸=80%)液脱除苄叉, 得到4, 6-二-羟基二糖化合物; 接着, 对6位伯羟基进行TEMPO氧化和苄酯化, 顺利得到二糖受体11和12.在合成二糖11和12时, 裸露的4位羟基可以促进TEMPO氧化, 该反应在4.5 h内完成.而在合成化合物10时, 底物中的4位羟基被苄基保护, 相应的TEMPO氧化反应进行得非常缓慢.使用正常试剂量(nBAIB/n11S or 12S=2.5, nTEMPO /n11S or 12S=0.03), 过夜反应才能完全.若增加醋酸碘苯至nBAIB/n11S or 12S=4, 则3 h即能反应完全.
成功得到糖砌块之后, 我们准备尝试[2+2]糖苷化反应, 探索最佳反应条件来获得高产率和优秀的α选择性.糖苷化反应是聚糖合成中最重要的反应, 糖化学家已发展出多种高效的糖苷化方法, 其中有很多糖苷化方法被成功应用于肝素类寡糖的化学合成中[15].常用的糖苷化反应需要两步操作, 即先由糖基半缩醛进行1位羟基衍生化得到糖基给体, 再在促进剂活化下与亲核性受体进行糖苷化反应.与之相比, 异头位裸露的糖基半缩醛直接进行脱水糖苷化反应, 在形式上更加方便和高效.近年来, 多种脱水试剂组合的开发, 使得这种糖苷化方法也得到了较大的发展[11, 16, 17]. 2000年, Gin小组发现在亚砜或者硫醚以及Tf2O的作用下可以顺利地完成一步脱水糖苷化, 并且取得较好的α选择性[17].随后, 荷兰莱顿大学的van der Marel等将这种脱水糖苷化方法应用于肝素寡糖的合成[18], 采用“一锅法”合成策略, 以较好的α选择性得到了保护的肝素五糖.几乎在相同时间, 我们也开展了类似的工作[19].
在这里, 我们把该脱水糖苷化方法应用于目标寡糖1~4的合成.首先将二糖半缩醛10溶于干燥的二氯甲烷(DCM)中, 加入二苯亚砜(Ph2SO)、2, 4, 6-三叔丁基吡啶(TTBP)和4Å分子筛(MS), 充分搅拌后, 冷却至-60 ℃, 慢慢滴加三氟甲磺酸酐(Tf2O), 滴加完毕后升至-40 ℃反应1 h.然后, 将受体11的二氯甲烷溶液滴加进反应液, 最后自然升温至室温反应, 薄层层析(TLC)监测, 约3 h后滴加三乙胺(NEt3)淬灭反应.该糖苷化反应的选择性非常好, 只得到α产物, 但是, 产率只有32 %(Table 1, entry 1). TCL监测发现, 体系中出现一个荧光很强、极性较小的副产物, 怀疑是给体10异头位发生消除的糖烯产物.但是, 该副产物不稳定, 柱层析分离后即发生分解.为了减少这一副产物的生成, 反应在升温至-40 ℃后, 尝试只搅拌15 min, 即加入受体11的二氯甲烷溶液.结果糖苷化仍然只得到α产物, 但是, 产率反而下降至22%(Table 1, entry 2).考虑到有时候用甲苯做溶剂可以显著提高糖苷化反应的收率[12b], 我们更换溶剂为甲苯, 确实, 糖苷化产率提高到65%, 而α选择性稍有下降(α/β=8.3/1.0)(Table 1, entry 3).
 下载:
导出CSV
下载:
导出CSV
 |
|||
| Entry | Solvent | Yield | α/β |
| 1a | CH2Cl2 | 32% | α only |
| 2b | CH2Cl2 | 22% | α only |
| 3a | Toluene | 65% | 8.3/1.0 |
| a室温条件下, 将二糖10, 二苯亚砜(Ph2SO), 2, 4, 6-三叔丁基嘧啶(TTBP)溶于溶剂中, 加入4 Å分子筛, 搅拌30 min; -60 ℃下加入三氟甲磺酸酐(Tf2O), 升至-40 ℃反应1 h; 加入二糖12, 自然升至室温反应(约3 h). b室温条件下, 将10, 二苯亚砜, 2, 4, 6-三叔丁基嘧啶溶于溶剂中, 加入4 Å分子筛, 搅拌30 min; -60 ℃条件下加入三氟甲磺酸酐, 升至-40 ℃反应15 min; 加入12, 自然升至室温反应(约3 h). | |||
随后, 我们利用优化的脱水糖苷化条件(Ph2SO, TTBP, 4Å MS, Tf2O, toluene), 使用单糖给体9[12b]、二糖给体10分别与二糖受体11和12进行糖苷化反应, 顺利合成了三糖5、6和四糖7(图式 2).糖苷化反应的产率都在65%以上, α/β选择性都高于5.4/1.值得一提的是, 该脱水糖苷化反应的α选择性优于采用N-苯基三氟乙酰亚胺酯给体对类似底物的糖苷化[12].

肝素类寡糖的化学合成中的一大难点是需要对多个羟基和氨基的完全硫酸化操作[15].在肝素合成的文献中, 脱保护和硫酸化的条件很多[20].在成功构建了肝素酶底物三糖和四糖骨架后, 我们尝试探索最优的保护基脱除和硫酸化修饰的操作步骤.
水解脱除酯基保护基(Bn)和酰基保护基(Bz):在文献中[15, 20a, 20b, 21], 一般采用甲酯保护糖醛酸的6位羧基, 为了防止碱性条件下脱除甲酯的同时发生糖醛酸的消除反应, 通常需要采用现制的氢过氧化锂(LiOOH)将甲酯和部分酯基脱除; 再加入氢氧化钾或者氢氧化钠水溶液, 脱除剩余的酰基.我们采用Bn作为羧基保护基, 避免了上述繁琐操作.直接采用3 mol/L氢氧化钾水溶液, 室温反应24 h, 可以一步96%的收率得到O-硫酸化前体13.在这步反应中, 未检测到葡萄糖醛酸发生消除反应的副产物.
O-硫酸化: O-硫酸化的一般条件有: SO3•pyridine, DMF, r.t., >3 h[12b, 20a, 21a, 21b]; SO3•Me3N或者SO3•Et3N, DMF, 55 ℃, 24 h[12a, 20b, 20d]; 氯磺酸, DMF, 8 h; 也可以采用吡啶作为溶剂.氯磺酸较危险; 而SO3•Me3N或者SO3•Et3N作为硫酸化试剂的反应需要加热, 并且, 位阻较大的糖醛酸2位羟基经常无法完全硫酸化.经过摸索, 我们采用SO3•pyridine作为硫酸化试剂, 无水DMF作为溶剂, 反应5 h, 便以定量的收率得到O-硫酸化产物14.该反应采用TLC监测(RP-18反相板), 显示只有一个新产物点生成.后处理时, 先加入甲醇淬灭反应, 然后加入三乙胺调节pH至碱性.减压浓缩后, 经过Sephadex LH-20或者反相柱层析分离, 最后经Dowex 50WX4钠离子柱交换, 便得到所需的O-硫酸化产物.
叠氮还原:在文献中, 还原叠氮有很多方法: Staudinger还原[22]; 丙二硫醇还原[23]; Lindlar催化[24]; Pd催化[25]; SmI2还原[26]等等.我们经过多次尝试发现:如果采用Lindlar催化剂氢化, 101.3 kPa H2条件不足以将寡糖2位叠氮基还原完全; 而用SmI2还原叠氮的效果也不理想; 丙二硫醇还原叠氮效果很好, 然而, 硫醇具有恶臭气味, 后处理比较麻烦.综合考虑, 我们采用Staudinger还原叠氮, 具体操作是加入1 mol/L的三甲基膦(PMe3)的THF溶液和0.1 mol/L NaOH水溶液, 反应过夜.反应结束后, 将体系旋干, 无需提纯, 可以直接进行下一步氨基保护.体系中加入碱的原因是[27]:在Staudinger还原时, 由于羧酸根裸露, 会形成稳定的电荷分离中间体, 加入氢氧化钠溶液破坏这类中间体, 可以顺利得到还原产物.在这里选择三甲基膦作为还原试剂, 是因为其沸点较低, 后处理简单, 直接旋干反应溶剂, 以便进行下一步N-硫酸化.
N-硫酸化:在肝素合成中, N-硫酸化的常用条件有: SO3•pyridine, Et3N, pyridine[21, 28]和SO3•pyridine, Et3N和0.1 mol/L NaOH, MeOH[12b, 20a].我们采用第二种条件, 这也是用于选择性N-硫酸化的条件.将上一步粗品溶于甲醇中, 冰浴下分别加入三乙胺, 0.1 mol/L NaOH, 将体系调节到碱性, 每隔0.5 h加入SO3•pyridine(氨基摩尔数的5倍), 分三批加入, 并且保持在0 ℃下反应约5 h.实验发现, 在这种方法操作下, N-硫酸化反应体系简单, TLC监测原料点消失后, 旋干溶剂, 进行反相柱层析和钠离子柱交换, 可以88%的产率得到N-硫酸化产物15.
还原氢化脱除苄基:还原氢化一般是肝素合成的最后一步, 在几乎所有的文献中, 还原氢化脱除苄基这一步都被忽视, 给出的实验操作非常简单, 例如Seeber- ger小组(Pd/C, 101.3 kPa H2, MeOH/H2O)[21]; 黄雪飞小组(Pd(OH)2/C or Pd/C, 101.3 kPa H2, MeOH/H2O)[20d, 29]; Boons小组两步策略(Pd/C, 101.3 kPa H2, MeOH/H2O; Pd(OH)2/C, 101.3 kPa H2, H2O)[20a]等等.然而, 肝素寡糖中的氨基对于氢化反应有很大的影响, 采用上述常压氢化条件, 甲醇和水作为溶剂, 反应从1 d延长到3 d, 苄基仍然大量存在.查阅台湾清华大学洪上程教授小组的博士论文[30], 我们发现, 虽然发表文献中的氢化操作很简单, 在实际操作中却都有类似的问题, 即无法将苄基完全脱除.洪上程小组的解决方法是:一、采取两次氢化, 先加入质量为原料质量的两倍的Pd/C(wPd=10%), 常压氢化1 d, 之后过滤旋干, 再次投料反应1 d, 可以将苄基基本脱除; 二、尝试同时还原叠氮基团和苄基时, 他们注意到氨基的影响, 故添加了乙酸, 仍然效果不佳.最后, 通过筛选氢化条件发现, 在344.7 kPa H2条件下, Pd/C(wPd=10%)催化氢化可以将苄基和叠氮还原完全.最近, 很多小组也开始尝试pH=7的缓冲水作为溶剂进行还原氢化脱除苄基[12b, 20b, 31].考虑到缓冲水作为溶剂, 会产生非常多的盐类物质, 需要采用凝胶柱多次层析才能去除, 增加了操作步骤.我们最终决定采用高压氢化条件(1013 kPa H2, Pd(OH)2/C(wPd=10%), MeOH/H2O), 反应3 d可以将苄基完全脱除, 以几乎定量的收率得到最终的肝素四糖3.高压氢化这个方法的后处理简单, 只需要过滤除去钯催化剂, 即能快速得到最终的肝素寡糖.
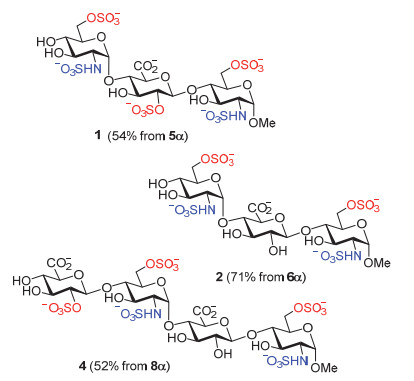
按照上述优化的保护基脱除和硫酸化的实验步骤, 我们顺利制备得到了另外三个目标寡糖1, 2和4.最后五步的总收率分别为54%, 71%和52%(图式 4).肝素酶底物三糖和四糖1~4的核磁和质谱数据与文献报道的相符合[12].
乙酰肝素酶是哺乳动物体内的内切葡萄糖醛酸水解酶, 在肿瘤细胞的增殖与转移中起着非常重要的作用.当人体中发生细胞的癌突, 在其癌变部位的乙酰肝素酶的表达会发生异常.定量质谱分析肝素酶水解能力的基础是需要实现还原端单糖B与氘代标准物单糖C[20i]的定量质谱检测.为此, 我们对于合成的单糖B和C进行了不同比例下的定量质谱测试.
下载:
导出CSV
| Sample No. | Molar ratio(B/C) | Observed m/z for [M-Na]-(B)a | Intensity(B) | Observed m/z for [M-Na]-(C) | Intensity(C)b | Intensity ratio(B/C) |
| 1 | NAc | NA | NA | NA | NA | NA |
| 2 | 9:1 | 374.13 | 49411 | 377.07 | 6486 | 7.6181 |
| 3 | 8:2 | 374.27 | 77031 | 377.20 | 22613 | 3.4065 |
| 4 | 7:3 | 374.27 | 65635 | 377.20 | 35878 | 1.8294 |
| 5 | 6:4 | 374.27 | 49172 | 377.20 | 42720 | 1.1510 |
| 6 | 5:5 | 374.20 | 31805 | 377.13 | 42525 | 0.7479 |
| 7 | 4:6 | 374.27 | 27158 | 377.13 | 54446 | 0.4988 |
| 8 | 3:7 | 374.33 | 22614 | 377.20 | 84840 | 0.2665 |
| a Calculated value m/z for [M-Na]- is 374.30; b Calculated value m/z for [M-Na]- is 377.32; c Not available. | ||||||
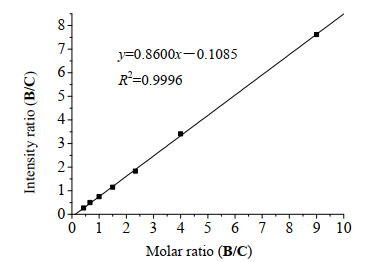
根据不同比例单糖的定量质谱丰度比做出量化曲线图(图 4), 建立了最佳的回归曲线和回归方程y=0.8600x-0.1085, 相关性系数R2=0.9996.这一结果为定量测试肝素酶水解能力打下坚实的基础.
我们报道了一条高效的合成肝素寡糖的路线:采用苯甲酰基保护待硫酸化的羟基、苄基保护羧基和裸露的羟基、叠氮基保护氨基; 应用脱水糖苷化方法高效构建关键的GlcN-(1α→4)-GlcA糖苷键; 采用标准的保护基脱除和硫酸化操作, 高效、便捷地获得肝素酶底物三糖和四糖1~4.最后五步操作总收率超过52%.这些寡糖的合成为我们进一步研究肝素酶的底物选择性, 并建立一套肝素酶活性定量测试方法打下了基础.而高效灵敏的测活方法可用于肝素酶抑制剂的筛选和肿瘤等相关疾病的诊断.
未特别注明的商业化试剂并未进行进一步纯化.常用的溶剂使用微波炉活化的4 Å分子筛进行干燥.核磁共振数据由Bruker AMX-300和BrukerAV-400型核磁共振仪测定(以TMS作为内标); 高分辨质谱由HP5989A和VG Quattro MS/MS质谱仪测定; 比旋光度由Perkin-Elmer 241 MC型自动比旋光仪测定, 光源为钠源, 589 nm; 板层析监测采用10%浓硫酸乙醇加热显色或是UV254 nm显色.
(Benzyl 2-O-benzoyl-3, 4-di-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α/β-D-glucopyranose(10):
二糖化合物13[12b](2.6 g, 2.7 mmol)溶于干燥的四氢呋喃(3 mL), 冰浴冷却, 加入BH3•THF(1 mol/L, 27 mL), 搅拌5 min, 加入Bu2B•OTf的二氯甲烷溶液(1 mol/L, 5.4 mL).冰浴下反应3 h, TLC显示反应完全.加入三乙胺(1.5 mL)后, 缓慢滴加甲醇淬灭反应, 减压浓缩, 再分别加入甲醇稀释, 浓缩三次.柱层析(V石油醚:V乙酸乙酯=6:1)得到tert-Butyldimethylsilyl(2-O-benzoyl-3, 4-O- benzyl-β-D-glucopyranosyl)-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-β-D-glucopyranoside无色糖浆(2.4 g, 2.5 mmol), 产率93%. Rf =0.45(V石油醚:V乙酸乙酯=4:1); 1H NMR(400 MHz, CDCl3) δ: 8.01 (d, 2H, J=6.8 Hz), 7.91 (d, 2H, J=7.2 Hz), 7.57 (t, 1H, J=7.2 Hz), 7.44~7.24 (m, 15H), 7.12~7.08 (m, 5H), 5.29 (t, 1H, J=9.2 Hz), 4.95 (d, 1H, J=11.2 Hz), 4.83~4.70 (m, 3H), 4.62~4.58 (m, 3H), 4.47~4.42 (m, 2H), 4.35 (dd, 1H, J=10.8 Hz, 6.0 Hz), 3.80~3.74 (m, 2H), 3.63~3.58 (m, 2 H), 3.47~3.43 (m, 1H), 3.39~3.30 (m, 3H), 3.26~3.22 (m, 1H), 0.83 (s, 9H), 0.057 (s, 3H), 0.034 (s, 3H); HRMS (ESI) calcd for C53H61N3O12SiNa: 982.3917; Found: 982.3942.
将上一步得到的无色糖浆(1.53 g, 1.59 mmol)溶于二氯甲烷和水的混合溶液(15 mL, V:V=2:1), 加入TEMPO (5 mg, 0.03 mmol)和BAIB (1.3 g, 4.0 mmol).室温下剧烈搅拌过夜(约12 h), TLC显示反应完全.饱和NaHSO3水溶液淬灭反应, 二氯甲烷稀释, 分层, 收集有机相.加入5%的稀盐酸酸化水层, 二氯甲烷萃取水相三次, 合并有机相, 无水硫酸钠干燥, 过滤, 用旋转蒸发仪旋干溶剂, 得到粗产品tert-Butyldimethylsilyl (2-O-benzoyl-3, 4-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-β-D-glucopyranoside为浅黄色糖浆.
将上一步的浅黄色糖浆溶解于无水DMF (15 mL)中, 加入KHCO3 (640 mg, 6.4 mmol), 冰浴冷却, 加入苄溴(0.3 mL, 2.4 mmol).恢复室温, 反应过夜, TLC显示反应完成.加入水和乙酸乙酯稀释, 分出有机相, 水相用乙酸乙酯萃取三次, 合并有机相, 饱和食盐水溶液洗一次, 无水硫酸钠干燥.柱层析纯化(V石油醚:V乙酸乙酯=5:1)得到tert-Butyldimethylsilyl(benzyl 2-O-benzoyl- 3, 4-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-β-D-glucopyranoside白色固体(1.5 g, 1.4 mmol), 两步产率88%.
称取上一步白色固体(410 mg, 0.39 mmol)溶于四氢呋喃(12 mL)和醋酸(0.07 mL, 1.2 mmol), 室温磁力搅拌下, 加入TBAF的四氢呋喃溶液(1 mol/L, 0.8 mL, 0.8 mmol).自然升至室温反应1.5 h, TLC跟踪监测反应完全后, 加入二氯甲烷稀释, 水洗, 饱和碳酸氢钠溶液洗, 食盐水洗, 无水硫酸钠干燥.过滤, 浓缩, 柱层析(V石油醚:V乙酸乙酯=2:1), 得无色糖浆10 (344 mg, 0.4 mmol), 收率94%. ESI-MS (m/z): 891.8[M+NH4]+, 896.7 [M+Na]+.
Methyl(benzyl 2-O-benzoyl-3-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside(11):
二糖化合物14[12b](1.2 g, 1.6 mmol)溶于冰乙酸(20 mL)中, 加热至80 ℃, 慢慢滴加入5 mL H2O, 继续回流反应2 h, TLC检测反应完全, 冷至室温, 旋干, 柱层析(V石油醚:V乙酸乙酯=1:1)得到脱除苄叉的无色糖浆(1.0 g, 1.4 mmol), 产率96%. 1H NMR (400 MHz, CDCl3) δ: 8.10 (d, 2H, J=6.8 Hz), 7.95 (d, 2H, J=6.8 Hz), 7.59 (t, 1H, J=7.2 Hz), 7.49~7.15 (m, 15H), 5.31 (t, 1H, J=8.8 Hz), 5.04 (d, 1H, J=10.8 Hz), 4.82 (d, 1H, J=10.8 Hz), 4.71~4.38 (m, 6H), 3.94~3.61 (m, 6H), 3.44~3.41 (dd, 1H, J=3.6, 9.6 Hz), 3.34 (s, 3H), 3.34~3.16 (m, 2H), 2.54~2.52 (m, 1H), 1.87~1.83 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 166.1, 165.0, 138.2, 137.7, 133.4, 129.8, 129.5, 129.2, 128.7, 128.6, 128.5, 128.0, 127.93, 127.90, 127.4, 101.0, 98.4, 82.7, 78.1, 77.7, 75.7, 75.3, 74.8, 73.9, 70.6, 68.9, 63.2, 62.6, 62.0, 55.4; HRMS (MALDI/DHB) calcd for C41H43N3O12Na: 792.2739; Found: 792.2735.
将上一步无色糖浆(1.0 g, 1.4 mmol)溶于二氯甲烷和水的混合溶液(15 mL, V:V=2:1), 加入TEMPO(4 mg, 0.03 mmol), BAIB(1.0 g, 3.2 mmol), 室温下剧烈搅拌4.5 h, TLC显示反应完全.饱和NaHSO3水溶液淬灭反应, 二氯甲烷稀释, 分层, 收集有机相.加入质量分数5%的稀盐酸酸化水层, 二氯甲烷萃取水相三次, 合并有机相, 无水硫酸钠干燥, 过滤, 用旋转蒸发仪旋干溶剂, 得到Methyl(2-O-benzoyl-3-O-benzyl-β-D- glucopyranosyluronate)-(1→4)-2-azido-6-O-benzoyl-3-O- benzyl-2-deoxy-α-D-glucopyranoside粗产品为浅黄色糖浆.
上一步得到的糖浆溶解于DMF(12 mL)中, 加入KHCO3(0.5 g, 5.1 mmol), 冰浴冷却, 加入苄溴(0.23 mL, 1.9 mmol), 恢复室温反应过夜. TLC显示反应完成.加入水和乙酸乙酯稀释, 分出有机相, 水相用乙酸乙酯萃取三次, 合并有机相, 饱和食盐水溶液洗一次, 无水硫酸钠干燥.柱层析纯化(V石油醚:V乙酸乙酯=4:1)得到化合物11 (0.7 g, 0.8 mmol), 为无色糖浆, 两步产率62%.[α]25 D=80.1 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 8.02 (d, 2H, J=7.6 Hz), 7.88 (d, 2H, J=7.2 Hz), 7.52 (t, 1H, J=7.6 Hz), 7.45 (t, 1H, J=7.2 Hz), 7.38~7.26 (m, 14H), 7.12 (s, 5H), 5.33 (t, 1H, J=8.8 Hz), 5.09~4.95 (m, 3H), 4.78~4.65 (m, 5H), 4.49~4.45 (m, 1H), 4.35 (dd, 1H, J=12.0 Hz, 4.0 Hz), 4.06 (t, 1H, J=9.6 Hz), 3.97~3.90 (m, 2H), 3.78~3.76 (m, 1H), 3.71 (d, 1H, J=10.0 Hz), 3.64 (t, 1H, J=9.2 Hz), 3.33~3.30 (m, 4H), 2.87 (bs, 1H); 13C NMR (100 MHz, CDCl3) δ: 168.39, 165.97, 164.96, 138.25, 137.68, 134.86, 133.39, 133.31, 129.85, 129.56, 129.53, 129.18, 128.64, 128.59, 128.51, 128.34, 128.29, 127.99, 127.60, 101.14, 98.51, 80.97, 77.96, 77.79, 75.33, 74.75, 74.44, 73.24, 72.14, 68.69, 67.51, 63.25, 62.55, 55.41; HRMS (ESI) calcd for C48H47N3O13Na: 896.3001; Found: 896.3040.
Methyl(benzyl 2, 3-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-2-azido-3-O-benzyl-2-deoxy-6-O-benzoyl-α-D-glucopyranoside(12):
二糖化合物15[12b](0.9 g, 1.0 mmol)溶于冰乙酸(10 mL)中, 加热至80 ℃, 慢慢滴加入6 mL H2O, 继续回流反应2 h, TLC检测反应完全, 冷至室温, 旋干, 柱层析(VPE:VEA=1:1)得到脱除苄叉的无色糖浆(0.65 g, 0.86 mmol), 产率82%. ESI-MS(m/z): 773.5 [M+NH4]+, 778.3[M+Na]+.
将上一步无色糖浆(0.62 g, 0.82 mmol)溶于二氯甲烷和水的混合溶液(15 mL, V:V=2:1), 加入TEMPO(3 mg, 0.02 mmol), BAIB(0.66 g, 2.05 mmol), 室温下剧烈搅拌4.5 h, TLC显示反应完全.饱和NaHSO3水溶液淬灭反应, 二氯甲烷稀释, 分层, 收集有机相.加入质量分数5%的稀盐酸酸化水层, 二氯甲烷萃取水相三次, 合并有机相, 无水硫酸钠干燥, 过滤, 用旋转蒸发仪旋干溶剂, 得到粗产品Methyl(2, 3-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-2-azido-3-O-benzyl-2-deoxy-6-O-benzoyl-α-D-glucopyranoside为浅黄色糖浆.
上一步得到的糖浆溶解于DMF(10 mL)中, 加入KHCO3(0.33 g, 3.3 mmol), 冰浴冷却, 加入苄溴(0.15 mL, 1.23 mmol), 恢复室温反应过夜. TLC显示反应完成.加入水和乙酸乙酯稀释, 分出有机相, 水相用乙酸乙酯萃取三次, 合并有机相, 饱和食盐水溶液洗一次, 无水硫酸钠干燥.柱层析纯化(VPE:VEA=4:1)得到化合物12(0.6 g, 0.65 mmol), 为无色糖浆, 两步产率79%. 1H NMR (400 MHz, CDCl3) δ: 7.95 (d, 2H, J=7.6 Hz), 7.49 (t, 1H, J=7.2 Hz), 7.41~7.25 (m, 22H), 5.09~4.98 (m, 3H), 4.87~4.79 (m, 4H), 4.73~4.66 (m, 3H), 4.57 (d, 1H, J=6.8 Hz), 4.44~4.39 (dd, 1H, J=12.4 Hz, 4.8 Hz), 4.02 (t, 1H, J=9.2 Hz), 3.94~3.85 (m, 2H), 3.78 (dd, 1H, J=10.0 Hz, 3.2 Hz), 3.69 (d, 1H, J=9.6 Hz), 3.45~3.42 (m, 5H), 3.36 (dd, 1H, J=10.4 Hz, 3.6 Hz), 2.67 (d, 1H, J=2.4 Hz); HRMS (ESI) calcd for C48H49N3O12Na: 882.3209; Found: 882.3228.
General Procedure A: 脱水糖苷化方法具体步骤:
给体(1 equiv., 0.02 mol/L in Tol)溶于适量干燥甲苯中, 加入Ph2SO (2.8 equiv.), TTBP (3.0 equiv.), 4 Å MS, 室温下搅拌0.5 h.降温至-60 ℃, 滴加入Tf2O (1.4 equiv.), 升温至-40 ℃反应1 h后, 滴加入受体(1.5~2.0 equiv.)的甲苯溶液(0.1 mol/L), 然后自然升温到室温反应.大约1到3 h后, TLC(VTol:VEA=20:1)检测反应完全.加入三乙胺淬灭反应.硅藻土过滤.滤液先后用饱和NaHCO3溶液, NaCl溶液洗涤, 无水Na2SO4干燥.浓缩后通过柱层析(甲苯/乙酸乙酯)提纯可以分离出α/β异构体.
Methyl(2-azido-3, 4-O-benzyl-2-deoxy-6-O-benzoyl-α-D-glucopyranosyl)-(1→4)-(benzyl 2-O-benzoyl-3-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-2-azido-3-O-benzyl-2-deoxy-6-O-benzoyl-α-D-glucopyranoside(5):
Procedure A:给体9(81 mg, 0.165 mmol), 受体11(290 mg, 0.330 mmol); 柱层析(VTol:VEA=30:1), 三糖产物5: 175 mg, 79%, α:β=24.0:1.0.
[α]D23=64.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 8.08 (d, 2H, J=7.2 Hz), 8.04 (d, 2H, J=7.2 Hz), 7.94 (d, 2H, J=7.2 Hz), 7.52~7.14 (m, 34H), 5.49~5.45 (m, 2H), 5.11~5.00 (m, 3H), 4.89~4.39 (m, 14H), 3.99~3.91 (m, 4H), 3.84 (t, 1H, J=9.2 Hz), 3.77~3.75 (m, 2H), 3.66 (t, 1H, J=9.6 Hz), 3.37~3.30 (m, 5H); 13C NMR (100 MHz, CDCl3) δ: 167.35, 166.13, 165.97, 164.80, 137.96, 137.53, 137.41, 137.20, 134.60, 133.54, 133.35, 133.15, 129.85, 129.78, 129.72, 129.55, 129.04, 128.99, 128.69, 128.62, 128.56, 128.51, 128.48, 128.43, 128.40, 128.32, 128.24, 128.09, 127.73, 127.67, 125.32, 101.19, 98.51, 97.34, 82.24, 80.26, 78.09, 77.90, 75.66, 75.59, 75.31, 74.75, 73.63, 70.06, 68.73, 67.82, 63.44, 63.17, 62.66, 62.44, 55.40; HRMS (ESI) calcd for C75H72N6O18Na: 1367.4795; Found: 1367.4803.
Methyl(2-azido-3, 4-O-benzyl-2-deoxy-6-O-benzoyl-α-D-glucopyranosyl)-(1→4)-(benzyl 2, 3-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-2-azido-3-O-benzyl-2-deoxy-6-O-benzoyl-α-D-glucopyranoside(6):
Procedure A:给体9(49 mg, 0.1 mmol), 受体12(172 mg, 0.2 mmol); 柱层析(VTol:VEA=30:1), 三糖产物6: 102 mg, 76%, α:β=6.3:1.0.
[α]D23=58.0 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 8.03 (d, 2H, J=7.2 Hz), 7.96 (d, 2H, J=7.2 Hz), 7.53~7.20 (m, 36H), 5.55 (d, 1H, J=3.2 Hz), 5.51 (t, 1H, J=9.6 Hz), 5.35 (t, 1H, J=8.7 Hz), 5.05~5.03 (m, 2H), 5.00 (d, 2H, J=1.5 Hz), 4.83~4.70 (m, 5H), 4.64~4.50 (m, 4H), 4.24~4.24 (m, 3H), 4.15~3.98 (m, 4H), 3.84~3.77 (m, 5H), 3.72~3.66 (m, 2H), 3.53 (dd, 1H, J=10.2 Hz, 3.6 Hz), 3.45~3.41 (m, 1H), 3.34~3.28 (m, 5H); 13C NMR (100 MHz, CDCl3) δ: 167.74, 166.22, 165.87, 138.20, 137.85, 137.83, 137.62, 137.50, 134.70, 133.36, 133.17, 129.96, 129.79, 129.73, 129.59, 129.08, 128.64, 128.62, 128.60, 128.53, 128.49, 128.18, 128.13, 128.10, 128.02, 127.94, 127.65, 127.39, 102.98, 98.48, 97.40, 84.08, 82.59, 80.35, 78.11, 77.95, 77.73, 75.93, 75.67, 75.56, 75.34, 75.29, 74.96, 74.58, 69.96, 69.05, 67.83, 63.51, 63.09, 62.79, 62.35, 55.55; HRMS (ESI) calcd for C75H74N6O17Na: 1353.5003; Found: 1353.5027.
Methyl(benzyl 2-O-benzoyl-3, 4-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-(2-azido-3-O-benzyl-2-deoxy-6-O-benzoyl-α-D-glucopyranosyl)-(1→4)-(benzyl 2-O-benzoyl-3-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-2-azido-3-O-benzyl-2-deoxy-6-O-benzoyl-α-D-glucopyranoside(7):
Procedure A:给体10(171 mg, 0.18 mmol), 受体11(315 mg, 0.36 mmol); 柱层析(VTol:VEA=25:1), 四糖产物7: 237 mg, 73%, α:β=5.4:1.0.
[α]D23=68.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 8.06~7.99 (m, 6H), 7.89 (d, 2H, J=8.0 Hz), 7.48~7.04 (m, 48H), 5.45~5.37 (m, 3H), 5.16 (d, 1H, J=11.2 Hz), 4.99~4.97 (m, 3H), 4.79 (d, 1H, J=8.4 Hz), 4.73~4.34 (m, 14H), 4.37 (dd, 1H, J=12.0 Hz, 3.6 Hz), 4.28 (t, 1H, J=9.6 Hz), 4.05~3.97 (m, 2H), 3.93~3.83 (m, 5H), 3.78~3.71 (m, 3H), 3.66 (d, 1H, J=10.4 Hz), 3.28~3.25 (m, 5H); 13C NMR (100 MHz, CDCl3) δ: 167.49, 167.09, 165.97, 165.92, 164.86, 164.74, 138.06, 137.87, 137.82, 137.45, 137.30, 137.04, 134.95, 134.53, 133.54, 133.49, 133.37, 129.83, 129.75, 129.59, 129.55, 129.48, 129.46, 129.07, 128.92, 128.88, 128.77, 128.68, 128.62, 128.56, 128.55, 128.48, 128.46, 128.37, 128.35, 128.29, 128.27, 128.25, 127.94, 127.87, 127.76, 127.74, 127.69, 127.67, 125.32, 101.16, 101.15, 98.47, 96.87, 82.55, 81.98, 79.45, 77.97, 77.81, 77.72, 77.60, 75.68, 75.64, 75.35, 75.02, 74.93, 74.69, 74.38, 73.95, 73.63, 73.48, 69.45, 68.72, 67.43, 67.36, 63.13, 62.83, 62.34, 62.02, 55.38; ESI-MS (m/z): 1828.4 [M+Na]+.
Methyl(benzyl 2-O-benzoyl-3, 4-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-(2-azido-3-O-benzyl-2-deoxy-6-O-benzoyl-α-D-glucopyranosyl)-(1→4)-(benzyl 2, 3-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-2-azido-3-O-benzyl-2-deoxy-6-O-benzoyl-α-D-glucopyranoside(8):
Procedure A:给体10(192 mg, 0.2 mmol), 受体12(343 mg, 0.4 mmol); 柱层析(VTol:VEA=30:1), 四糖产物8: 236 mg, 65%, α:β=8.3:1.0.
1H NMR (400 MHz, CDCl3) δ: 7.98~7.95 (m, 4H), 7.89 (d, 2H, J=8.4 Hz), 7.48~7.06 (m, 49H), 5.50 (d, 1H, J=4.0 Hz), 5.38 (t, 1H, J=8.8 Hz), 5.13 (d, 1H, J=10.4 Hz), 5.01~4.92 (m, 4H), 4.79~4.73 (m, 4H), 4.69~4.41 (m, 13H), 4.34 (dd, 1H, J=12.4 Hz, 4.0 Hz), 4.11 (t, 1H, J=9.2 Hz), 4.03~3.90 (m, 3H), 3.83~3.61 (m, 8H), 3.46 (t, 1H, J=8.0 Hz), 3.40 (s, 2H), 3.31 (dd, 1H, J=10.4 Hz, 3.6 Hz), 3.24 (dd, 1H, J=10.4 Hz, 4.0 Hz); 13C NMR (100 MHz, CDCl3) δ: 167.59, 167.49, 166.08, 165.86, 164.87, 138.13, 138.06, 137.82, 137.77, 137.53, 137.38, 135.05, 134.62, 133.56, 133.46, 133.43, 129.93, 129.69, 129.68, 129.64, 129.54, 129.26, 129.03, 128.99, 128.73, 128.70, 128.69, 128.67, 128.64, 128.59, 128.55, 128.52, 128.49, 128.37, 128.07, 128.05, 128.00, 127.94, 127.86, 127.76, 127.70, 127.54, 102.95, 101.22, 98.44, 96.89, 84.20, 82.68, 82.11, 79.55, 78.07, 77.91, 77.70, 77.62, 77.36, 75.97, 75.81, 75.61, 75.48, 75.40, 75.17, 75.01, 74.29, 74.08, 73.52, 69.37, 69.07, 67.48, 63.05, 62.88, 62.20, 62.13, 55.60; ESI-MS (m/z): 1813.0 [M+Na]+.
肝素寡糖脱保护硫酸化的一般性步骤:
General Procedure B: 水解:
将化合物溶于四氢呋喃和甲醇的混合溶液(VTHF :VMeOH=1:1, 0.01 mol/L), 慢慢加入3 mol/L KOH水溶液(VKOH:VTHF=1:1), 室温反应24 h. TLC(VDCM:VMeOH=10:1)检测反应完全后, 加入酸性树脂调至中性.使用旋转蒸发仪旋去反应液, 凝胶柱LH-20(VDCM:VMeOH= 1:1)可分离出目标产物.
General Procedure C: O-硫酸化:
将起始原料溶于DMF溶液(0.02 mol/L)中, 加入三氧化硫吡啶(10 equiv. per hydroxyl), 室温下反应4~8 h. TLC(反相板, VMeOH:VH2O=3:1)检测反应完全后, 可以直接LH-20(VDCM:VMeOH=1:1)提纯; 或者加入甲醇搅拌15 min, 使用旋转蒸发仪旋去反应液, 反相柱RP-18提纯, MeOH和H2O作为洗脱剂, 梯度淋洗(0:100→75:25, V:V)分离出目标分子, 之后通过Dewex50WX4Na+柱交换成钠型.
General Procedure D: 叠氮还原以及N-硫酸化:
将起始原料溶于THF(0.005 mol/L)中, 加入0.1 mol/L NaOH水溶液(5 mol per —N3), 之后滴加入PMe3的THF溶液(4 mol per —N3), 室温下反应8 h. TLC(反相板, VMeOH:VH2O=2:1)检测反应完全后, 小心滴加0.1 mol/L HCl溶液调pH至7~8, 使用旋转蒸发仪旋去反应液.
残余物直接溶解在甲醇(0.005 mol/L)中, 加入三乙胺(VNEt3:VMeOH=1:3), 0.1 mol/L NaOH水溶液(2 mol per —NH2), 充分搅拌后, 降温至0 ℃, 将三氧化硫吡啶(10 mol per —NH2)加入, 冰浴下继续反应过夜. TLC(反相板, VMeOH:VH2O=1:1)检测反应完全后, 浓缩反应液, 反相柱RP-18提纯, MeOH和H2O作为洗脱剂, 梯度淋洗(0:100→50:50, V:V)分离出目标产物, 之后通过Dewex50WX4Na+柱交换成钠型.
General Procedure E: Pd(OH)2/C脱保护:
将起始原料溶于甲醇和水的混合溶液(VMeOH:VH2O=3:1)中, 室温1013 kPa H2下氢化3 d, 使用滤纸过滤掉Pd(OH)2/C, 旋干得到目标分子.如果产物中残余少量Pd(OH)2/C, 可使用凝胶柱G-10提纯.
Methyl(3, 4-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-(2-azido-3-O-benzyl-2-deoxy-α-D-glucopyranosyl)-(1→4)-(3-O-benzyl-β-D-glucopyranosyluronate)-(1→4)-2-azido-3-O-benzyl-2-deoxy-α-D-glucopyranoside(13):
Procedure B:起始原料7α(134 mg, 0.074 mmol); 产物13(85 mg, 96%).
[α]D24=46.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CD3OD) δ: 7.47~7.21 (m, 25H), 5.52 (d, 1H, J=2.8 Hz), 5.13~5.07 (m, 3H), 4.91~4.63 (m, 10H), 4.06~3.48 (m, 18H), 3.31 (s, 3H), 3.20 (dd, 1H, J=9.6 Hz, 2.8 Hz), 3.10~3.08 (m, 1H); 13C NMR (100 MHz, CD3OD) δ: 140.19, 140.06, 139.76, 139.71, 139.56, 130.01, 129.98, 129.39, 129.36, 129.31, 129.09, 129.04, 129.01, 128.77, 128.73, 128.71, 128.67, 128.64, 104.69, 104.56, 100.11, 99.19, 86.13, 85.48, 80.95, 79.55, 79.21, 78.74, 77.87, 76.74, 76.40, 76.36, 76.14, 75.99, 73.14, 72.77, 64.29, 64.16, 61.06, 60.56, 55.62; HRMS (ESI) calcd for C60H68N6O21Na: 1231.4330; Found: 1231.4300.
Methyl(3, 4-O-benzyl-2-O-sulfo-β-D-glucopyranosyluronate)-(1→4)-(2-azido-3-O-benzyl-6-O-sulfo-2-deoxy-α-D-glucopyranosyl)-(1→4)-(3-O-benzyl-2-O-sulfo-β-D-glucopyanosyluronate)-(1→4)-2-azido-3-O-benzyl-6-O-sulfo-2-deoxy-α-D-glucopyranoside(14):
Procedure C:起始原料13(22 mg, 0.018 mmol); 产物14(28 mg, 99%).
1H NMR (400 MHz, CD3OD) δ: 7.55~7.23 (m, 25H), 5.49 (d, 1H, J=3.6 Hz), 5.37 (d, 1H, J=9.6 Hz), 5.27~5.18 (m, 3H), 5.07~5.02 (m, 2H), 4.79~4.67 (m, 9H), 4.50 (m, 2H), 4.32~4.23 (m, 2H), 4.07~3.87 (m, 12H), 3.41 (s, 3H), 3.26~3.20 (m, 2H); ESI-MS (m/z): 509.1[M+3H]3-.
Methyl(3, 4-O-benzyl-2-O-sulfo-β-D-glucopyrano-syluronate)-(1→4)-(2-N-sulfo-3-O-benzyl-6-O-sulfo-2-deoxy-α-D-glucopyranosyl)-(1→4)-(3-O-benzyl-2-O-sulfo-β-D-glucopyanosyluronate)-(1→4)-2-N-sulfo-3-O-benzyl-6-O-sulfo-2-deoxy-α-D-glucopyranoside(15):
Procedure D:起始原料14(28 mg, 0.018 mmol); 产物15 (25 mg, 86%).
1H NMR (400 MHz, CD3OD) δ: 7.63~7.14 (m, 25H), 5.46 (d, 1H), 5.12~4.98 (m, 5H), 4.94~4.83 (m, 3H), 4.72~4.59 (m, 7H), 4.51~4.47 (m, 2H), 4.31~4.19 (m, 5H), 4.09~3.95 (m, 6H), 3.89~3.74 (m, 3H), 3.67~3.66 (m, 1H), 3.50~3.41 (m, 4H); ESI-MS (m/z): 545.1 [M+5H]3-.
Methyl(2-O-sulfo-β-D-glucopyranosyluronate)-(1→4)-(2-N-sulfo-6-O-sulfo-2-deoxy-α-D-glucopyrano-syl)-(1→4)-(2-O-sulfo-β-D-glucopyanosyluronate)-(1→4)-2-N-sulfo-6-O-sulfo-2-deoxy-α-D-glucopyranoside(3):
Procedure E:起始原料15(25 mg, 0.016 mmol); 产物3(19 mg, 99%).
1H NMR (400 MHz, D2O) δ: 5.51 (d, 1H, J=3.6 Hz), 5.03 (d, 1H, J=3.2 Hz), 4.79 (d, 1H), 4.59~4.54 (m, 2H), 4.27~4.09 (m, 4H), 4.04~3.95 (m, 3H), 3.85~3.69 (m, 9H), 3.59 (t, 1H, J=9.6 Hz), 3.42 (s, 3H), 3.31~3.28 (m, 2H); 13C NMR (100 MHz, D2O) δ: 100.06, 99.82, 98.58, 98.23, 79.78, 79.68, 78.79, 78.06, 77.58, 76.55, 75.66, 74.53, 74.46, 71.71, 69.43, 69.09, 68.04, 65.93, 65.83, 57.86, 57.19, 55.48, 55.44; ESI-MS (m/z): 394.8 [M+5H]3-.
Methyl(2-N-sulfo-6-O-sulfo-2-deoxy-α-D-gluco-pyranosyl)-(1→4)-(2-O-sulfo-β-D-glucopyanosyluro-nate)-(1→4)-2-N-sulfo-6-O-sulfo-2-deoxy-α-D-gluco-pyranoside(1):
Procedure E:起始原料5(67 mg, 0.05 mmol); 产物1(25 mg, 54%).
1H NMR (400 MHz, D2O) δ: 5.69 (d, 1H, J=2.4 Hz), 5.07 (d, 1H, J=2.8 Hz), 4.79 (d, 1H), 4.62 (d, 1H, J=10.0 Hz), 4.40 (d, 1H, J=10.4 Hz), 4.29 (d, 1H, J=11.2 Hz), 4.23~4.15 (m, 2H), 4.04~3.60 (m, 9H), 3.46 (s, 3H), 3.33~3.27 (m, 2H); 13C NMR (100 MHz, D2O) δ: 100.07, 98.31, 97.82, 79.93, 77.91, 76.47, 75.05, 71.30, 69.95, 69.48, 69.08, 68.10, 66.39, 65.97, 57.94, 57.26, 55.51; ESI-MS (m/z): 309.2 [M+3H]3-, 464.2 [M+4H]2-.
Methyl(2-N-sulfo-6-O-sulfo-2-deoxy-α-D-gluco-pyranosyl)-(1→4)-β-D-glucopyanosyluronate-(1→4)-2-N-sulfo-6-O-sulfo-2-deoxy-α-D-glucopyranoside(2):
Procedure B, C, D, E:起始原料6(75 mg, 0.056 mmol); 产物2(34 mg, 71%).
1H NMR (400 MHz, D2O) δ: 5.67 (d, 2H, J=3.6 Hz), 5.07 (d, 1H, J=3.6 Hz), 4.63 (d, 1H, J=8.0 Hz), 4.43~4.35 (m, 3H), 4.19 (d, 1H, J=10.8 Hz), 4.03~4.01 (m, 1H), 3.92~3.80 (m, 4H), 3.77~3.70 (m, 2H), 3.67~3.58 (m, 2H), 3.46 (s, 3H), 3.41 (t, 1H, J=8.4 Hz), 3.34~3.28 (m, 2H); 13C NMR (100 MHz, D2O) δ: 102.14, 98.24, 97.40, 78.53, 76.43, 76.28, 76.17, 72.99, 71.12, 69.79, 69.67, 69.04, 69.23, 66.52, 66.35, 57.93, 57.25, 55.52; ESI-MS (m/z): 282.3 [M+2H]3-; 424.2 [M+3H]2-.
Methyl(2-O-sulfo-β-D-glucopyranosyluronate)-(1→4)-(2-N-sulfo-6-O-sulfo-2-deoxy-α-D-glucopyrano-syl)-(1→4)-β-D-glucopyanosyluronate-(1→4)-2-N-sulfo-6-O-sulfo-2-deoxy-α-D-glucopyranoside(4):
Procedure B, C, D, E:起始原料8(30 mg, 0.017 mmol); 产物4(10 mg, 52%).
1H NMR (400 MHz, D2O) δ: 5.52 (d, 1H, J=3.6 Hz), 5.05 (d, 1H, J=3.2 Hz), 4.79 (d, 1H), 4.60 (d, 1H, J=8.0 Hz), 4.56 (d, 1H, J=10.8 Hz), 4.42 (d, 1H, J=10.8 Hz), 4.33 (dd, 1H, J=11.2 Hz, 3.6 Hz), 4.20 (d, 1H, J=10.8 Hz), 4.12 (t, 1H, J=8.8 Hz), 4.03~4.01 (m, 2H), 3.88~3.68 (m, 9H), 3.60 (t, 1H, J=9.6 Hz), 3.44 (s, 3H), 3.42~3.31 (m, 3H); 13C NMR (100 MHz, D2O) δ: 181.53, 175.60, 102.23, 100.81, 99.84, 98.21, 79.70, 78.81, 78.67, 77.58, 76.63, 75.74, 75.68, 74.48, 72.96, 71.74, 69.70, 69.36, 69.01, 68.25, 66.62, 65.87, 57.86, 57.24, 55.50; ESI-MS (m/z): 367.5 [M+4H]3-.
Molean, J. Am. J. Physiol. 1916, 41, 250. doi: 10.1152/ajplegacy.1916.41.2.250
Capila, R. J. Linhardt, D. Angew. Chem. Int. Ed. 2002, 41, 390. doi: 10.1002/1521-3773(20020201)41:3<390::AID-ANIE390>3.0.CO;2-B
(a) Petitou, M.; van Boeckel, C. A. A. Angew. Chem. Int. Ed. Engl. 1993, 32, 1671; (b) Petitou, M.; van Boeckel, C. A. A. Angew. Chem. Int. Ed. 2004, 43, 3118.
(a) Casu, B. Adv. Carbohydr. Chem. Biochem. 1985, 43, 51; (b) Gao, N. G.; Cheng, X. L.; Yang, J.; Zhang, S. Z. Prog. Biotechnol. 1999, 19(5), 4 (in Chinese). (高宁国, 程秀兰, 杨敬, 张树政, 生物工程进展, 1999, 19(5), 4.)
Poletti, L.; Lay, L. Eur. J. Org. Chem. 2003, 2999; (b) Karst, N. A.; Linhardt, R. J. Curr. Med. Chem. 2003, 10, 1993.
Vlodavsky, I.; Goldshmidt, O.; Zcharia, E.; Metzger, S.; Chajek-Shaul, T.; Atzmon, R.; Guatta-Rangini, Z.; Friedmann, Y. Biochimie 2001, 83, 831. doi: 10.1016/S0300-9084(01)01318-9
Elkin, M.; Ilan, N.; Ishai-Michaeli, R. FASEB 2001, 15, 1661. doi: 10.1096/fj.00-0895fje
Bartleet, M. R.; Underwood, P. A.; Parish, C. R. Immunol. Cell Biol. 1995, 73, 113. doi: 10.1038/icb.1995.19
Bingley, J. A.; Hayward, I. P.; Campbell, J. H. J. Vasc. Surg. 1998, 28, 308. doi: 10.1016/S0741-5214(98)70167-3
Okada, Y.; Yamada, S.; Toyoshima, M.; Dong, J.; Nakajima, M.; Sugahara, K. J. Biol. Chem. 2002, 277, 42488. doi: 10.1074/jbc.M206510200
For reviews, see: (a) Gin, D. J. Carbohydr. Chem. 2002, 21, 645; (b) O'Neill, S.; Rodriguez, J.; Walczak, M. A. Chem. Asian J. 2018, 13, 2978; (c) Ryan, D. A.; Gin, D. Y. Glycoside Synthesis from 1-Oxygen Substituted Glycosyl Donors. In Handbook of Chemical Glycosylation, Ed.: Demchenko, A. V., Wiley-Ver & Co. KGaA, Weinheim, 2008, pp. 95–143.
(a) Chen, J.; Zhou, Y.; Chen, C.; Xu, W.; Yu, B. Carbohydr. Res. 2008, 343, 2853; (b) Xu, P.; Xu, W.; Dai, Y.; Yang, Y.; Yu, B. Org. Chem. Front. 2014, 1, 405.
Jiang, L.; Chan, T. Tetrahedron Lett. 1998, 39, 355. doi: 10.1016/S0040-4039(97)10599-8
Epp, J. B.; Widlanski, T. S. J. Org. Chem., 1999, 64, 293. doi: 10.1021/jo981316g
(a) Yin, X.; Yan, J.; Ji, S.; Wang, F.; Cao, H. Chin. J. Org. Chem. 2012, 32, 1388; (b) Li, J.; Dai, Y.; Li, W.; Laval, S.; Xu, P.; Yu, B. Asian J. Org. Chem. 2015, 4, 756; (c) Mende, M.; Bednarek, C.; Wawryszyn, M.; Sauter, P.; Biskup, M. B.; Schepers, U.; Brase, S. Chem. Rev. 2016, 116, 8193
(a) Nishida, Y.; Shingu, Y.; Dohi, H.; Kobayashi, K. Org. Lett. 2003, 5, 2377; (b) Kim, K. S.; Fulse, D. B.; Baek, J. Y.; Lee, B.-Y.; Jeon, H. B. J. Am. Chem. Soc. 2008, 130, 8537; (c) Mossotti, M.; Panza, L. J. Org. Chem. 2011, 76, 9122; (d) Nogueira, J. M.; Nguyen, S. H.; Bennett, C. S. Org. Lett. 2011, 13, 2814; (e) Nogueira, J. M.; Bylsma, M.; Bright, D. K.; Bennett, C. S. Angew. Chem. Int. Ed. 2016, 55, 10088; (f) Zhou, M.-H.; Wilbur, D. J.; Kwan, E. E.; Bennett, C. S. J. Am. Chem. Soc. 2019, 141, 16743; (g) Dyapa, R.; Dockery, L. T.; Walczak, M. A. Org. Biomol. Chem. 2017, 15, 51; (h) Ghosh, T.; Mukherji, A.; Srivastava, H. K.; Kancharla, P. K. Org. Biomol. Chem. 2018, 16, 2870; (i) Manhas, S.; Taylor, M. S. Carbohydr. Res. 2018, 470, 42; (j) Cai, L.; Zeng, J.; Li, T.; Xiao, Y.; Ma, X.; Xiao, X.; Zhang, Q.; Meng, L.; Wan, Q. Chin. J. Chem. 2020, 38, 43.
(a) Garcia, B. A.; Poole, J. L.; Gin, D. Y. J. Am. Chem. Soc. 1997, 119, 7597; (b) Garcia, B. A.; Gin, D. Y. J. Am. Chem. Soc. 2000, 122, 4269.
(a) Codée, J. D. C.; van den Bos, L. J.; Litjens, R. E. J. N.; Overkleeft, H. S.; van Boom J. H.; van der Marel, G. A. Org. Lett. 2003, 5, 1947; (b) van den Bos, L. J.; Codée, J. D. C.; van Boom J. H.; Overkleeft, H. S.; van der Marel, G. A. Org. Biomol. Chem. 2003, 1, 4160; (c) van den Bos, L. J.; Codée, J. D. C.; van Boom, J. H.; van der Toorn, J. C.; Boltje, T. J.; van Boom J. H.; Overkleeft, H. S.; van der Marel, G. A. Org. Lett. 2004, 6, 2165; (d) Codée, J. D. C.; Stubba, B.; Schiattarella, M.; Overkleeft, H. S.; van Boeckel, C. A. A.; van Boom J. H.; van der Marel, G. A. J. Am. Chem. Soc. 2005, 127, 3767.
周映, 博士论文, 中国科学院上海有机化学研究所, 上海, 2005.Zhou, Y. Ph.D. Dissertation, Shanghai Institute of Organic Chemistry, CAS, Shanghai, 2005 (in Chinese).
(a) Arungundram, S.; Al-Mafraji, K.; Asong, J.; Leach III, F. E.; Amster, J., Venot, A.; Turnbull, J. E.; and Boons, G. J. Am. Chem. Soc. 2009, 131, 17394; (b) Hu, Y.; Lin, S.; Huang, C.; Zulueta, M. M. L.; Liu J.; Chang, W.; Hung, S.-C. Nat. Chem. 2011, 3, 557; (c) Zulueta, M.; Lin, S.; Lin, Y.; Huang, C.; Wang, C.; Ku, C.; Shi, Z.; Wong, C.-H.; Hung, S.-C. J. Am. Chem. Soc. 2012, 134, 8988; (d) Wang, Z.; Xu, Y.; Yang B., Tiruchinapally, G.; Sun, B.; Liu, R.; Dulaney, S.; Liu, J.; Huang, X. Chem. Eur. J. 2010, 16, 8365; (e) Haller, M.; Boons, G-J. J. Chem. Soc., Perkin Trans.1. 2001, 814; (f) Lubineau, A.; Lortat-Jacob, H.; Gavard, O.; Sarrazin, S.; Bonnaffé, D. Chem. Eur. J. 2004, 10, 4265; (g) Lin, F.; Lian, G.; Zhou, Y. Carbohydr. Res. 2013, 371, 32; (h) Li, T.; Ye, H.; Cao, X.; Wang, J.; Liu, Y.; Zhou, L.; Liu, Q.; Wang, W.; Shen, J. Zhao, W.; Wang, P. ChemMedChem 2014, 9, 1071; (i) Xu, P.; Laval, S.; Guo, Z.; Yu, B. Org. Chem. Front. 2016, 3, 103; (j) Dai, X.; Liu, W.; Zhou, Q.; Cheng, C.; Yang, C.; Wang, S.; Zhang, M.; Tang, P.; Song, H.; Zhang, D.; Qin, Y. J. Org. Chem. 2016, 81, 162; (k) Ding, Y.; Vara Prasad C. V. N. S.; Bai, H.; Wang, B. Bioorg. Med. Chem. Lett. 2017, 27, 2424; (l) Jin, H.; Chen, Q.; Zhang, Y.; Hao, K. Zhang, G.; Zhao, W. Org. Chem. Front. 2019, 6, 3116.
(a) Orgueira, H. A.; Bartolozzi, A.; Schell, P.; Litjens, R. E. J. N.; Palmacci, E. R.; Seeberger, P. H. Chem. Eur. J. 2003, 9, 140; (b) Noti, C.; de Paz, J. L.; Polito, L.; Seeberger, P. H. Chem. Eur. J. 2006, 12, 8664; (c) Zhang, L.; Xu, P.; Liu, B.; Yu, B. J. Org. Chem. 2020, DOI: 10.1021/acs.joc.0c01009.
(a) Mungall, W. S.; Greene, G. L.; Heavner, G. A.; Letsinger, R. L. J. Org. Chem. 1975, 40, 1659; (b) Alper, P. B.; Hendrix, M.; Sears, P.; Wong, C. H. J. Am. Chem. Soc. 1998, 120, 1965.
Bayley, H.; Standring, D. N.; Knowles, J. R. Tetrahedron Lett. 1978, 19, 3633.
Corey, E. J.; Nicolaou, K. C.; Balanson, R. D.; Machida, Y. Synthesis 1975, 590.
Lee, J.-C.; Lu, X.-A.; Kulkarni, S. S.; Wen, Y.-S.; Hung, S.-C. J. Am. Chem. Soc. 2003, 126, 476.
(a) Benati, L.; Montevecchi, P. C.; Nanni, D.; Spagnolo, P.; Volta, M. Tetrahedron Lett. 1995, 36, 7313; (b) Goulaouic-Dubois, C.; Hesse, M. Tetrahedron Lett. 1995, 36, 7427.
Brewer, M.; Rich, D. H. Org. Lett. 2001, 3, 945. doi: 10.1021/ol015612i
Gregory, J.; Lohman, S.; Seeberger, P. H. J. Org. Chem. 2004, 69, 4081. doi: 10.1021/jo035732z
Yang, B.; Yoshida, K.; Yin, Z.; Dai, H.; Kavunja, H.; El-Dakdouki, M. H.; Sungsuwan, S.; Dulaney, S. B.; Huang, X. Angew. Chem., Int. Ed. 2012, 51, 10185. doi: 10.1002/anie.201205601
(a) Lee, J.-C.; Lu, X.-A.; Kulkarni, S. S.; Wen, Y.-S.; Hung, S.-C. J. Am. Chem. Soc. 2003, 126, 476; (b) Lee, J. C. Ph.D. Dissertation, Tsing Hua University, Taiwan, 2005 (in Chinese). (李静琪, 博士论文, 台湾清华大学, 台湾, 2003.)
Dilhas, A.; Lucas, R.; Loureiro-Morais, L.; Hersant, Y.; Bonnaffé, D. J. Comb. Chem. 2008, 10, 166. doi: 10.1021/cc8000019

图 1 肝素酶底物四糖A的结构
Figure 1 Structure of tetrasaccharide A relevant to the substrate of heparinase

图 2 三糖和四糖1~4的水解和肝素酶活性定量分析
Figure 2 Hydrolysis of tri- and tetrasaccharides 1~4 and quantitative analysis of the activity of heparinase.

图 3 肝素酶底物三糖和四糖1~4的逆合成分析
Figure 3 Retrosynthetic analysis of tri- and tetrasaccharides 1~4

图式 2 脱水糖苷化合成三糖5和6和四糖7
Scheme 2 Dehydrative glycosylation for the synthesis of tri- and tetrasaccharides 5~7
表 1 二糖10和12脱水糖苷化反应的条件优化
Table 1. Optimization of dehydrative glycosylation of disaccharides 10 and 12
|
|||
| Entry | Solvent | Yield | α/β |
| 1a | CH2Cl2 | 32% | α only |
| 2b | CH2Cl2 | 22% | α only |
| 3a | Toluene | 65% | 8.3/1.0 |
| a室温条件下, 将二糖10, 二苯亚砜(Ph2SO), 2, 4, 6-三叔丁基嘧啶(TTBP)溶于溶剂中, 加入4 Å分子筛, 搅拌30 min; -60 ℃下加入三氟甲磺酸酐(Tf2O), 升至-40 ℃反应1 h; 加入二糖12, 自然升至室温反应(约3 h). b室温条件下, 将10, 二苯亚砜, 2, 4, 6-三叔丁基嘧啶溶于溶剂中, 加入4 Å分子筛, 搅拌30 min; -60 ℃条件下加入三氟甲磺酸酐, 升至-40 ℃反应15 min; 加入12, 自然升至室温反应(约3 h). | |||
 下载: 导出CSV
下载: 导出CSV
表 2 单糖B/C的电喷雾质谱测试结果
Table 2. ESI Test for monosaccharide B/C
| Sample No. | Molar ratio(B/C) | Observed m/z for [M-Na]-(B)a | Intensity(B) | Observed m/z for [M-Na]-(C) | Intensity(C)b | Intensity ratio(B/C) |
| 1 | NAc | NA | NA | NA | NA | NA |
| 2 | 9:1 | 374.13 | 49411 | 377.07 | 6486 | 7.6181 |
| 3 | 8:2 | 374.27 | 77031 | 377.20 | 22613 | 3.4065 |
| 4 | 7:3 | 374.27 | 65635 | 377.20 | 35878 | 1.8294 |
| 5 | 6:4 | 374.27 | 49172 | 377.20 | 42720 | 1.1510 |
| 6 | 5:5 | 374.20 | 31805 | 377.13 | 42525 | 0.7479 |
| 7 | 4:6 | 374.27 | 27158 | 377.13 | 54446 | 0.4988 |
| 8 | 3:7 | 374.33 | 22614 | 377.20 | 84840 | 0.2665 |
| a Calculated value m/z for [M-Na]- is 374.30; b Calculated value m/z for [M-Na]- is 377.32; c Not available. | ||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们