图式 1.
羟胺的Stieglitz重排反应
Scheme 1.
The Stieglitz rearrangement of hydroxylamines

羟胺类化合物具有丰富的生物学性能[1], 并且作为一种实用的合成子被广泛地应用于有机合成当中[2].近年来, 基于羟胺的转化得到了迅速的发展和应用[3], 但其中一个有趣的Stieglitz重排反应却没有得到很好的发展(图式 1).由于羟基较弱的离去能力, 目前已报道的Stieglitz重排反应均需要在比较剧烈的条件下进行, 且需要PCl5或P2O5作为试剂参与到反应当中.此外, 该反应的底物范围也非常窄, 基本上仅局限于三芳基甲基羟胺类底物[4], 限制了其进一步的应用.我们小组一直致力于发展C—C键断裂以及经过氮自由基的氮化反应[5], 对这一领域的研究兴趣驱使我们解决上述Stieglitz重排反应中面临的问题, 并发展一种能在温和条件下进行、且具有较好底物适用范围的Stieglitz重排反应, 这对于扩展羟胺类化合物的应用范围也具有重要的意义.
受O-取代羟胺于近年来被广泛用作氮自由基前体[6]及胺化试剂[7]的启发, 我们设想通过在羟基原位生成易离去基团的方式, 使羟胺的N—O键键能被弱化, 进而使得Stieglitz重排反应能够在更简单温和的条件下发生.然而, 该假设的实现需要解决以下三个问题.首先, 羟胺分子中氮原子和氧原子都具有亲核能力, 且在O-未取代羟胺类化合物参与的亲核反应当中, 一般氮原子表现出更强的亲核性, 故如何使活化试剂选择性地与羟基反应至关重要.其次, 必须生成合适的离去基团以削弱N—O键键能, 使得反应拥有更广的底物适用范围.最后, 所使用的活化试剂不能与形成的亚胺中间体以及随后生成的芳胺发生进一步的反应, 以保证目标产物芳胺的获得.
在此, 我们报道一例在温和条件下通过三氟乙酸酐(TFAA)促进的Stieglitz重排反应将羟胺转化为相应芳基伯胺的工作.该反应使用简单的TFAA作为活化试剂解决了上述传统Stieglitz重排反应存在的问题, 为羟胺的进一步转化和应用提供了实用方案.
根据设想, 我们以对苯基苯乙基羟胺(1a)作为模板底物, 利用Lewis酸如AlCl3和Cu(OTf)2作活化试剂对该反应进行了初步尝试(表 1, Entries 1~2).很遗憾Lewis酸并没有起到活化的效果.同样地, 使用Brönsted酸作为活化试剂的尝试也以失败告终(表 1, Entries 3~4).随后, 我们尝试加入三氟甲磺酸酐(Tf2O)或三氟甲磺酰氯(TfCl)作为活化试剂, 并十分幸运地在二氯甲烷(DCM)做溶剂的条件下观测到少量苯胺产物的生成(表 1, Entries 5~6).受此鼓舞, 我们随后尝试了其他各种酰氯或者酸酐作为活化试剂, 并最终发现在使用TFAA作为活化试剂时能够得到较好的产率(表 1, Entry 7).后续的溶剂筛选实验发现, 六氟异丙醇(HFIP)的高介电常数和低亲核性[8]有助于稳定亚胺中间体, 是最合适该反应的溶剂(表 1, Entries 8~11).对添加剂进行筛选的过程中, 我们受一些以羟胺衍生物作为氮源实现硼酸胺化反应的工作启发[9], 尝试加入当量的有机硼化合物使之与羟胺的氮原子形成配位键, 以此来提高TFAA对于羟基的活化效率.令人惊喜的是, 在使用BF3•Et2O作为添加剂时, 反应收率显著提升至83%(表 1, Entries 13~14).
 下载:
导出CSV
下载:
导出CSV
 |
|||
| Entry | Activator | Solvent | Yieldb/% |
| 1 | AICl3 | DCM | 0 |
| 2 | Cu(OTf)2 | DCM | 0 |
| 3 | H2SO4 | DCM | 0 |
| 4 | HCI | DCM | 0 |
| 5 | Tf2O | DCM | trace |
| 6 | TfCI | DCM | 11 |
| 7 | TFAA | DCM | 34 |
| 8 | TFAA | DCE | 30 |
| 9 | TFAA | Toluene | 10 |
| 10 | TFAA | HFIP | 54 |
| 11 | TFAA | i-PrOH | 0 |
| 12c | TFAA | HFIP | 42 |
| 13d | TFAA | HFIP | 70 |
| 14e | TFAA | HFIP | 83 |
| a Reaction conditions: 1a (0.2 mmol), activator (0.22 mmol), solvent (2 mL), stirred for 1 h at room temperature, then quenched by 4 mL 2 mol/L NaOH (aq.). b Yeild was determined by 1H NMR using 1, 1, 2, 2-Tetrachlorothane as an internal standard. c Reaction time extended to 12 h. d BF3•2H2O (0.2 mmol) was added. e BF3•Et2O (0.2 mmol) was added. | |||
基于上述实验结果, 我们确定了最优反应条件为:底物物质的量的1.1倍TFAA为活化剂, 底物物质的量的1.0倍BF3•Et2O为添加剂, HFIP为溶剂, 最佳反应温度为室温.
在最优反应条件下(表 1, Entry 14), 我们对反应底物的适用范围进行了考察(表 2).我们发现邻、间、对位取代的苯乙基羟胺(1)均适用于此反应体系, 以中等到较高的产率生成对应的芳基伯胺(2).其中, 支链或长链烷基取代的底物(1c, 1d)以及具有给电子基团如苯基、甲氧基、甲硫基和三氟甲氧基的底物(1a, 1l~1j)均能够以优良的产率得到对应的芳基伯胺.除此之外, 该反应对于卤素取代的苯乙基羟胺也有很好的的兼容性(1f~1i).双取代底物以及邻位和间位取代的底物也能够以中等收率得到目标产物(1m~1t).另外一个有趣的现象是使用全氟丙酸酐(PFPA)替代TFAA时可以提高2-溴苯乙基羟胺(1s)约10%的收率, 但比较遗憾的是PFPA对于其他底物的产率并没有提升作用.
此外, 我们还探索了N-(1-苯基烷基)羟胺类化合物的适用范围(表 3).当R2基团为支链烷基, 长链烷基, 环烷基以及氢原子时均可以完成由羟胺到苯胺的转化(3a~3f).但遗憾的是由于这些底物在被TFAA活化后无法通过重排完全转化为亚胺, 故N-(1-苯基烷基)羟胺类化合物的产率要低于取代的N-(1-苯乙基)羟胺.
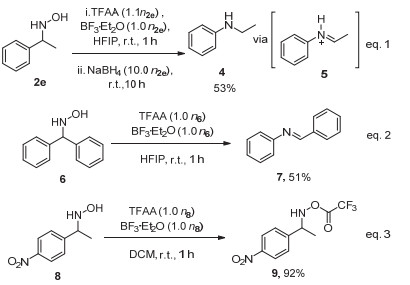
随后我们对反应历程进行了进一步探究.首先, 我们使用2e作为底物在标准条件下进行反应, 并在反应完成后向其中添加NaBH4作为氢化试剂, 随后继续反应10 h.反应结束后我们以53%的分离收率得到N-乙基苯胺4并且检测不到苯胺的生成(图式 2, eq.1), 说明该反应可能经历了亚胺中间体的过程, 并且最终在后处理过程中水解生成苯胺.此外, 当二苯甲基羟胺6在标准条件下进行反应并直接分离时, 由于产物较稳定且不易水解, 因此可以分离得到(E)-N, 1-二苯基甲亚胺7, 进一步证明亚胺中间体的存在(图式 2, eq.2).
以N-(1-(4-硝基苯基)乙基)羟胺8作为底物时, 由于吸电子的硝基会影响芳基迁移, 使得我们可以检测到O-(三氟乙酰基)苄基羟胺9的存在.提示我们活性的三氟乙酸酯可能是该反应初始中间体(图式 2, eq.3).
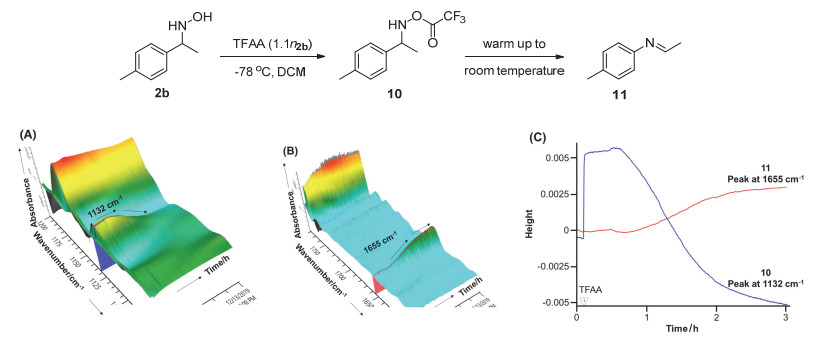
为了进一步探究中间体的形成过程, 我们还对反应的过程进行了在线红外监测(图 1).从3D-FTIR谱图中可以看出, 当将TFAA加入2b的DCM溶液中时, 会立即产生初始中间体10, 其在1132 cm-1处存在C—N键的特征吸收峰(图 1, A).随后在逐渐升温的过程中, 中间体发生Stieglitz重排反应并产生相应的亚胺中间体11, 其在1655 cm-1处有特征吸收峰(图 1, B).两个中间体的反应动力学行为也清晰地表明了这一趋势(图 1, C).进一步证明了底物先被TFAA活化并立即形成初始中间体O-(三氟乙酰基)苯乙基羟胺, 随后再转化为亚胺中间体的反应历程.
本工作成功实现了TFAA促进的Stieglitz重排反应, 能在相对温和的反应条件下, 完成由各种苯乙基羟胺到相应芳基伯胺的转化, 拓展了Stieglitz重排反应的底物适用范围.机理研究实验还证明O-(三氟乙酰基)苯乙基羟胺是该反应的活性中间体.目前以羟胺为直接氮源, 以简单的烷基芳烃为底物在一步反应中构建苯胺的方法仍正在进一步探究中.
(a) Piechnick, R.; Heck, M.; Sommer, M. E. Biochemistry 2011, 50, 7168; (b) Chen, Y.; Wang, X.; Xiang, W.; He, L.; Tang, M.; Wang, F.; Wang, T.; Yang, Z.; Yi, Y.; Wang, H.; Niu, T.; Zheng, L.; Lei, L.; Li, X.; Song, H.; Chen, L. J. Med. Chem. 2016, 59, 5488.
(a) Rappoport, Z.; Liebman, J. F., The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids, Vol. 1, John Wiley & Sons, 2008; (b) Zhang, Y.; Wang, M.; Cao, P.; Liao, J. Acta Chim. Sinica 2017, 75, 794 (in Chinese). (张涌灵, 王敏, 曹鹏, 廖建, 化学学报, 2017, 75, 794); (c) He, Y.; Teng, J.; Tian, C.; Borzov, M.; Hu, Q.; Nie, W. Acta Chim. Sinica 2018, 76, 774 (in Chinese). (何云清, 滕金伟, 田冲, Borzov Maxim, 胡启山, 聂万丽, 化学学报, 2018, 76, 774).
For some examples in recent years: (a) Nguyen, T. B.; Martel, A.; Dhal, R.; Dujardin, G. Org. Lett. 2008, 10, 4493; (b) Guimond, N.; MacDonald, M. J.; Lemieux, V.; Beauchemin, A. M. J. Am. Chem. Soc. 2012, 134, 16571; (c) Pusterla, I.; Bode, J. W. Angew. Chem. Int. Ed. 2012, 51, 513; (d) Li, J.; He, Y.; Ren, X.; Shi, X.; Yang, S.; Gao, X.; Huang, G. Chin. J. Chem. 2013, 31, 1003; (e) Hesp, C. R.; MacDonald, M. J.; Zahedi, M. M.; Bilodeau, D. A.; Zhao, S. B.; Pesant, M.; Beauchemin, A. M. Org. Lett. 2015, 17, 5136; (f) Sun, H. B.; Gong, L.; Tian, Y. B.; Wu, J. G.; Zhang, X.; Liu, J.; Fu, Z.; Niu, D. Angew. Chem. Int. Ed. 2018, 57, 9456.
(a) Stieglitz, J.; Leech, P. N. Ber. 1913, 46, 2147; (b) Stieglitz, J.; Leech, P. N. J. Am. Chem. Soc. 1914, 36, 272. (c) Morgan, A. F. J. Am. Chem. Soc. 1916, 38, 2095; (d) Newman, M. S.; Hay, P. M. J. Am. Chem. Soc. 1953, 75, 2322; (e) Stolyarov, B. V.; Krylov, A. I.; Ioffe, B. V. Zh. Org. Khim. 1977, 13, 2004.
For some selected examples in recent years: (a) Qin, C.; Zhou, W.; Chen, F.; Ou, Y.; Jiao, N. Angew. Chem. Int. Ed. 2011, 50, 12595; (b) Qin, C.; Shen, T.; Tang, C.; Jiao, N. Angew. Chem. Int. Ed. 2012, 51, 6971; (c) Qin, C.; Feng, P.; Ou, Y.; Shen, T.; Wang, T.; Jiao, N. Angew. Chem. Int. Ed. 2013, 52, 7850; (d) Liang, Y.; Liang, Y.-F.; Jiao, N. Org. Chem. Front. 2015, 2, 403; (e) Song, S.; Feng, P.; Zou, M.; Jiao, N. Chin. J. Chem. 2017, 35, 845; (f) Shen, T.; Zhu, B.; Lin, F.; Pan, J.; Wei, J.; Luo, X.; Liu, J.; Jiao, N. Chin. J. Chem. 2018, 36, 815; (g) Liu, J.; Qiu, X.; Huang, X.; Luo, X.; Zhang, C.; Wei, J.; Pan, J.; Liang, Y.; Zhu, Y.; Qin, Q.; Song, S.; Jiao, N. Nat. Chem. 2019, 11, 71; (h) Lin, F.; Liang, Y.; Li, X.; Song, S.; Jiao, N. Acta Chim. Sinica 2019, 77, 906 (in Chinese). (林凤闺蓉, 梁宇杰, 郦鑫耀, 宋颂, 焦宁, 化学学报, 2019, 77, 906). For some reviews, see: (i) Sivaguru, P.; Wang, Z.; Zanoni, G.; Bi, X. Chem. Soc. Rev. 2019, 48, 2615; (j)Yu, X. Y.; Chen, J. R.; Xiao, W. J. Chem. Rev. 2020, DOI: 10.1021/acs.chemrev.0c00030.
For some reviews, see: (a) Davies, J.; Morcillo, S. P.; Douglas, J. J.; Leonori, D. Chem. - Eur. J. 2018, 24, 12154; (b) Xiao, L.; Li, J.; Wang, T. Acta Chim. Sinica 2019, 77, 841 (in Chinese). (肖丽, 李嘉恒, 王挺, 化学学报. 2019, 77, 841). For recent examples, see: (c) Allen, L. J.; Cabrera, P. J.; Lee, M.; Sanford, M. S. J. Am. Chem. Soc. 2014, 136, 5607; (d) Qin, Q.; Yu, S. Org. Lett. 2014, 16, 3504; (e) Chen, J. R.; Hu, X. Q.; Lu, L. Q.; Xiao, W. J. Chem. Soc. Rev. 2016, 45, 2044; (f) Xiong, T.; Zhang, Q. Chem. Soc. Rev. 2016, 45, 3069; (g) Svejstrup, T. D.; Ruffoni, A.; Julia, F.; Aubert, V. M.; Leonori, D. Angew. Chem. Int. Ed. 2017, 56, 14948; (h) Wu, K.; Du, Y.; Wang, T. Org. Lett. 2017, 19, 5669; (i) Yu, X.; Zhou, F.; Chen, J.; Xiao, W. Acta Chim. Sinica 2017, 75, 86 (in Chinese). (余晓叶, 周帆, 陈加荣, 肖文精, 化学学报, 2017, 75, 86); (j) An, X.-D.; Zhang, H.; Xu, Q.; Yu, L.; Yu, S. Chin. J. Chem. 2018, 36, 1147; (k) Jin, J.; Zhang, F.; Wang, Y. Acta Chim. Sinica 2019, 77, 889 (in Chinese). (靳继康, 张凤莲, 汪义丰, 化学学报. 2019, 77, 889).
For some reviews, see: (a) Sabir, S.; Kumar, G.; Jat, J. L. Org. Biomol. Chem. 2018, 16, 3314; (b) Xu, L.; Xu, H.; Lin, H.; Dai, H. Chin. J. Org. Chem. 2018, 38, 1940 (in Chinese). (徐琳琳, 徐辉, 林海霞, 戴辉雄, 有机化学, 2018, 38, 1940). For recent examples, see: (c) Legnani, L.; Prina Cerai, G.; Morandi, B. ACS Catal. 2016, 6, 8162; (d) Paudyal, M. P.; Adebesin, A. M.; Burt, S. R.; Ess, D. H.; Ma, Z.; Kürti, L.; Falck, J. R. Science 2016, 353, 1144; (e) Zou, M.; Liu, J.; Tang, C.; Jiao, N. Org. Lett. 2016, 18, 3030; (f) Liu, J.; Wu, K.; Shen, T.; Liang, Y.; Zou, M.; Zhu, Y.; Li, X.; Li, X.; Jiao, N. Chem. - Eur. J. 2017, 23, 563; (g) D'Amato, E. M.; Borgel, J.; Ritter, T. Chem. Sci. 2019, 10, 2424.
Colomer, I.; Chamberlain, A. E. R.; Haughey, M. B.; Donohoe, T. J. Nat. Rev. Chem. 2017, 1, 0088. doi: 10.1038/s41570-017-0088
(a) Mlynarski, S. N.; Karns, A. S.; Morken, J. P. J. Am. Chem. Soc. 2012, 134, 16449; (b) Xiao, Q.; Tian, L.; Tan, R.; Xia, Y.; Qiu, D.; Zhang, Y.; Wang, J. Org. Lett. 2012, 14, 4230; (c) Zhu, C.; Li, G.; Ess, D. H.; Falck, J. R.; Kurti, L. J. Am. Chem. Soc. 2012, 134, 18253; (d) Voth, S.; Hollett, J. W.; McCubbin, J. A. J. Org. Chem. 2015, 80, 2545.

图 1 (A) 1100~1200 cm-1处3D FTIR谱图(B) 1600~1750 cm-1处3D FTIR谱图(C) 1655 cm-1和1132 cm-1处吸光度随时间变化曲线
Figure 1 (A) the 3D FTIR profile (1100~1200 cm-1) of the reaction of 2b (0.2 mmol) and TFAA (B) the 3D FTIR profile (1600~1750 cm-1) of the reaction of 2b (0.2 mmol) and TFAA (C) Kinetic profile (absorbance height vs time) of the peak at 1655 cm-1 and 1132 cm-1.
表 1 反应条件优化a
Table 1. Optimization of reaction condition a
|
|||
| Entry | Activator | Solvent | Yieldb/% |
| 1 | AICl3 | DCM | 0 |
| 2 | Cu(OTf)2 | DCM | 0 |
| 3 | H2SO4 | DCM | 0 |
| 4 | HCI | DCM | 0 |
| 5 | Tf2O | DCM | trace |
| 6 | TfCI | DCM | 11 |
| 7 | TFAA | DCM | 34 |
| 8 | TFAA | DCE | 30 |
| 9 | TFAA | Toluene | 10 |
| 10 | TFAA | HFIP | 54 |
| 11 | TFAA | i-PrOH | 0 |
| 12c | TFAA | HFIP | 42 |
| 13d | TFAA | HFIP | 70 |
| 14e | TFAA | HFIP | 83 |
| a Reaction conditions: 1a (0.2 mmol), activator (0.22 mmol), solvent (2 mL), stirred for 1 h at room temperature, then quenched by 4 mL 2 mol/L NaOH (aq.). b Yeild was determined by 1H NMR using 1, 1, 2, 2-Tetrachlorothane as an internal standard. c Reaction time extended to 12 h. d BF3•2H2O (0.2 mmol) was added. e BF3•Et2O (0.2 mmol) was added. | |||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们