Received Date:
26 May 2020 Available Online:
15 August 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21971134, 21631007, 21225103)
Abstract:
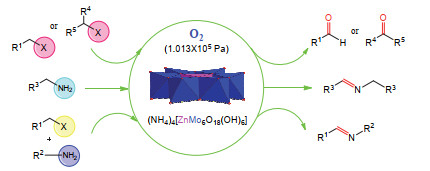
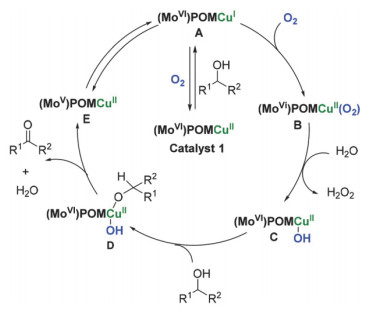
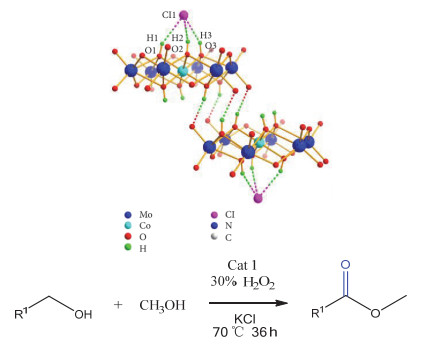
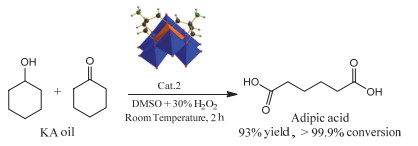
Anderson type heteropoly acids, also known as Anderson type polyoxometalates, are a kind of important structures in polyoxometalates. Their general structural formula can be expressed as [XM6O24]n-, in which the core heteroatom X can almost be replaced by almost any metal or nonmetal element in the periodic table. Due to unique structure easy to be modified with organic ligands and designability, as well as their potential applications in materials, catalysis and medicines, Anderson type heteropoly acids have been widely concerned by researchers. In recent years, the application of Anderson type heteropoly acids in organic synthesis has gradually shown great significance for the study of green catalytic process. In this paper, the catalytic application of Anderson type heteropoly acids in organic synthesis has been reviewed and summarized according to the structure classification of Anderson type polyoxometalates. This will be helpful for the researchers to further study the catalytic application of Anderson heteropoly acids and provides new ideas for the research of green catalysis.
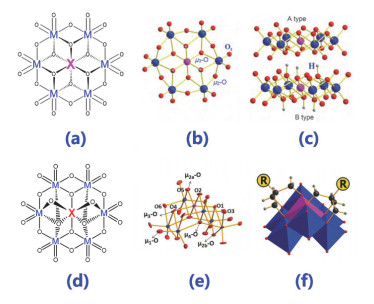
Figure 1.
(a) Structure diagram of α-Anderson type heteropoly acids; (b, c) model diagram of α-Anderson type heteropoly acids Club; (d) structure diagram of β-Anderson type heteropoly acids; (e) model diagram of β-Anderson type heteropoly acids Club; (f) organically modified β-Anderson type heteropoly acids
Wei, Y. G. New progress of Polyacid Imine Derivatives, Abstracts of the 6th annual academic meeting and member congress of China Crystal Society (functional molecular crystal branch), China Crystal Society, 2016, p. 28.
Li, J. J.; Wu, F. Textbook of introduction to green chemistry, Wuhan University Press, WuHan, 2015, p. 8.
[20]
He, Y. M.; Sun, Y. H.; Han, B. X. Chin. Sci. Bull.2015, 16, 1421.
[21]
Song, J. L.; Han, B. X. Natl. Sci. Rev.2015, 3, 255.
[22]
SD, K.; Gokavi, G. S. Res. J. Chem. 2016, 6, 17.
[23]
Yu, H.; Zhai, Y. Y.; Dai, G. Y.; Ru, S.; Han, S.; Wei, Y. G. Chem.-Eur. J. 2017, 23, 13883. doi: 10.1002/chem.201703185
[24]
Yu, H.; Ru, S.; Zhai, Y. Y.; Dai, G. Y.; Han, S.; Wei, Y. G. ChemCatChem2018, 10, 1253. doi: 10.1002/cctc.201701599
[25]
Zhai, Y. Y.; Zhang, M. Q.; Fang, H. B.; Ru, S.; Yu, H.; Zhao, W. S.; Wei, Y. G. Org. Chem. Front. 2018, 5, 3454. doi: 10.1039/C8QO00833G
[26]
Zhang, M. Q.; Zhai, Y. Y.; Ru, S.; Zang, D. J.; Han, S.; Yu, H.; Wei, Y. G. Chem. Commun. 2018, 54, 10164. doi: 10.1039/C8CC03722A
[27]
Wang, J. J.; Zhai, Y. Y.; Wang, Y.; Yu, H.; Zhao, W. S.; Wei, Y. G. Dalton Trans. 2018, 47, 13323. doi: 10.1039/C8DT03003K
[28]
Sawant, J. D.; Patil, K. K.; Gokavi, G. S. Transition Met. Chem. 2019, 44, 153. doi: 10.1007/s11243-018-0279-4
[29]
Wei, Z. Y.; Ru, S.; Zhao, Q. X.; Yu, H.; Zhang, G.; Wei, Y. G. Green Chem. 2019, 21, 4069. doi: 10.1039/C9GC01248F
[30]
Zhou, Z. H.; Dai, G. Y.; Ru, S.; Yu, H.; Wei, Y. G. Dalton Trans. 2019, 48, 14201. doi: 10.1039/C9DT02997D
[31]
Yu, H.; Wang, J. J.; Wu, Z. K.; Zhao, Q. X.; Dan, D. M.; Han, S.; Tang, J. J.; Wei, Y. G. Green Chem. 2019, 21, 4550. doi: 10.1039/C9GC02053E
[32]
Yu, H.; Wu, Z. K.; Wei, Z. Y.; Zhai, Y. Y.; Ru, S.; Zhao, Q. X.; Wang, J. J.; Han, S.; Wei, Y. G. Commun. Chem. 2019, 2, 1. doi: 10.1038/s42004-018-0104-1
[33]
Wu, Z. K.; Zhai, Y. Y.; Zhao, W. S.; Wei, Z. Y.; Yu, H.; Han, S.; Wei, Y. G. Green Chem. 2020, 22, 737. doi: 10.1039/C9GC03564H
[34]
Xu, J. J.; Zhu, Z. G.; Yuan, Z. L.; Su, T.; Zhao, Y. C.; Ren, W. Z.; Zhang, Z. H.; Lu, H. Y. J. Taiwan Inst. Chem. E. 2019, 104, 8. doi: 10.1016/j.jtice.2019.08.006
[35]
Wang, J. J.; Yu, H.; Wei, Z. Y.; Qi, L.; Xuan, W. M.; Wei, Y. G. Research,2020, 1, 3875920.
[36]
Luo, J. H.; Huang, Y. C.; Ding, B.; Wang, P. M.; Geng, X. F.; Zhang, J. W.; Wei, Y. G. Catalysts2018, 8, 121. doi: 10.3390/catal8030121
[37]
Yu, H.; Ru, S.; Dai, G. Y.; Zhai, Y. Y.; Lin, H. L.; Han, S.; Wei, Y. G. Angew. Chem. Int. Ed. 2017, 56, 3867. doi: 10.1002/anie.201612225
[38]
She, S.; Mu, L.; Li, Q.; Huang, Z. H.; Wei, Y. G.; Yin, P. C. ChemPlusChem2019, 11, 84.
Figure 1
(a) Structure diagram of α-Anderson type heteropoly acids; (b, c) model diagram of α-Anderson type heteropoly acids Club; (d) structure diagram of β-Anderson type heteropoly acids; (e) model diagram of β-Anderson type heteropoly acids Club; (f) organically modified β-Anderson type heteropoly acids

 下载:
下载:




 下载:
下载:












