引用本文:
封博谞, 庄小东. 碳基介熵材料:理论与实验[J]. 化学学报,
2020, 78(9): 833-847.
doi:
10.6023/A20050167

Citation: Feng Boxu, Zhuang Xiaodong. Carbon-Enriched meso-Entropy Materials: from Theory to Cases[J]. Acta Chimica Sinica, 2020, 78(9): 833-847. doi: 10.6023/A20050167
Citation: Feng Boxu, Zhuang Xiaodong. Carbon-Enriched meso-Entropy Materials: from Theory to Cases[J]. Acta Chimica Sinica, 2020, 78(9): 833-847. doi: 10.6023/A20050167
碳基介熵材料:理论与实验
摘要:
富碳型材料,包括纯碳材料的各种同素异形体、碳基骨架的稠环芳香分子、聚合物、框架材料等,已成为当今材料领域最重要的研究领域之一.在这些研究当中,很大一部分工作都是研究材料本身的结构与性质,而忽略了这些材料之间的内在联系.课本中的很多概念,如同分异构体、同素异形体和拓扑缺陷,已经无法用于深入理解种类和数量繁多的富碳型材料之间的构效关系.这就使得通过改变已知材料的有限结构来调控材料的性质变得工作重复而繁重,且基础理解受限于研究个体上.作者将从材料"熵"的概念入手,尝试理解富碳型材料之间熵的相对高低,并建议基于"介熵"的认识开发新型富碳型材料、开发新型介熵富碳型材料的全新性质.基于对具体的不同的新型富碳型材料的讨论,将"介熵"这一概念引入到同素异形体、同分异构体以及广泛存在于碳材料中的拓扑缺陷的理解上.类似富碳型材料的关系不再模糊地停留在几何结构层面上,为今后介熵富碳型材料及其他介熵材料的开发提供参考.
English
Carbon-Enriched meso-Entropy Materials: from Theory to Cases
Abstract:
Carbon-enriched materials, including carbon allotropes, polycyclic aromatic hydrocarbons, polymers, frameworks, etc., are rising as stars in functional materials field. Large amount of reported work focused on development of new structures with typical features for novel applications, and has long ignored the intrinsic relationship between similar structures. The superficial relationships of those carbon-enriched materials in textbook, e.g., isomers, allotropes and topological defects, are no longer enough for fundamental understanding the structure-property relationship study due to more and more carbon-enriched materials have been developed. Such disadvantage has long hindered development of new materials based on well-established material systems. In this work, meso-entropy concept is proposed for understanding and development of different kinds of carbon-enriched materials by comparing their relative entropy values. Based on theoretical models and case-to-case discussion, meso-entropy concept has been found compatible with the concept of isomers, allotropes and topological defects in carbon-enriched materials. From now on, hopefully, the meso-entropy defined relationship for carbon-enriched materials will be no longer staying at the geometric level, and provide new thinking development of new carbon-enriched materials and other meso-entropy materials.
-
Key words:
- meso-entropy
- / carbon
- / polymer
- / structure-property relationship
- / preparation
-
1. 富碳型材料和熵
自 1789 年 Lavoisier 发现碳元素后, 人们对于富碳型材料(Carbon-enriched materials)的研究就从未停止过, 所涉及的诺贝尔奖已包括液晶[1]与聚合物(1991)[2]、富勒烯(1996)[3]、导电聚合物(2000)[4]、石墨烯(2010)[5]等诸多方向. 尽管如此, 富碳型材料至今仍在被陆续开发当中. 为避免概念混淆, 在文中与碳相关的概念, 如“碳材料”, “富碳型材料” 和“碳基材料”, 在这里进行区分说明: (1)碳材料: 元素组成上只含有碳或以碳元素为绝对主导的形式, 如富勒烯、碳纳米管[6]、金刚石[7]、石墨、石墨烯、多孔碳[8]等; (2)富碳型材料: 是对“碳材料”概念的拓展, 即除了“碳材料”外, 还包含以碳基骨架小分子为构筑单元的稠环芳香分子[9]、线性聚合物、纳米带[10]、二维聚合物[11]、多孔聚合物[12]、框架材料[13-20]等; (3)碳基材料: 在文中, 与“富碳型材料”具有相同概念, 不仅仅是“碳材料”.
在1960年之前, 碳材料主要包括石墨、活性炭、炭黑以及天然钻石等. 随着技术的发展, 在1960~1985年这段时间里出现了由聚丙烯腈制备的碳纤维、由气象化学沉积(Chemical vapor deposition, 简称CVD)技术制备的热解石墨(Pyrolytic carbon)以及由热固性树脂(Thermosetting resins)制备的玻璃碳(Glass-like carbon)等. 对比于1960年以前的经典碳材料, 碳纤维、热解石墨和玻璃碳被称为新碳材料. 1985年在氦气流中以激光汽化蒸发石墨制备出了由60个碳组成的碳原子簇结构分子C60 (Buckminsterfullerene), 随后一系列相似结构的碳材料被开发出来, 如: C70、C86等. 在1991年多壁碳纳米管(Multi-walled carbon nanotube)和单壁碳纳米管(Single-walled carbon nanotubes)也相继被开发出来, 之后“纳米碳”的概念被提出[21]. 自此, 碳材料家族研究呈现爆发式增长.
1.1 碳材料的种类
根据碳原子间共价键的主要类型, 可以将碳材料的纳米结构分为两大类. 第一类涉及石墨烯纳米结构, 尽管它们在缺陷部位或边缘也可能含有一些sp3碳原子, 但它们主要由紧密堆积在六角形蜂窝晶格中的sp2杂化的碳原子组成. 这类碳材料主要包括石墨烯(Graphene)[22]、石墨烯纳米片(Graphene flake)[23]、碳纳米管(Carbon nanotube, CNT)[24, 25]、纳米角(Nanohorn)[26]、洋葱状碳纳米球(Onion-like carbon nanosphere)[27]等. 从理论上讲, 石墨烯可以被视为其他石墨/石墨同素异形体的“基石”. 例如, 原则上可以将一块适当切割的石墨烯片(Graphene sheet)包裹起来以形成一维的碳纳米管, 或与其他石墨烯片堆叠以形成多层的二维碳纳米片或石墨.
第二类碳纳米结构包含各种比例的sp3和sp2杂化的碳原子, 并具有无定形和类石墨烯部分的混合物, 也可能主要由sp3碳原子组成. 目前, 纳米金刚石(Nanodiamond)[28]是此类碳材料的最重要成员, 但某些具有非石墨结构的碳点[24]也可以归入其中.
此外, 也可以根据碳材料的形貌特征对碳的同素异形体进行分类. 这种分类方式中的第一类包含具有内部空间的纳米结构, 例如富勒烯、碳纳米管和纳米角. 这些空心纳米结构的内部空隙可以容纳客体分子、金属、原子或其他纳米结构. 在某些情况下, 它们甚至可以提供有助于特定反应的纳米环境. 另一类则是不含有空心纳米结构的碳材料, 如: 纳米金刚石、碳点、洋葱状碳纳米球以及石墨烯.
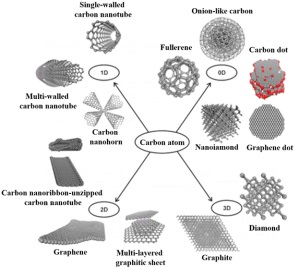
最后一种分类方式是基于碳材料纳米结构的维度(图 1). 在该分类方式下将碳材料分为: (1)零维的碳纳米结构, 例如富勒烯、洋葱状碳纳米球、碳点和纳米金刚石; (2)一维的碳纳米带、碳纳米管、碳纳米纤维和单壁纳米角; (3)二维石墨烯、石墨烯纳米带和多层堆积的石墨烯. 这类纯几何学上的分类, 在材料领域比较受欢迎, 但本身并不具有重要的物理和化学意义.
图 1
1.2 课本中的相关概念
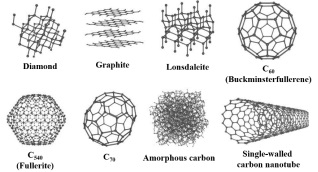
尽管可以通过不同方式对碳材料进行分类, 但在直观上所有纯碳材料都互为同素异形体(Allotropes). 同素异形体的概念最初是由Berzelius于1841年提出[29]. 如图 2所示, 以不同形式存在的碳单质如富勒烯、金刚石、石墨、无定形碳、碳纳米管等均属于碳的同素异形体. 碳的同素异形体均由同样的单一碳元素组成, 因碳原子排列方式不同, 而具有不同性质的单质, 其中最为典型的例子是金刚石和石墨. 在金刚石晶体中, 每个碳原子都以sp3杂化轨道与另外4个碳原子形成共价键, 构成正四面体. 由于所有的价电子都参与共价键的形成, 没有自由电子, 所以钻石不导电. 而石墨由多层片状结构堆积而成, 单层内的碳原子排列成平面六边形, 每个碳原子以sp2共价键与其它碳原子结合, 同层中的离域电子可以在整层活动, 层间碳原子以分子间作用力(范德华力)相结合. 石墨是一种灰黑色、不透明、有金属光泽的晶体. 天然石墨耐高温, 热膨胀系数小, 导热、导电性好, 摩擦系数小. 富勒烯, 由于同时具有五元环、六元环及其球状结构, 其特殊的空间构型带来的丰富性质使得其在光电材料与器件领域大放异彩. 但是, 无定形碳, 同样可能存在丰富的非六元环及其内在孔道曲率, 也有诸多应用, 但是其构效关系研究大多停留在理论层面. 显然, 同素异形体可以用于区分不同碳材料, 但无法用于深入研究材料的性质及其之间的关系.
图 2
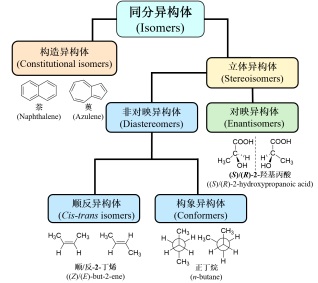
在化学中, 同分异构体(Isomers)是指具有相同分子式和元素组成而结构不同的分子和离子[30]. 如图 3所示, 同分异构体分为构造异构体(Constitutional isomers)与立体异构体(Stereoisomers). 构造异构体是指因分子中原子的连接次序或键合方式不同引起的异构体, 如萘(Naphthalene)和薁(Azulene)就属于构造异构体中的碳架异构体. 立体异构体是指分子中原子或原子团互相连接次序相同, 但空间排列不同而引起的异构体. 立体异构体可以分为对映异构体(Enantisomers)和非对映异构体(Diastereomers), 如(S)/(R)-2-羟基丙酸. 非对映异构体中包括顺反异构体(Cis-trans isomers)和构象异构体(Conformers). 顺反异构是指化合物分子中由于具有限制自由旋转的因素, 使各个基团在空间的排列方式不同而出现的非对映异构现象, 如顺式和反式2-丁烯; 构象异构体是指仅由单键的旋转而引起的立体异构现象, 如图 3中所示正丁烷中的两个甲基由于碳碳单键的旋转可以形成两个甲基处于对位和邻位的两种构象. 这些分类和定义大大促进了有机合成化学的发展, 也为生物化学注入了活力. 但是, 这些分类和定义完全基于几何构型, 而非内在关联, 分子之间的本征性质预测依赖于经验而非构效关系理解.
图 3
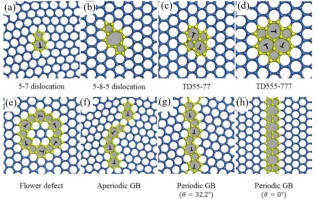
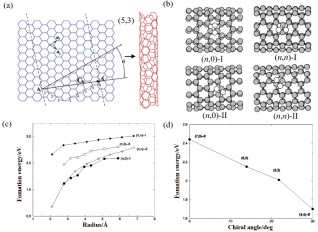
碳原子呈高度规律排布的碳材料(如金刚石、石墨烯等)在结构上会存在各种各样的缺陷. 其中碳材料中的拓扑缺陷碳会以同素异形体和同分异构体的形式出现. 例如, 完美的石墨烯[31]中碳原子以sp2共价键紧密排列在六方晶格中, 但当其中碳原子发生重排时就会出现各种形式的拓扑缺陷, 这些原子的重新排布破坏了二维晶格的六边形对称性. 如图 4所示, 石墨烯中拓扑缺陷类型主要包括位错[32](Dislocation)和向错[33] (Disclination), 向错和位错分别破坏了晶格的旋转和平移对称性, 其中以5-7位错和5-8-5位错较为常见. 扩大以上两种缺陷结构可以得到簇状拓扑缺陷, 如555-777簇状拓扑缺陷(555-777 clustered topological defect)和闭环的花形拓扑缺陷(Closed loop flower topological defect)以及具有不同晶界角(Misorientation angle, θ)的周期性和非周期性晶界[32, 34]. 不限于这里所列类型, 这类材料的构型非常丰富, 由于原子级的分辨下的研究非常具有挑战, 因此这一领域的实验报道有限, 而理论研究相对丰富.
图 4
 图 4. 石墨烯中的拓扑缺陷: (a) 5-7位错; (b) 5-8-5位错; (c) 55-77簇状拓扑缺陷; (d) 555-777簇状拓扑缺陷; (e) 闭环的花形拓扑缺陷; (f) 非周期晶界; (g) θ=32.2°周期晶界; (h) θ=0°周期平动晶界[35]Figure 4. Topological defects in graphene: (a) 5-7 dislocation; (b) 5-8-5 dislocation; (c) 55-77 clustered topological defect; (d) 555-777 clustered topological defect; (e) closed loop flower topological defect; (f) aperiodic grain boundary; (g) θ=32.2° periodic grain boundary; (h) θ=0° periodic translational grain boundary[35]
图 4. 石墨烯中的拓扑缺陷: (a) 5-7位错; (b) 5-8-5位错; (c) 55-77簇状拓扑缺陷; (d) 555-777簇状拓扑缺陷; (e) 闭环的花形拓扑缺陷; (f) 非周期晶界; (g) θ=32.2°周期晶界; (h) θ=0°周期平动晶界[35]Figure 4. Topological defects in graphene: (a) 5-7 dislocation; (b) 5-8-5 dislocation; (c) 55-77 clustered topological defect; (d) 555-777 clustered topological defect; (e) closed loop flower topological defect; (f) aperiodic grain boundary; (g) θ=32.2° periodic grain boundary; (h) θ=0° periodic translational grain boundary[35]综上, 无论是同素异形体、同分异构体还是拓扑缺陷, 本质上都是在说明碳材料内部的原子排布的不同, 只能用于区分不同碳材料的不同结构, 而预测、区分碳材料性质及其构效关系仍然具有一定的挑战.
1.3 如何计算和测试材料的熵
为了避免在区分碳材料的时候只考虑几何因素, 引入量子力学、统计力学、热力学等因素的考虑显得尤其重要. 在统计力学中, 熵表示体系内所包含不同状态的多少. 统计力学中的熵主要包括振动熵[36, 37](Vibrational entropy)、转动熵[37, 38](Rotational entropy)、构象熵[37, 39, 40] (Configuration entropy)和信息熵[41](Information entropy).
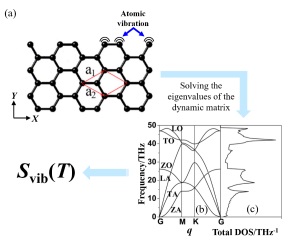
在固体物理学中, 结晶态固体中的原子或分子是按一定的规律排列在晶格上的. 在晶体中, 原子间有相互作用, 原子并非是静止的, 它们总是在其平衡位置不断地振动(图 5(a)), 这种晶体内原子振动的自由度可以通过振动熵来衡量. 振动熵和材料的带隙(Bandgap)、热导率(Thermal conductivity)、相稳定性(Phase stability)和振动光谱(Vibrational spectra)相关. 振动熵(Svib)的计算公式为[37]
$ {S_{{\rm{vib}}}}{\rm{ = - }}{k_{\rm{B}}}\sum\limits_{q,s} {\frac{h}{{2\pi }}} \omega (q,s) + \frac{1}{T}\sum\limits_{q,s} {\frac{{h\omega (q,s)}}{{2\pi {\rm{exp}}\left( { - \frac{{h\omega (q,s)}}{{2\pi {{\rm{k}}_{\rm{B}}}T}}} \right) - 1}}} $
(1) 图 5

其中kB为玻尔兹曼常数, h为普朗克常数, T为绝对温度, ω(q, s)为声子频率. 尽管相关计算理论在半个多世纪以前就被提出, 但直到近几年, 高性能计算机的出现才使耗时且复杂的第一性原理计算变得易于实现. 振动熵的计算方式主要有晶格动力学(Lattice dynamics)和分子动力学(Molecular dynamics, MD)计算, 其中晶格动力学方法已被应用于计算振动熵对晶体多晶型[42]、分子晶体的相稳定性[43]以及合金形成[44, 45]的影响. 分子动力学的方法已被应用于计算振动熵在有机聚合物晶体相变中的作用[46]. 和晶格动力学的方法相比分子动力学的方法需要更高的运算成本, 因为分子动力学计算需要数千个步骤来获得良好的平衡量, 而且热力学积分需要许多(通常是几十到几百个)单独的分子动力学计算运行.以图 5(a)所示的石墨烯为例, Svib可以通过计算声子态密度(Phonon density of states)得到. 通过求解石墨烯体系的动态矩阵(Dynamic matrix)可以得到一组特征向量(Eigenvectors)和声子频率(Phonon frequencies, ω), 从而得到声子谱(Phonon spectra). 声子态密度可以通过对声子谱积分处理得到.
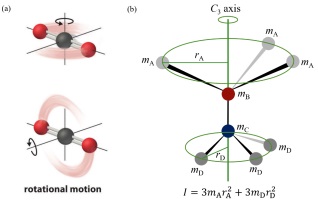
除了振动, 分子在非零度条件下可以以某个旋转轴转动从而产生一定的转动自由度, 例如图 6(a)所示的二氧化碳分子. 转动熵通常会影响体系的相稳定性、热导率以及电子极化(Electronic polarisation), 软物质模拟已经证实转动熵对碳纳米管和水分子[48]、蛋白和配体[49]之间的相互作用具有显著影响. 分子的转动自由度可以由转动熵(Srot)来衡量, 对于一个分子单元, 转动熵的解析表达式为[37]
$ {S_{{\rm{rot}}}} = R{\rm{In}}\left[ {\frac{{{\pi ^{1/2}}}}{\sigma }\prod\limits_{j = 1}^3 {{{(\frac{{8{\pi ^2}{I_j}{k_{\rm{B}}}T}}{{{h^2}}})}^{1/2}}} } \right] $
(2) 其中kB为玻尔兹曼常数, h为普朗克常数, T为绝对温度, Ij为分子的惯性矩, σ为分子的对称数. 分子的惯性矩可以通过
$ {I_j} = \sum\limits_i {{m_i}r_{ij}^2} $
(3) 计算. 其中mi为分子中某个原子的质量, rij表示第i个原子的原子核到第j个主旋转轴之间的垂直距离. 如图 6(b)所示, 处于C3旋转轴上两个相连的原子B和C分别和三个相同的原子A和D相连, 原子A和D的质量分别为mA和mD, 原子A和D的原子核到C3旋转轴的垂直距离分别为rA和rD, 则该分子对应于C3旋转轴的惯性矩为3mArA2+3mDrD2. 直接计算转动熵需要在足够长的时间尺度上运行分子动力学计算来收集体系旋转的统计学数据, 这往往需要数十万甚至数百万步的运算. 此外, 典型的分子动力学运算的时间步长在飞秒尺度, 而转动可以发生在皮秒至纳秒尺度[50], 利用偏置分子动力学计算(biased-MD calculations)[51]可以使上述问题得到解决从而获得精确的转动熵信息.
图 6

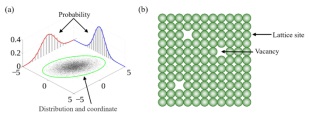
在统计力学中, 构象熵是系统熵的一部分, 与系统组成粒子的离散位置有关, 它可以指原子或分子在混合物、合金或玻璃中堆积在一起的方式的数目以及分子构象的数目或磁体中自旋结构的数目. 构象熵在材料体系中的作用早在1968年就被Navrotsky和Kleppa[53]所证实, 一般来说, 构象熵和材料的带隙、磁序(Magnetic ordering)、热导率、相稳定性和电子极化相关. 如图 7(a)所示, 粒子以不同的概率分布于二维平面的直角坐标系内, 纵坐标表示粒子在平面坐标系中不同坐标下的分布概率, 通过对体系的概率密度函数(Probability density function)积分可以得到构象熵, 其表达式为[54]
$ {S_{{\rm{exac}}}} = - {k_{\rm{B}}}\int {{\rm{d}}{\bm{q}}{\rho _q}({\bm{q}}){\rm{In}}{\rho _q}({\bm{q}})} $
(4) 图 7
 图 7. (a)构象熵的理论模型; (b)含有晶格空位的单原子晶体的构象熵Figure 7. (a) Theoretical model of conformational entropy; (b) conformational entropy of monatomic crystal with lattice vacancies
图 7. (a)构象熵的理论模型; (b)含有晶格空位的单原子晶体的构象熵Figure 7. (a) Theoretical model of conformational entropy; (b) conformational entropy of monatomic crystal with lattice vacancies其中, kB是玻尔兹曼常数, ρq(q)表示体系所处的坐标空间q的概率密度函数. 原则上, 可以通过对公式的右侧进行数值积分来计算熵Sexac, 但实际上这几乎是无法实现的, 因为数据的统计准确性较低, 并且需要高维数组(High dimensional array)来存储函数ρq(q)的直方图. 为了解决这个问题, 研究人员已经开发出了各种估算构型熵的方法, 其中应用最为广泛的是相干势近似法(Coherent potential approximation methods)[55]、特殊准随机结构法(Special quasi-random structure approach)[56]和团簇展开法(Cluster expansion method)[57]等.
计算构象熵最简单的一个例子是含有晶格空位的单原子晶体. 假设如图 8(b)所示的晶格中含有N个晶格位, N足够大并假设所有的晶格位都是相同的, 该体系的构象熵可以通过简化的公式
$ S = {k_{\rm{B}}}{\rm{ln}}{\Omega _n} $
(5) 图 8

计算. 其中, Ωn表示空位所处位置的可能数量, n表示晶格中空位的数量. 当晶格不含缺陷时, 体系只存在一种构象, 则构象熵S=kBln1=0; 当晶格中只有一个空位时, 这个空位可以出现在任何一个晶格位上, 因此S=kBlnN; 当晶格中有两个空位时并假设这两个空位是相同的, 则Ω2=1/[2N(N-1)].
信息熵的概念对信息理论[58]以及自然科学具有重要的意义, 如Trucco[59]在1956年的研究证明了信息熵能够恰当地描述图表中的所包含的信息容量. 由于分子在一定程度上和图形具有相似之处, 因此信息熵也被应用于描述有机化合物的结构[60-68]、化学反应[68-71]和开放体系的物理化学过程[72-76]的多样性. 不同于克劳修斯(Clausius)所提出的热力学熵和玻尔兹曼(Boltzmann)熵, 信息熵的概念所包括的范围要更为广泛. 由于热力学熵的概念局限于粒子热运动这种特定的物质运动方式, 与能量(热量)的分配有特定的比例关系, 因此对于不涉及热量、能量转换的非热力学过程是不适用的. 玻尔兹曼熵具有热力学熵的所有特征, 且玻尔兹曼熵还可以延拓到非热力学系统和远离平衡态的热力学系统的非平衡态, 但是为了保持熵函数的特征, 要加入等概率的条件. 信息熵可以与热量、能量转换的多少无关, 也不受到等概率的条件的限制. 因此, 热力学熵的概念包含于玻尔兹曼熵的概念之中, 玻尔兹曼熵的概念又包含于信息熵的概念之中[77].
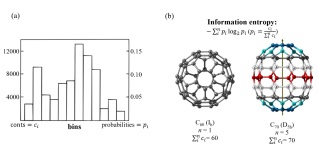
Hartley[78]和Shannon[79]将信息定义为不确定性的对数形式. 如图 8(a)所示, 对于一个包含n个研究对象的概率分布直方图, 每个研究对象所对应的概率为p1, p2, …, pi, 根据Shannon的理论每个结果的不确定性为-log pi, 当用信息熵表示一个可能的结果集的不确定性时, 信息熵(h)的计算公式为[41]
$ h = - \sum\limits_i^n {{p_i}\log {p_i}} $
(6) 其中每个研究对象所对应的概率为
$ {p_i} = {\rm{ }}\frac{{{c_i}}}{{\sum\nolimits_i^n {{c_i}} }} $
(7) 将式(7)带入式(6),
$ h = - \sum\limits_i^n {\frac{{{c_i}}}{{\sum {_i^n{c_i}} }}} {\log _2}\frac{{{c_i}}}{{\sum {_i^n{c_i}} }} $
(8) Shannon的方法也适用于研究各种纳米体系(如富勒烯[80]和碳纳米管[81])及其体系发生的变化. Sabirov及其合作者[82]利用简便的方式对富勒烯分子的信息熵进行了计算, 以图 8(b)中所示的C60和C70为例, 高度对称的C60由60个相同的碳原子组成, 对应公式中的n=1, p1=1, 因此C60的信息熵为0. 如图 8(b)所示, 对称性不如C60的C70分子包含五种不同的碳原子, 对应公式中的n=5, ca=cb=ce=10, cb=cc=20, 通过计算这五种碳原子在C70中所占比例可以得到每种碳原子的pi, 代入公式后求得C70的信息熵为2.236. 这个简单的例子说明了信息熵如何定量描述两种最为常见的富勒烯的结构差异.
除了利用统计力学方法来计算碳材料的熵还可以利用量子力学中的纠缠熵(Entanglement entropy)量化碳材料间的熵关系. 碳的同素异形体尽管都由同种元素组成但表现出十分不同的物理和化学性质. 根据凝聚态物理[83]和涌现理论[84], 材料的不同性质来源于原子在材料中的不同组织方式. 原子以多种方式组织起来, 从而形成了许多不同的顺序和许多不同类型的材料, 不同的顺序实际上对应于原子的组织中的不同对称性[85]. 这种体系内的不对称性可以利用量子力学中的纠缠熵来描述. 纠缠熵表示两个量子力学系统之间的相关性.
如图 9(a)所示, 对于一个被分为两部分的希尔伯特空间(Hilbert space,
$ {\cal H} $ )等于两个子系统(A和B)的直积(Directed product),$ {\cal H} = {{\cal H}_{\rm{A}}} \otimes {{\cal H}_{\rm{B}}} $
(9) 图 9

对于一个给定的纯态
$ \left| \phi \right\rangle \in {\cal H} $ , 体系的密度矩阵$ \rho = \left| \phi \right\rangle \left\langle \phi \right| $ , 约化密度矩阵(Reduced density matrix)$ {\rho _{\rm{A}}}{\rm{ = }}tr{{\cal H}_{\rm{B}}}\rho $ ,$ {\rho _{\rm{B}}}{\rm{ = }}tr{{\cal H}_{\rm{A}}}\rho $ , 纠缠熵(SEE)定义为冯诺依曼熵(von Neumann entropy),$ {S_{{\rm{EE}}}}{\rm{ = }} - t{r_{{{\cal H}_{\rm{A}}}}}{\rho _{\rm{A}}}{\rm{log}}{\rho _{\rm{A}}} $
(10) 纠缠熵的计算可以通过一个简单的2量子比特系统说明. 如图 9(b)所示, 在同一时间下两个子系统A和B均有两种状态
$ \left| \uparrow \right\rangle $ 和$ \left| \downarrow \right\rangle $ , 当整个系统的态为$ \left| \phi \right\rangle {\rm{ = }}\frac{1}{2}\left[ {{{\left| \uparrow \right\rangle }_{\rm{A}}} + {{\left| \downarrow \right\rangle }_{\rm{A}}}} \right] \otimes \left[ {{{\left| \uparrow \right\rangle }_{\rm{B}}} + {{\left| \downarrow \right\rangle }_{\rm{B}}}} \right] $
时,
$ {\rho _{\rm{A}}}{\rm{ = }}T{r_{\rm{B}}}\left[ {\left| \phi \right\rangle \langle \phi |} \right]{\rm{ = }}\frac{1}{2}\left[ {{{\left| \uparrow \right\rangle }_{\rm{A}}} + {{\left| \downarrow \right\rangle }_{\rm{A}}}} \right]\left[ {\langle \uparrow {|_{\rm{A}}} + \langle \downarrow {|_{\rm{A}}}} \right] $
此时SA=0, 表明子系统A和B之间不存在纠缠. 意味着即便在子系统A的状态是已知的情况下, 子系统B的状态仍然未知. 而当整个系统的态为
$ \left| \phi \right\rangle {\rm{ = }}\frac{1}{{\sqrt 2 }}\left[ {{{\left| \uparrow \right\rangle }_{\rm{A}}} \otimes {{\left| \downarrow \right\rangle }_{\rm{B}}}} \right] + \left[ {{{\left| \downarrow \right\rangle }_{\rm{A}}} \otimes {{\left| \uparrow \right\rangle }_{\rm{B}}}} \right] $
时,
$ {\rho _{\rm{A}}}{\rm{ = }}T{r_{\rm{B}}}\left[ {\left| \phi \right\rangle \langle \phi |} \right]{\rm{ = }}\frac{1}{2}\left[ {{{\left| \uparrow \right\rangle }_{\rm{A}}}\left\langle { \uparrow {|_{\rm{A}}}} \right. + {{\left| \downarrow \right\rangle }_{\rm{A}}}\left\langle { \downarrow \left| {_{\rm{A}}} \right.} \right.} \right] $
此时SA=log2, 表明子系统A和B在一定程度上纠缠. 这种情况的一个典型例子是同一轨道内的电子自旋, 根据泡利不相容原理, 同一轨道内的两个自旋电子的自旋方向是不同的, 意味着当第一个电子的自旋方向确定后, 另一个电子的自旋方向必然相反, 这两个自旋电子间存在纠缠.
对于二维体系, Holzhey等[86]和Cardy等[87]首先研究了二维共形场理论中的纠缠熵. 根据Kitaev[88]和Levin[89]的开创性工作, 二维系统的一个分区的纠缠熵可以用公式表达为
$ S({\rm{A}}) = \alpha \ell - \gamma + ... $
(11) 其中A表示一个圆盘状区域, 其边界(纠缠表面)的长度为l, 其中省略号表示在l→∞极限内消失的项. 由于边界附近的短程物理作用, 系数α是非通用的. γ是表征基态中长程纠缠的通用附加常数, 可以量化为γ=ln D, D是介质的总量子尺寸. 对于长度为L的有限间隔(Finite interval of length)和UV隔断(UV cut-off) ε给出的空间区域(如图 9(c)所示), 此间隔(Interval)的纠缠熵为
$ {S_L} = \frac{c}{3}\log \frac{L}{\epsilon} + C $
(12) 式中对数项是通用的, 仅取决于共形场理论(Conformal field theory, CFT)的中心电荷c, 而常数C不是通用的, 通常取决于所研究的体系. 对于通过拓扑界面的纠缠熵(The entanglement entropy through topological interfaces)[90]
$ S = \frac{c}{3}\log L - \sum\limits_{i,\bar l} {Trp_i^{\rm{A}}\log \frac{{p_i^{\rm{A}}}}{{p_i^{{\rm{id}}}}}} $
(13) 其中, 指数(Indices) i和
$ \overline l $ (通常是不同的)是共同子代数的两个手性部分的不可约最高权重表示(Irreducible highest weight representations of the two chiral parts of the common subalgebra), 黑体的指数i是在两个相邻的CFT中不可约表示的一对左右乘积(A pair of leftright products of irreducible representations in the two adjacent CFTs),$ i \equiv (i,\overline l ,;\alpha ,\beta ) $ , 矩阵$ p_i^{\rm{A}} $ 是半正定埃尔米特矩阵(Positive-semidefinite Hermitian matrix),$ p_i^{{\rm{id}}} $ 是CFT1中同一性缺陷对应的分布(The distribution corresponding to the identity defect in CFT1).对于左/右纠缠熵(Left/right entanglement entropy)[90],
$ S = \frac{c}{6}{\rm{log}}L - \sum\limits_i {{{\left| {{S_{bi}}} \right|}^2}} \log \frac{{{{\left| {{S_{bi}}} \right|}^2}}}{{{S_{i0}}}} $
(14) 除了上述提及的几种熵, 也可以用能量来量化同素异形体、同分异构体以及碳材料中缺陷间的关系. 2010年, Louie及其合作者[32]通过第一性原理计算建立了石墨烯中不同位错和晶界间的能量关系(图 10). 如图 10(a)~10(d)所示, 石墨烯中的位错和晶界缺陷所对应的单位长度能量可以用Read-Shockley方程
$ \gamma (\theta ') = \frac{{\mu \left| {\vec b} \right|}}{{4\pi (1 - \nu )}}\theta '(1 + {\rm{ln}})\frac{{\left| {\vec b} \right|}}{{2\pi {r_0}}} - \ln \theta ') $
(15) 图 10
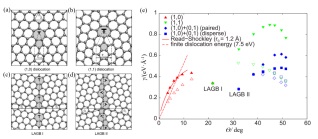
 图 10. (a), (b)分别为(1, 0)和(1, 1)位错的原子结构; (c), (d)分别为θ=21.8° (LAGB I)和θ=32.2° (LAGB II)对称的大角度晶界的原子结构; (e)各种平面(实心符号)和弯曲(空心符号)晶界结构的单位长度γ的晶界能量与取向角θ的关系[32]Figure 10. (a), (b) Atomic structures of (1, 0) and (1, 1) dislocations, respectively; (c), (d) atomic structures of the θ=21.8° (LAGB I) and the θ=32.2° (LAGB II) symmetric large-angle grain boundaries, respectively; (e) grain-boundary energy per unit length γ as a function of misorientation angle θ for various flat (filled symbols) and buckled (open symbols) grain-boundary structures[32]
图 10. (a), (b)分别为(1, 0)和(1, 1)位错的原子结构; (c), (d)分别为θ=21.8° (LAGB I)和θ=32.2° (LAGB II)对称的大角度晶界的原子结构; (e)各种平面(实心符号)和弯曲(空心符号)晶界结构的单位长度γ的晶界能量与取向角θ的关系[32]Figure 10. (a), (b) Atomic structures of (1, 0) and (1, 1) dislocations, respectively; (c), (d) atomic structures of the θ=21.8° (LAGB I) and the θ=32.2° (LAGB II) symmetric large-angle grain boundaries, respectively; (e) grain-boundary energy per unit length γ as a function of misorientation angle θ for various flat (filled symbols) and buckled (open symbols) grain-boundary structures[32]计算, 其中γ表示单位长度的能量, 单位为eV/Å,
$ \overrightarrow b $ 为博格斯向量(Burgers vector), 是反映位错引起晶格畸变的大小和方向的拓扑不变量, μ为剪力模数(Shear modulus), ν为泊松比(Poisson’s ratio), θ为取向角(Misorientation angle), r0是位错的核心半径. 如图 10(a)所示, 沿晶界线对齐(1, 0)位错会产生一组离散的错位角, 其符合弗兰克方程(Frank’s equation)$ \theta {\rm{ = }}2\alpha rc\sin \frac{{\left| {{{\vec b}_{(1,0)}}} \right|}}{{2{d_{(1,0)}}}} $
(16) 其中d(1, 0)是相邻位错之间可能的距离值之一. 对于图 10(b)的(1, 1)位错
$ \theta {\rm{ = 60}}^\circ {\rm{ }} - 2\alpha rc\sin \frac{{\left| {{{\vec b}_{(1,1)}}} \right|}}{{2{d_{(1,1)}}}} $
(17) Read-Shockley方程中的剪力模数(μ)可以通过
$ \mu = \frac{E}{{2(1 + v)}} $
(18) 求得, 其中E为杨氏模量(Young’s modulus).
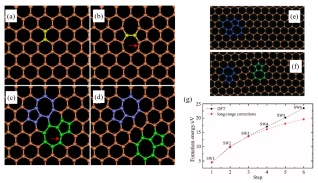
石墨烯中最常见的Stone-Wales缺陷可以经过Stone-Wales键的多次旋转和五/七元环在二维晶格内的移动形成一对5-7位错. 如图 11(a)所示, 石墨烯中(标黄色的)两个碳原子间的键经旋转形成图 11(b)中的Stone-Wales缺陷, 接着图 11(b)中的箭头所指的化学键经旋转形成图 11(c)中的位错对结构(紫色和绿色部分). 类似地, 图 11(c)中箭头所指的键发生旋转以使位错沿着滑动面蠕动形成图 11(d)中的结构. 缺陷演化过程中各步骤结构间的关系可以通过形成能来量化, 形成能的计算公式为[95]
$ {E_{{\rm{form}}}}\left[ {N - n} \right] = {E_{{\rm{tot}}}}\left[ {N - n} \right] -( N - n){\mu _{{\rm{gra}}}} $
(19) 图 11
 图 11. (a)~(d)由一系列Stone-Wales键旋转形成的石墨烯中的位错; (e), (f)分别为一个和四个Stone-Wales键旋转得到的DFT原子模型(步骤1和步骤4); (g)每一步的形成能[96]Figure 11. (a)~(d) Dislocations in graphene formed by rotations of a series of Stone-Wales bond; (e), (f) atomistic models obtained within DFT for one and four Stone-Wales bond rotations (steps 1 and 4); (g) Formation energy at each step[96]
图 11. (a)~(d)由一系列Stone-Wales键旋转形成的石墨烯中的位错; (e), (f)分别为一个和四个Stone-Wales键旋转得到的DFT原子模型(步骤1和步骤4); (g)每一步的形成能[96]Figure 11. (a)~(d) Dislocations in graphene formed by rotations of a series of Stone-Wales bond; (e), (f) atomistic models obtained within DFT for one and four Stone-Wales bond rotations (steps 1 and 4); (g) Formation energy at each step[96]其中N是原始石墨烯单胞(Unit cell)中碳原子的总数, n为空位数, Etot[N-n]为缺陷石墨烯中数量为N-n个碳原子的总能量, μgra为原始石墨烯每个碳原子的能量. 利用密度泛函理论(Density functional theory, DFT)的局部密度近似(Local-density approximation), Kirkland及其合作者[81]计算了Stone-Wales缺陷演化步骤中不同缺陷结构的形成能. 如图 11(g)所示, 图 11(f)所对应的5-7位错对的形成能比图 11(e)所对应的Stone-Wales缺陷的形成能高约13 eV, 并且缺陷演化过程中每一步的形成能较前一步更高(最多高出5.4 eV).
1.4 部分已知碳材料的热力学熵
标准摩尔熵(
$ S^\circ $ )是标准状态下1 mol物质的规定熵. 根据热力学第三定律, 纯晶体结构的熵在0 K时为0 J•mol-1•K-1, 前提是材料形成了“完美的晶体”而没有任何因结构缺陷而产生的熵. 但晶体总是在特定的温度下生长, 所以在晶体中出现各种缺陷是难以避免的. 温度为0 K的1 mol物质通过热交换达到298 K, 则总摩尔熵为所有N个升温阶段贡献的总和[97], 表示为$ S^\circ = \sum\limits_{k = 1}^N {\int {\Delta {S_k} = } } \sum\limits_{k = 1}^N {\int {\frac{{{\rm{d}}{q_k}}}{T}{\rm{d}}T} } $
(20) 其中dqk/T表示在温度T处极小的热能交换. 总摩尔熵是摩尔熵的多个阶段微小变化的总和, 其中每个阶段都可以视为可逆过程. 目前部分已知碳材料的标准摩尔熵如表 1所示.
表 1

 下载:
导出CSV
下载:
导出CSV
Standard
Enthalpy of
Formation/
(J•mol-1•K-1)Standard
Gibbs Energy
of Formation/
(J•mol-1•K-1)Standard Molar
Entropy/
(J•mol-1•K-1)Diamond +1.895 +2.900 2.377 Impurity-free
nanocrystalline
diamond— — 3.08 Nanocrystalline
diamond— — 3.83 diamond — — 3.83 Graphite 0 0 5.740 Multi-walled
carbon
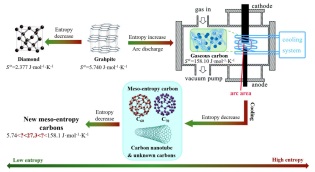
nanotube— — 6.439 Fullerene-C60 — — 27.3 Gaseous carbon +716.68 +671.26 158.10 金刚石晶体中的每个碳原子都与四面体中的四个其他碳原子共价键合, 这些四面体在椅式构象中形成六元碳环(类似于环己烷)的三维网络. 正是因为金刚石晶体内碳原子的高度有序排列, 使得金刚石的标准摩尔熵是目前已知物质中最低的(2.377 J•mol-1•K-1)[98]. 熵值的大小往往和物质的稳定性相对应, 尽管一直以来石墨在标准实验室条件(273或298 K, 1.013×105 Pa)下被认为是最稳定的碳同素异形体, 但最近的一项计算研究表明[99], 在理想条件下(T=0, P=0), 金刚石的能量比石墨低1.1 kJ•mol-1. 但当金刚石晶体中的高度有序的碳原子排布被一定程度上打破后, 材料的标准摩尔熵就会上升, 根据Duda及其合作者[100]的研究, 纯净的纳米晶金刚石(Nanocrystalline diamond)的标准摩尔熵为3.08 J•mol-1•K-1, 含有杂质的纳米晶金刚石的标准摩尔熵为3.83 J•mol-1•K-1. 尽管石墨由多层石墨烯通过范德华力结合而成, 但其碳原子的有序排列被局限在二维平面内, 和金刚石相比具有更高的标准摩尔熵(5.74 J•mol-1• K-1)[100].
单从结构上看, 单壁碳纳米管可以认为是将石墨烯进行剪切并沿着六边形晶格的一个布拉菲晶格矢量(Bravais lattice vectors)滚动而形成的空心圆柱体. 由于破坏了在二维平面范围内的碳原子有序排布, 由多层单壁碳纳米管组成的多壁碳纳米管的标准摩尔熵[101]为6.439 J•mol-1•K-1, 相比于石墨更高. C60的标准摩尔熵[97]为27.3 J•mol-1•K-1, 高于多壁碳纳米管, 这是因为C60由60个碳原子构成像足球一样的32面体, 包括20个六边形和12个五边形. 在电弧放电条件下对石墨进行升温处理得到的气态碳的标准摩尔熵[98]为158.1 J•mol-1•K-1, 明显高出其他碳材料的标准摩尔熵. 从表中数据来看, 可以将低熵的金刚石和高熵的气态碳分别定义为低熵和高熵碳材料, 标准摩尔熵处于二者之间的定义为介熵碳材料. 因此, 通过计算或测量熵值便可以在不同碳材料中区分低熵和高熵以及介熵碳材料.
1.5 熵增熵减法制备和发现新的碳材料
新的碳材料可以通过改变体系的熵值得到. 以富勒烯的制备过程为例(如图 12所示), 在高直流感应电弧处理中使石墨急剧升温, 可以得到气态碳. 此过程是典型的熵增步骤, 但高熵的气态碳并不能稳定存在, 与外部空间进行热交换后, 高熵的气态碳重新结合并形成新的碳物质, 包括富勒烯C60, 富勒烯C70, 碳纳米管和一些未知或结构不确定的碳残基. C60的标准摩尔熵[97]为27.3 J•mol-1•K-1, 介于石墨和气态碳的标准摩尔熵之间. 理论上来讲, 可以用类似的手段处理所有类型的碳并通过调控条件使其熵值尽可能降低, 例如可以在足够的大气压力下将石墨(
$ S^\circ $ =5.740 J•mol-1•K-1)转化为金刚石($ S^\circ $ =2.377 J•mol-1•K-1). 尽管如此, 由于目前的碳材料结构的准确表征技术所限, 这种合成新型碳材料的方法并未得到足够的重视. 但随着结构表征技术的发展, 利用改变碳材料的熵的方法势必会发现大量新型碳材料.图 12

2. 拓扑学缺陷与熵
根据Mermin-Wagner理论, 二维晶体结构的长程有序性可能会因长波长波动而被破坏[104, 105], 另外, 弹性理论表示二维薄膜在非绝对温度(即>0 K)下表现出不稳定性(例如弯曲)[106]. 基于此, 长期以来人们一直认为在正常环境中不存在“没有晶格缺陷的”二维晶体, 这意味着在现实中所实际存在的碳材料所具有的熵要比其具有完美晶体结构的碳材料更高. 因此, 现实存在的绝大多数材料在结构上保持着一定程度上的有序, 介于绝对有序和无序之间, 属于介熵材料.
2.1 理想石墨烯中的拓扑学缺陷
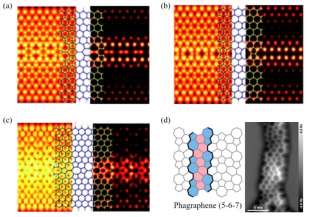
石墨烯是一种新型的二维蜂窝结构材料, 由单层sp2杂化轨道碳原子形成, 其厚度为约0.335 nm, 对应于一个碳原子的厚度. 石墨烯可以通过包裹、堆叠等方法形成三维石墨、一维纳米管[107]和零维富勒烯[108]. 由于石墨烯具有独特的微观结构, 因此它具有许多优异的物理和化学特性, 例如超高比重、表面积, 优异的刚度[109]和强度, 高电子迁移率和导热率[110]. 另外, 它具有半整数量子霍尔效应和完美的量子隧穿效应[111]. 这些特性使石墨烯成为继富勒烯和碳纳米管之后最受欢迎的低维功能碳材料. 然而, 热力学第二定律表示晶体材料中会存在一定数量的缺陷和无序, 在制备过程中不可避免地会产生各种石墨烯缺陷. 缺陷对晶体和纳米结构的性能影响很大. 不同类型的缺陷可以改变拓扑或曲率, 进而改变石墨烯的结构[112]. 在大多数情况下, 这些不可避免的缺陷会影响石墨烯和基于石墨烯的纳米复合材料[113]的机械性能、导热性和导电性[114]. 因此, 对石墨烯中的缺陷的深入理解有助于进一步改进基于石墨烯的纳米工程以及开发新的碳材料. Charlier及其合作者[100]对石墨烯中一维线性的五元环、七元环和八元环缺陷(图 13(a)~13(c))进行了局域态密度和量子传输计算, 发现t5t7类型的线性拓扑缺陷能够产生额外的传导通道和局域态, 从而增强石墨烯的化学反应性. 局部密度近似(Local-density approximations, LDA)计算表明在三种线性缺陷中t5t7类型的缺陷最为稳定(形成能为0.171 eV•atom-1), 585和d5d7类型的缺陷的形成能分别为0.258和0.264 eV•atom-1, 因此线性拓扑缺陷的石墨烯是理想石墨烯的高熵物种, 稳定性居中的t5t7型缺陷石墨烯是理想石墨烯和具有585线性缺陷的石墨烯之间的介熵物种.
图 13
为了调控石墨烯的电荷传输性质, 研究人员开发了一系列具有非六元环结构的石墨烯同素异形体. 然而, 结构明确的石墨烯同素异形体的合成难度阻碍了该领域的快速发展. 直到最近, Gottfried及其合作者[115]报道了Phagraphene(5-6-7)可以通过表面合成(2, 6-polyazulene链的脱氢C-C偶联)的方法制备(图 13(d)). 在实验过程中, 作者发现所合成的Phagraphene不如石墨烯稳定, 并且Phagraphene中的薁(Azulene)单元有转化为萘单元的趋势. 通过计算表明 Phagraphene更不稳定是因为每个萘单元的能量比薁单元低1.62 eV, 这可能是难以合成Phagraphene的内在原因. 因此, 可以将含有五/七元环结构的Phagraphene看作是石墨烯的高熵物种.
2.2 碳纳米管中的拓扑学缺陷
自从被发现以来, 碳纳米管[117-119]已吸引了各领域的许多研究小组进行深入研究. 碳纳米管在纳米电子器件[120]、氢存储[121]、透明导电膜[122]和能量存储器件[123]等技术应用中显示出巨大潜力. 如图 14(a), 单壁碳纳米管可以被看作是卷起的单层石墨烯片, 不同直径和手性的单壁碳纳米管可表现出金属或者半导体的性质[124]. 由于应力或特定合成方法, 碳纳米管会在结构中存在缺陷, 这些缺陷已在实验观察中被证实[125, 126]. 根据缺陷的方向, 纳米管中的拓扑缺陷通常会引起碳纳米管的宽度甚至手性的变化[127, 128]. 由拓扑缺陷(例如五边形-七边形缺陷对[125])形成的结构可以描述为两个不同的碳纳米管连接在一起而形成的具有异质结[129]的碳纳米管, 这样的异质结不仅提供了设计具有不同电学特性的电路的有效方法, 而且还提供了碳纳米管在纳米器件[130]和整流二极管[131]中用作功能组件的潜力.
图 14
碳纳米管中缺陷的形成能对理解缺陷的形成以及缺陷所带来的性质变化至关重要. Yang及其合作者[132]通过对四种不同的5-77-5缺陷进行计算表明(图 14(b), 14(c)) 5-77-5缺陷的形成能取决于碳纳米管的手性和半径以及管中缺陷的取向, 当缺陷管的半径大于1.5 nm时, 缺陷的形成能会随着半径的增加而逐渐增加. 此外, 如果5-77-5缺陷减小了碳纳米管的曲率, 则该缺陷的碳纳米管比曲率增加的碳纳米管更稳定.
3. 同分异构体与熵
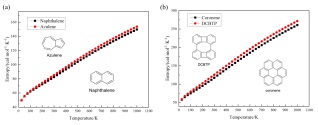
互为同分异构体的分子在有机化学领域内极为常见, 不仅如此, 石墨烯、碳纳米管等碳材料中的拓扑缺陷也经常以苯同系物的同分异构体形式出现. 其中最为常见的Stone-Wales缺陷是一种晶体学缺陷, 两个苯环间共用的π键发生旋转而生成两个稠和在一起的五/七元环. 这种非六元环的缺陷结构会导致体系的熵值增加, 如图 15(a)所示, 通过计算对比薁(Azulene)和其同分异构体萘的温度-熵曲线[134]发现在特定的温度下五/七元环的薁的熵值高于萘的熵值. 因此, 五/七元环的薁属于其同分异构体萘的高熵物种.
图 15

如图 15(b)所示, 另一对典型的同分异构体是晕苯(Coronene, 又名: 六苯并苯, 蒄)和DCBTP (Dicyclobuta[def, pqr]tetraphenylene). 根据有机化学的芳香性理论, 仅由六元芳香环组成的晕苯应比由两个四元环和一个八元环组成的DCBTP更稳定. 通过模拟二者的温度-熵曲线[134](如图 15(b))发现晕苯和DCBTP的熵随温度升高而增加, 在可控温度范围内DCBTP的熵总是高于晕苯的熵, 表明晕苯的结构比DCBTP更加稳定. 这是因为晕苯包含六个具有高度对称的稠合苯环, 而DCBTP作为联苯的二聚体, 对称性会被苯环之间的四元环和中心位置的八元环破坏, 从而在特定温度下比DCBTP具有更高的熵值.
4. 高分子中熵的引入
如上一节所述, 在特定温度下薁与萘相比有更高的熵值, 这意味着薁是其同分异构体萘的高熵物种. 由于其独特的电学和光学特性[135-138], 薁已被应用于高分子材料骨架的构建. 这种高熵单元的引入势必会导致高分子材料熵值的上升从而影响材料的特定性质, 如导电性、磁性、光吸收等. 含有薁单元的高分子聚合物除了被应用为刺激响应材料[139], 还在有机场效应晶体管[140, 141], 太阳能电池[142]等诸多领域被应用. 几乎所有这些已报道的工作, 均围绕基本光电性质及其应用展开研究, 不涉及熵的讨论, 但这不影响我们把这些案例用于本文的讨论.
4.1 线性聚合物
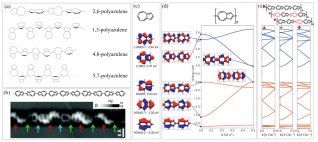
以薁作为基本连接单元的线性聚合物根据连接位点的不同主要分为四类(图 16(a)): 2, 6-聚薁、1, 3-聚薁, 4, 8-聚薁和5, 7-聚薁. 薁单元的不同连接方式会对化合物的光学和电学性质产生显著的影响[143], Dias[144]通过理论计算证实在p型掺杂后1, 3-和2, 6-聚薁比4, 8-和5, 7-聚薁表现出更高的导电性. 另一个例子是2, 6':2', 6''-三薁(2, 6':2', 6''-terazulene), 由于其最低为占用轨道在整个分子范围内形成电子离域, 增强了每个薁单元间的连结性从而表现出n型半导体性质[145]. 基于2, 6位相连的薁单元在场效应晶体管和质子导电领域的应用[141, 146], Fasel及其合作者[147]利用表面合成手段在金基底上合成了2, 6-聚薁, 并利用高分辨隧道扫描显微镜和非接触原子力显微镜对其进行了结构表征(图 16(b)), 在生成的产物中薁单元有三种连接方式: 五元环连接七元环(蓝色箭头处)、七元环连接七元环(红色箭头处)和五元环连接五元环(绿色箭头处). DFT理论计算表明(图 16(d)), 首尾相连的2, 6-聚薁的HOMO-2和LUMO+1轨道相对平坦, 而由于相邻薁单元的轨道重叠, 2, 6-聚薁的 HOMO-1和LUMO轨道在能量上更加分散. 通过对比图 16(e)中三种聚合物发现薁单元之间的不同连接方式并不会对聚合物的电子性质产生显著的影响, 这三种结构的电子带隙均为(0.94±0.03) eV, 并具有相似的能带结构.
图 16
4.2 多孔聚合物
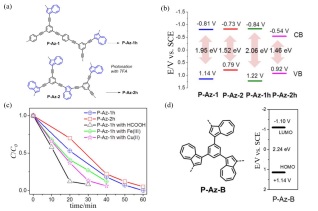
含有薁单元的多孔聚合物并不像聚薁一样被广泛研究, 一个可能的原因是用于构建多孔聚合物的薁单体的合成难度限制了此类多孔聚合物的开发. 尽管如此, 含薁的多孔聚合物已在光催化领域表现出应用前景. 由于传统多孔聚合物光催化剂的高度疏水性, 使得催化体系中难以避免要使用有毒的有机溶剂. 水作为一种廉价的、可持续的、易于循环利用的绿色有机反应溶剂[148], 一直以来研究人员希望开发出能在水相体系中进行光催化反应的多孔聚合物催化剂. 2016年, Zhang及其合作者[149]通过Sonogashira交叉偶联反应合成了两种含有不同比例薁单元的多孔聚合物PAz-1和P-Az-2(图 17(a)). 相比于PAz-1(图 17(b)), 有更高五/七元环比例的P-Az-2具有更窄的带隙(1.52 eV). P-Az-1和P-Az-2具有和某些成熟的光催化剂接近的价带和导带位置, 如[Ru(bpy)3(+1.29 V vs. SCE, bpy=2, 2'-吡啶)、[Ru(bpy)3]2+]3+(-0.81 V vs. SCE)和Eosin(+0.83 and -1.06 V vs. SCE)[150], 用三氟乙酸质子化后的多孔聚合物PAz-1h 和P-Az-2h能在60 min内定量地将高毒的CrVI还原成CrIII. 由于具有位置更低的导带(-0.54 eV)和更窄的带隙, P-Az-1h相比于PAz-2h表现出了更高的光催化活性. 通过向催化体系中加入Fe(III)或 Cu(II)作为共催化剂使其形成级联催化循环, 可以使PAz-1h的光催化活性得到显著提高(如图 17(c)所示). 除了加入共催化剂, 向聚合物中引入更高比例的高熵的五/七元环从而调节催化剂的能带位置也可以作为提高催化活性的一种途径. 2017年, 相同课题组[151]利用Suzuki交叉偶联反应合成了另一种同时含有苯环和五/七元环的多孔聚合物(图 17(d)). 和P-Az-1、P-Az-2不同的是, P-Az-B的六元苯环和五/七元环之间并没有共轭的碳碳三键而是直接相连, 这在一定程度上破坏了苯环和五/七元环间的电子云共轭, 使P-Az-B具有比P-Az-1和P-Az-2更宽的带隙(2.24 eV). P-Az-B的LUMO轨道处于-1.1 eV, 光生激发电子可以从LUMO轨道转移至取代的碘苯从而催化Stille偶联反应.
图 17
 图 17. (a) P-Az-1和P-Az-2的结构; (b)价带和导带的位置; (c)通过在级联催化循环中使用P-Az-1h或P-Az-2h以及FeIII或CuII作为助催化剂, 并使用甲酸作为水中的额外电子供体, 将CrVI光催化还原为CrIII; (d) P-Az-B的结构以及HOMO和LUMO谱带位置[149, 151]Figure 17. (a) Structures of P-Az-1 and P-Az-2; (b) positions of the valence bands and conduction bands; (c) photocatalytic reduction of CrVI to CrIII by using P-Az-1h or P-Az-2h, FeIII or CuII as co-catalyst in a cascade catalytic cycle, and formic acid as an extra electron donor in water; (d) structure, HOMO and LUMO band positions of P-Az-B[149, 151]
图 17. (a) P-Az-1和P-Az-2的结构; (b)价带和导带的位置; (c)通过在级联催化循环中使用P-Az-1h或P-Az-2h以及FeIII或CuII作为助催化剂, 并使用甲酸作为水中的额外电子供体, 将CrVI光催化还原为CrIII; (d) P-Az-B的结构以及HOMO和LUMO谱带位置[149, 151]Figure 17. (a) Structures of P-Az-1 and P-Az-2; (b) positions of the valence bands and conduction bands; (c) photocatalytic reduction of CrVI to CrIII by using P-Az-1h or P-Az-2h, FeIII or CuII as co-catalyst in a cascade catalytic cycle, and formic acid as an extra electron donor in water; (d) structure, HOMO and LUMO band positions of P-Az-B[149, 151]4.3 配位聚合物框架
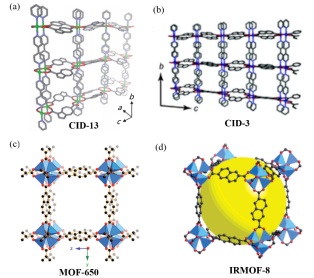
配位聚合物框架因其永久的孔隙而被应用于分子吸附领域, 其吸附性能很大程度上和其配体有关. 相比于含萘的配位聚合物框架, 含高熵薁单元的配位聚合物框架表现出对极性分子更强的吸附性. 在2012年, Kitagawa及其合作者[152]利用2, 7-萘二甲酸酯的异构体1, 6-薁二甲酸酯合成了配位聚合物框架CID-13. 如图 18(a)和(b)所示, 以2, 7-萘二甲酸酯为配体合成的CID-3尽管和CID-13具有相似的结构(空间群和晶胞参数), 但由于分子内极化显著增大了薁的偶极矩(1.08 D)[153], 使含有五/七元环的配位聚合物框架和水分子之间产生较强的相互作用, 因此比含有非极性萘单元的CID-3对极性的水分子有更强的吸附性. 此外, 引入高熵的薁单元同样能提高配位聚合物框架对非极性分子的吸附性[154]. 经过理论计算发现, 薁的极化作用使以2, 6-薁二甲酸酯为配体制备的MOF-650比以2, 6-萘二甲酸酯为配体制备的IRMOF-8[155-157]具有更高的氢分子结合能(结构如图 18(c)和(d)), 在77 K和0.1 MPa压力条件下对氢气的吸附量为14.8 mg•g-1.
图 18
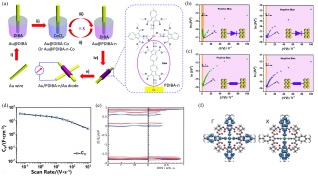
薁单元除了可以增强配位聚合物框架对特定分子的吸附, 还可以通过π电子和金属中心的d轨道偶联而增强电荷转移[159]. 2017年, 本课题组利用逐层浸涂法制备了含有薁单元的金属配位框架薄膜(图 19(a)), 由薄膜制成的整流器的平均整流比高达5.7[160]. PDIBA-5中的异氰基和钴金属中心配位, Au/PDIBA-5/Au表现出高达6.5的整流比, 而Au/DIBA/Au的整流比只有1.08. 通过对比电荷在Au/PDIBA-5/Au和Au/DIBA/Au中的传输过程发现(图 19(b)和(c)), 在正负偏压下Au/PDIBA-5/Au均呈现两种电荷传输过程, 其过渡电压(Vth)分别为0.31 V和-0.62 V, 表明Au/PDIBA-5/Au在正负偏压下有两种不同的电荷传输模式, 而Au/DIBA/Au在正负偏压下的电荷传输过程相同. 因此, 金属-配体电荷转移与正负偏压区的不同电子隧穿过程的协同作用使Au/PDIBA-5/Au结(Junction)具有高整流比.
图 19
 图 19. (a) 制造基于BCCF分子的二极管的示意图; (b) PDIBA-5和(c) DIBA的I-V曲线的Fowler-Nordheim图; (d)不同扫描速率下微超级电容器的叠层电容;(e)模拟的能带结构和状态密度; (f)越过费米能级的较低能带的电荷密度[160, 161]Figure 19. (a) Schematic fabrication of BCCF-based molecular diode. Fowler-Nordheim plots for I-V curves of junctions based on (b) PDIBA-5 and (c) DIBA; (d) stack capacitance of the MSCs at different scan rates; (e) simulated band structure and density of states (DOS); (f) charge density of the lower band crossing the Fermi level[160, 161]
图 19. (a) 制造基于BCCF分子的二极管的示意图; (b) PDIBA-5和(c) DIBA的I-V曲线的Fowler-Nordheim图; (d)不同扫描速率下微超级电容器的叠层电容;(e)模拟的能带结构和状态密度; (f)越过费米能级的较低能带的电荷密度[160, 161]Figure 19. (a) Schematic fabrication of BCCF-based molecular diode. Fowler-Nordheim plots for I-V curves of junctions based on (b) PDIBA-5 and (c) DIBA; (d) stack capacitance of the MSCs at different scan rates; (e) simulated band structure and density of states (DOS); (f) charge density of the lower band crossing the Fermi level[160, 161]薁基团的引入能使配位聚合物骨架具有超低带隙和良好的载流子迁移率, 使其成为储能和光电材料. 2017年, 本课题组[161]开发了一种基于含有薁基团金属有机框架的微型超级电容器, 含有薁基团的金属配位骨架PiCBA的载流子迁移率高达5×10-3 cm2•V-1•s-1, 所制造的微超级电容器在50 mV•s-1时可提供高达34.1 F•cm-3的比电容(图 19(d)), 以及1323 W•cm-3的高体积功率密度. 通过理论计算PiCBA的能带结构和状态密度发现(图 19(e)), 两个自旋向上的能带穿过费米能级, 表明PiCBA框架在具有和金属类似的导电性. 计算表明, 这些能带的电荷密度分布在整个薁单元上(图 19(f)), 并且分布比较集中, 这可能是由于金属中心和薁单元之间的扭转所导致的. 对含薁单元的金属配位聚合物膜的储能能力的理论研究表明PiCBA膜由于薁单元具有高的电荷亲和力而具有高电容.
5. 结论与展望: 介熵物质与介熵材料
从传统的金刚石、石墨, 到富勒烯、碳纳米管、石墨烯, 再到石墨炔、稠环芳香分子、聚合物、框架材料等, 这些富碳型材料已经成为当今材料领域最重要的研究领域之一. 虽然传统的IUPAC分类大大促进了这一学科的发展, 但是随着新材料的不断涌现, 传统的一些分类已经在某些领域受到了限制. 如同分异构体、同素异形体和拓扑缺陷, 更多的是在强调材料或分子之间空间几何结构的不同, 而具体性质如何不同, 因不同的案例而异, 很难构建明确的构效关系. 这就使得通过改变已知材料的结构来调控材料的性质变得困难. 因此, 避免纯几何角度的理解, 对富碳型材料不同结构之间的关系研究将具有重要意义. 作者尝试从材料“熵”的概念入手, 将力学(热力学、量子力学、统计力学等)作为重要考虑因素, 将熵从其他材料概念引入富碳型材料; 对可能可以及其已经应用于部分富碳型材料的几种类型的熵, 从含义和公式方面进行了总结. 通过理解富碳型材料之间熵的相对高低, 尝试基于“介熵”概念认识和开发新型富碳型材料、开发新型介熵富碳型材料的全新性质. 基于对具体的不同的新型富碳型材料的讨论, 将“介熵”这一概念引入到同素异形体、同分异构体以及广泛存在于碳材料中的拓扑缺陷的理解上, 具体如下:
(1) 同素异形体、同分异构体、拓扑缺陷以及不同结构的富碳型材料间的关系均可以用熵来说明. 熵是描述系统状态的函数, 其通用的表达式为
$ S = k1{\rm{n}}\Omega $ , 其中k为玻尔兹曼常数, Ω为该宏观状态中所包含的微观状态数量. 不同类型的熵是体系某一方面的状态数的度量, 如振动熵所描述的是体系中振动所产生的量子化的弹性波的数量.(2) 富碳型材料虽然可以基于上述理论计算其绝对熵, 但具体案例的相对熵值更加重要, 这也是“介熵”的主要含义. 正如在特定实验条件下可以转化的富碳型材料, 如利用石墨通过熵增再熵减可以获得介熵物种富勒烯及其他新型介熵富碳型材料, 可以通过类似手段获得新的介熵富碳型材料. 因此“介熵”不仅是类似富碳型材料之间相互关系的定义, 还是开发新型富碳型材料的方法.
(3) 向已知富碳型材料中引入高熵或低熵单元, 可以得到具有性质可调的新型富碳型材料. 如将薁引入聚合物中所获得的材料在微型超级电容器和分子整流器中分别表现出更高的比电容和整流比等.
材料合成手段、结构表征技术以及理论模拟计算技术的协同发展下, “介熵”概念的提出, 对富碳型材料的再开发将会起到极大的推动作用. 理论模拟计算材料的熵值日益变得简单易得, 将其结构和性质, 如导电性、带隙等物理化学性质, 联系起来, 可以系统预测“介熵”材料性能. 在已知富碳型材料的结构基础上, 可以通过调节材料的熵值实现对性能的多维调控. 总而言之, 我们希望基于新的“介熵”概念的运用, 使具有相似结构的富碳型材料之间的关系不再模糊地停留在几何结构层面上, 为全新介熵富碳型材料及其它介熵材料的开发提供理论和实验依据.
-
-
[1]
de Gennes, P. G. Angew. Chem., Int. Ed. 1992, 31, 842. doi: 10.1002/anie.199208421
-
[2]
Cowie, J. M. G.; Arrighi, V. Polymers:Chemistry and Physics of Modern Materials, CRC press, New York, 2007. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[3]
Smalley, R. E. Angew. Chem., Int. Ed. 1997, 36, 1594. doi: 10.1002/anie.199715941
-
[4]
Heeger, A. Chem. Soc. Rev. 2010, 39, 2352. doi: 10.1039/c005384h
-
[5]
Geim, A.; Novoselov, K. Nat. Phys. 2010, 6, 836. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[6]
Baughman, R. H.; Zakhidov, A. A.; De Heer, W. A. Science 2002, 297, 787. doi: 10.1126/science.1060928
-
[7]
Yang, N.; Yu, S.; Macpherson, J. V.; Einaga, Y.; Zhao, H.; Zhao, G.; Swain, G. M.; Jiang, X. Chem. Soc. Rev. 2019, 48, 157. doi: 10.1039/C7CS00757D
-
[8]
Wu, J.; Xu, F.; Li, S.; Ma, P.; Zhang, X.; Liu, Q.; Fu, R.; Wu, D. Adv. Mater 2019, 31, 1802922. doi: 10.1002/adma.201802922
-
[9]
Scott, L. T. Chem. Soc. Rev. 2015, 44, 6464. doi: 10.1039/C4CS00479E
-
[10]
Dodiuk, H.; Goodman, S. H. Handbook of Thermoset Plastics:1. Introduction, William Andrew, Boston, 2013. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[11]
Sakamoto, J.; van Heijst, J.; Lukin, O.; Schlüter, A. D. Angew. Chem., Int. Ed. 2009, 48, 1030. doi: 10.1002/anie.200801863
-
[12]
庞楚明, 罗时荷, 郝志峰, 高健, 黄召昊, 余家海, 余思敏, 汪朝阳, 有机化学, 2018, 38, 2606. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498Pang, C.-M.; Luo, S.-H.; Hao, Z.-F.; Gao, J.; Huang, Z.-H.; Yu, J.-H.; Yu, S.-M.; Wang, C.-Y. Chin. J. Org. Chem. 2018, 38, 2606(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[13]
Yuan, S.; Feng, L.; Wang, K.; Pang, J.; Bosch, M.; Lollar, C.; Sun, Y.; Qin, J.; Yang, X.; Zhang, P.; Wang, Q.; Zou, L.; Zhang, Y.; Zhang, L.; Fang, Y.; Li, J.; Zhou, H.-C. Adv. Mater 2018, 30, 1704303. doi: 10.1002/adma.201704303
-
[14]
Lee, J.; Farha, O. K.; Roberts, J.; Scheidt, K. A.; Nguyen, S. T.; Hupp, J. T. Chem. Soc. Rev. 2009, 38, 1450. doi: 10.1039/b807080f
-
[15]
James, S. L. Chem. Soc. Rev. 2003, 32, 276. doi: 10.1039/b200393g
-
[16]
Zhang, X.-R.; Wang, X.; Fan, W.-D.; Sun, D.-F. Chin. J. Chem. 2020, 38, 509. doi: 10.1002/cjoc.201900493
-
[17]
曾锦跃, 王小双, 张先正, 卓仁禧, 化学学报, 2019, 77, 1156. https://www.zhangqiaokeyan.com/academic-journal-cn_acta-chimica-sinica_thesis/0201273995763.htmlZeng, J.-Y.; Wang, X.-S.; Zhang, X.-Z.; Zhuo, R.-X. Acta Chim. Sinica 2019, 77, 1156(in Chinese). https://www.zhangqiaokeyan.com/academic-journal-cn_acta-chimica-sinica_thesis/0201273995763.html
-
[18]
陈之尧, 刘捷威, 崔浩, 张利, 苏成勇, 化学学报, 2019, 77, 242. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract346942.shtmlChen, Z.-Y.; Liu, J.-W.; Cui, H.; Zhang, L.; Su, C.-Y. Acta Chim. Sinica 2019, 77, 242(in Chinese). http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract346942.shtml
-
[19]
Cao, L.; Wang, T.; Wang, C. Chin. J. Chem. 2018, 36, 754. doi: 10.1002/cjoc.201800144
-
[20]
刘治鲁, 李炜, 刘昊, 庄旭东, 李松, 化学学报, 2019, 77, 323. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract346957.shtmlLiu, Z.-L.; Li, W.; Liu, H.; Zhuang, X.-D.; Li, S. Acta Chim. Sinica 2019, 77, 323(in Chinese). http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract346957.shtml
-
[21]
Inagaki, M.; Radovic, L. R. Carbon 2002, 40, 2279. doi: 10.1016/S0008-6223(02)00204-X
-
[22]
Allen, M. J.; Tung, V. C.; Kaner, R. B. Chem. Rev. 2010, 110, 132. doi: 10.1021/cr900070d
-
[23]
James, D. K.; Tour, J. M. Acc. Chem. Res. 2013, 46, 2307. doi: 10.1021/ar300127r
-
[24]
Iijima, S.; Ichihashi, T. Nature 1993, 363, 603. doi: 10.1038/363603a0
-
[25]
Bethune, D.; Kiang, C. H.; De Vries, M.; Gorman, G.; Savoy, R.; Vazquez, J.; Beyers, R. Nature 1993, 363, 605. doi: 10.1038/363605a0
-
[26]
Zhu, S.; Xu, G. Nanoscale 2010, 2, 2538. doi: 10.1039/c0nr00387e
-
[27]
Mykhailiv, O.; Zubyk, H.; Plonska-Brzezinska, M. E. Inorg. Chim. Acta 2017, 468, 49. doi: 10.1016/j.ica.2017.07.021
-
[28]
Georgakilas, V.; Perman, J. A.; Tucek, J.; Zboril, R. Chem. Rev. 2015, 115, 4744. doi: 10.1021/cr500304f
-
[29]
Jensen, W. B. J. Chem. Edu. 2006, 83, 838. doi: 10.1021/ed083p838
-
[30]
Petrucci, R. H.; Harwood, W. S.; Herring, F. G. General Chemistry:Principles and Modern Applications, Vol. 1, Prentice Hall, New York, 2002. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[31]
Novoselov, K. S.; Geim, A. K.; Morozov, S. V.; Jiang, D.; Zhang, Y.; Dubonos, S. V.; Grigorieva, I. V.; Firsov, A. A. Science 2004, 306, 666. doi: 10.1126/science.1102896
-
[32]
Yazyev, O. V.; Louie, S. G. Phys. Rev. B 2010, 81, 195420. doi: 10.1103/PhysRevB.81.195420
-
[33]
Liu, Y.; Yakobson, B. I. Nano Lett. 2010, 10, 2178. doi: 10.1021/nl100988r
-
[34]
Huang, P. Y.; Ruiz-Vargas, C. S.; Van Der Zande, A. M.; Whitney, W. S.; Levendorf, M. P.; Kevek, J. W.; Garg, S.; Alden, J. S.; Hustedt, C. J.; Zhu, Y. Nature 2011, 469, 389. doi: 10.1038/nature09718
-
[35]
Lu, J.; Bao, Y.; Su, C. L.; Loh, K. P. ACS Nano 2013, 7, 8350. doi: 10.1021/nn4051248
-
[36]
Fultz, B. Prog. Mater. Sci. 2010, 55, 247. doi: 10.1016/j.pmatsci.2009.05.002
-
[37]
Butler, K. T.; Walsh, A.; Cheetham, A. K.; Kieslich, G. Chem. Sci. 2016, 7, 6316. doi: 10.1039/C6SC02199A
-
[38]
Wei, J. Ind. Eng. Chem. Res. 1999, 38, 5019. doi: 10.1021/ie990588m
-
[39]
Karplus, M.; Kushick, J. N. Macromolecules 1981, 14, 325. doi: 10.1021/ma50003a019
-
[40]
Levy, R. M.; Karplus, M.; Kushick, J.; Perahia, D. Macromolecules 1984, 17, 1370. doi: 10.1021/ma00137a013
-
[41]
Shannon, C. E. Bell Syst. Tech. J. 1948, 27, 379. doi: 10.1002/j.1538-7305.1948.tb01338.x
-
[42]
Kieslich, G.; Kumagai, S.; Butler, K. T.; Okamura, T.; Hendon, C. H.; Sun, S.; Yamashita, M.; Walsh, A.; Cheetham, A. K. Chem. Commun. 2015, 51, 15538. doi: 10.1039/C5CC06190C
-
[43]
Nyman, J.; Day, G. M. CrystEngComm 2015, 17, 5154. doi: 10.1039/C5CE00045A
-
[44]
Doak, J. W.; Wolverton, C.; Ozoliņš, V. Phys. Rev. B 2015, 92, 174306. doi: 10.1103/PhysRevB.92.174306
-
[45]
Rhein, R. K.; Dodge, P. C.; Chen, M.-H.; Titus, M. S.; Pollock, T. M.; Van der Ven, A. Phys. Rev. B 2015, 92, 174117. doi: 10.1103/PhysRevB.92.174117
-
[46]
Vela, S.; Mota, F.; Deumal, M.; Suizu, R.; Shuku, Y.; Mizuno, A.; Awaga, K.; Shiga, M.; Novoa, J. J.; Ribas-Arino, J. Nat. Commun. 2014, 5, 1. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[47]
Ma, F.; Zheng, H.; Sun, Y.; Yang, D.; Xu, K.; Chu, P. K. Appl. Phys. Lett. 2012, 101, 111904. doi: 10.1063/1.4752010
-
[48]
Pascal, T. A.; Goddard, W. A.; Jung, Y. Proc. Natl. Acad. Sci. 2011, 108, 11794. doi: 10.1073/pnas.1108073108
-
[49]
Chia-en, A. C.; Chen, W.; Gilson, M. K. Proc. Natl. Acad. Sci. 2007, 104, 1534. doi: 10.1073/pnas.0610494104
-
[50]
Besara, T.; Jain, P.; Dalal, N. S.; Kuhns, P. L.; Reyes, A. P.; Kroto, H. W.; Cheetham, A. K. Proc. Natl. Acad. Sci. 2011, 108, 6828. doi: 10.1073/pnas.1102079108
-
[51]
Laio, A.; Gervasio, F. L. Rep. Prog. Phys. 2008, 71, 126601. doi: 10.1088/0034-4885/71/12/126601
-
[52]
Silberberg, M. Principles of General Chemistry, McGraw-Hill Education, New York, 2012. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[53]
Navrotsky, A.; Kleppa, O. J. Inorg. Nucl. Chem. 1968, 30, 479. doi: 10.1016/0022-1902(68)80475-0
-
[54]
Nguyen, P. H. Chem. Phys. Lett. 2009, 468, 90. doi: 10.1016/j.cplett.2008.11.061
-
[55]
Gyorffy, B. Phys. Rev. B 1972, 5, 2382. doi: 10.1103/PhysRevB.5.2382
-
[56]
Zunger, A.; Wei, S.-H.; Ferreira, L.; Bernard, J. E. Phys. Rev. Lett. 1990, 65, 353. doi: 10.1103/PhysRevLett.65.353
-
[57]
Sanchez, J. M.; Ducastelle, F.; Gratias, D. Phys. A 1984, 128, 334. doi: 10.1016/0378-4371(84)90096-7
-
[58]
Nielsen, M. A.; Chuang, I. L. Phys. Today 2001, 54, 60. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[59]
Trucco, E. Bull. Math. Biophys. 1956, 18, 129. doi: 10.1007/BF02477836
-
[60]
Lin, S.-K. Mol. Online 1996, 1, 57. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[61]
Agrafiotis, D. K. J. Chem. Inf. Comput. Sci. 1997, 37, 576. doi: 10.1021/ci960156b
-
[62]
Godden, J. W.; Stahura, F. L.; Bajorath, J. J. Chem. Inf. Comput. Sci. 2000, 40, 796. doi: 10.1021/ci000321u
-
[63]
Godden, J. W.; Bajorath, J. J. Chem. Inf. Comput. Sci. 2001, 41, 1060. doi: 10.1021/ci0102867
-
[64]
Graham, D. J. J. Chem. Inf. Comput. Sci. 2002, 42, 215. doi: 10.1021/ci0102923
-
[65]
Dehmer, M.; Varmuza, K.; Borgert, S.; Emmert-Streib, F. J. Chem. Inf. Model. 2009, 49, 1655. doi: 10.1021/ci900060x
-
[66]
Graham, D. J.; Malarkey, C.; Schulmerich, M. V. J. Chem. Inf. Comput. Sci. 2004, 44, 1601. doi: 10.1021/ci0400213
-
[67]
Tarko, L. J. Math. Chem. 2011, 49, 2330. doi: 10.1007/s10910-011-9889-1
-
[68]
Zhdanov, Y. A. Information Entropy in Organic Chemistry, Rostov University, Rostov, Russia, 1979, p. 56. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[69]
Karreman, G. Bull. Math. Biophys. 1955, 17, 279. doi: 10.1007/BF02477754
-
[70]
Levine, R. Annu. Rev. Phys. Chem. 1978, 29, 59. doi: 10.1146/annurev.pc.29.100178.000423
-
[71]
Nguyen, P. H. Chem. Phys. Lett. 2009, 468, 90. doi: 10.1016/j.cplett.2008.11.061
-
[72]
H, M.; Schmider, H. L.; Weaver, D. F.; Smith Jr., V. H.; Sagar, R. P.; Esquivel, R. O. Int. J. Quantum Chem. 2000, 77, 376. doi: 10.1002/(SICI)1097-461X(2000)77:1<376::AID-QUA37>3.0.CO;2-3
-
[73]
Kobozev, N. Russ. J. Phys. Chem. A 1966, 40, 281. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[74]
Haken, H. Information and Self-Organization. A Macroscopic Approach to Complex Systems, Springer-Verlag, Berlin, 1988, p. 240. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[75]
García-Garibay, M. A. Photochem. Photobiol. Sci. 2010, 9, 1574. doi: 10.1039/c0pp00248h
-
[76]
Nosonovsky, M. Philos. Trans. R. Soc., A 2010, 368, 4755. doi: 10.1098/rsta.2010.0179
-
[77]
李鹤龄, 大学物理, 2004, 12, 37. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498Li, H.-L. College Physics 2004, 12, 37(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[78]
Hartley, R. V. L. Bell Syst. Tech. J. 1928, 7, 535. doi: 10.1002/j.1538-7305.1928.tb01236.x
-
[79]
Shannon, C. E. Bell Syst. Tech. J. 1948, 27, 379. doi: 10.1002/j.1538-7305.1948.tb01338.x
-
[80]
Nemcsics, A.; Nagy, S.; Mojzes, I.; Schwedhelm, R.; Woedtke, S.; Adelung, R.; Kipp, L. Vacuum 2009, 84, 152. doi: 10.1016/j.vacuum.2009.04.060
-
[81]
Torrens, F.; Castellano, G. Microelectron. J. 2007, 38, 1109. doi: 10.1016/j.mejo.2006.04.004
-
[82]
Sabirov, D. S.; Ōsawa, E. J. Chem. Inf. Model. 2015, 55, 1576. doi: 10.1021/acs.jcim.5b00334
-
[83]
Kohn, W. Rev. Mod. Phys. 1999, 71, S59.
-
[84]
Goldstein, J. Emergence 1999, 1, 49. doi: 10.1207/s15327000em0101_4
-
[85]
Chen, X.; Gu, Z.-C.; Wen, X.-G. Phys. Rev. B 2010, 82, 155138. doi: 10.1103/PhysRevB.82.155138
-
[86]
Holzhey, C.; Larsen, F.; Wilczek, F. Nucl. Phys. B 1994, 424, 443. doi: 10.1016/0550-3213(94)90402-2
-
[87]
Calabrese, P.; Cardy, J. J. Stat. Mech.:Theory Exp. 2004, 2004, P06002. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[88]
Kitaev, A.; Preskill, J. Phys. Rev. Lett. 2006, 96, 110404. doi: 10.1103/PhysRevLett.96.110404
-
[89]
Levin, M.; Wen, X.-G. Phys. Rev. Lett. 2006, 96, 110405. doi: 10.1103/PhysRevLett.96.110405
-
[90]
Brehm, E.; Brunner, I.; Jaud, D.; Schmidt-Colinet, C. Fortschr. Phys. 2016, 64, 516. doi: 10.1002/prop.201600024
-
[91]
Moore, G.; Seiberg, N. Nucl. Phys. B 1989, 313, 16. doi: 10.1016/0550-3213(89)90511-7
-
[92]
Verlinde, E. Nucl. Phys. B 1988, 300, 360. doi: 10.1016/0550-3213(88)90603-7
-
[93]
Fendley, P.; Fisher, M. P. A.; Nayak, C. J. Stat. Phys. 2007, 126, 1111. doi: 10.1007/s10955-006-9275-8
-
[94]
Jaud, D. Ph.D. Dissertation, Ludwig-Maximilians-Universitt München, München, 2002. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[95]
Jeong, B. W.; Ihm, J.; Lee, G.-D. Phys. Rev. B 2008, 78, 165403. doi: 10.1103/PhysRevB.78.165403
-
[96]
Warner, J. H.; Margine, E. R.; Mukai, M.; Robertson, A. W.; Giustino, F.; Kirkland, A. I. Science 2012, 337, 209. doi: 10.1126/science.1217529
-
[97]
Jin, Y.; Cheng, J.; Varma-Nair, M.; Liang, G.; Fu, Y.; Wunderlich, B.; Xiang, X. D.; Mostovoy, R.; Zettl, A. K. J. Phys. Chem. 1992, 96, 5151. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[98]
Atkins, P.; De Paula, J.; Friedman, R. Quanta, matter, and change:a molecular approach to physical chemistry, Oxford University Press, Oxford, 2009. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[99]
Grochala, W. Angew. Chem., Int. Ed. 2014, 53, 3680. doi: 10.1002/anie.201400131
-
[100]
Vasiliev, O. O.; Muratov, V. B.; Kulikov, L. M.; Garbuz, V. V.; Duda, T. I. J. Superhard Mater. 2015, 37, 388. doi: 10.3103/S1063457615060039
-
[101]
Muratov, V. B.; Vasil'ev, O. O.; Kulikov, L. M.; Garbuz, V. V.; Nesterenko, Y. V.; Duda, T. I. J. Superhard Mater. 2012, 34, 173. doi: 10.3103/S1063457612030045
-
[102]
Lebedev, B. V.; Bykova, T. A.; Lobach, A. S. J. Therm. Anal. Calorim. 2000, 62, 257. doi: 10.1023/A:1010139501374
-
[103]
Zhang, Y.; Zhao, J.; Fang, Y.; Liu, Y.; Zhao, X. Nanoscale 2018, 10, 17824. doi: 10.1039/C8NR05465G
-
[104]
Mermin, N. D. Phys. Rev. 1968, 176, 250. doi: 10.1103/PhysRev.176.250
-
[105]
Mermin, N. D.; Wagner, H. Phys. Rev. Lett. 1966, 17, 1307. doi: 10.1103/PhysRevLett.17.1307
-
[106]
Le Doussal, P.; Radzihovsky, L. Phys. Rev. Lett. 1992, 69, 1209. doi: 10.1103/PhysRevLett.69.1209
-
[107]
Iijima, S. Nature 1991, 354, 56. doi: 10.1038/354056a0
-
[108]
Kroto, H. W.; Heath, J. R.; O'Brien, S. C.; Curl, R. F.; Smalley, R. E. Nature 1985, 318, 162. doi: 10.1038/318162a0
-
[109]
Zandiatashbar, A.; Lee, G.-H.; An, S. J.; Lee, S.; Mathew, N.; Terrones, M.; Hayashi, T.; Picu, C. R.; Hone, J.; Koratkar, N. Nat. Commun. 2014, 5, 3186. doi: 10.1038/ncomms4186
-
[110]
Tian, W.-C.; Zhang, X.-Y.; Chen, Z.-Q.; Ji, H.-Y. Recent Pat. Nanotechnol. 2016, 10, 3. doi: 10.2174/187221051001160322151412
-
[111]
Novoselov, K. S.; Jiang, Z.; Zhang, Y.; Morozov, S. V.; Stormer, H. L.; Zeitler, U.; Maan, J. C.; Boebinger, G. S.; Kim, P.; Geim, A. K. Science 2007, 315, 1379. doi: 10.1126/science.1137201
-
[112]
Terrones, M.; Botello-Méndez, A. R.; Campos-Delgado, J.; López-Urías, F.; Vega-Cantú, Y. I.; Rodríguez-Macías, F. J.; Elías, A. L.; Muoz-Sandoval, E.; Cano-Márquez, A. G.; Charlier, J.-C.; Terrones, H. Nano Today 2010, 5, 351. doi: 10.1016/j.nantod.2010.06.010
-
[113]
Berman, D.; Erdemir, A.; Sumant, A. V. Mater. Today 2014, 17, 31. doi: 10.1016/j.mattod.2013.12.003
-
[114]
Araujo, P. T.; Terrones, M.; Dresselhaus, M. S. Mater. Today 2012, 15, 98. doi: 10.1016/S1369-7021(12)70045-7
-
[115]
Botello-Méndez, A. R.; Declerck, X.; Terrones, M.; Terrones, H.; Charlier, J. C. Nanoscale 2011, 3, 2868. doi: 10.1039/c0nr00820f
-
[116]
Fan, Q.; Martin-Jimenez, D.; Ebeling, D.; Krug, C. K.; Brechmann, L.; Kohlmeyer, C.; Hilt, G.; Hieringer, W.; Schirmeisen, A.; Gottfried, J. M. J. Am. Chem. Soc. 2019, 141, 17713. doi: 10.1021/jacs.9b08060
-
[117]
Sun, Y.-P.; Fu, K.; Lin, Y.; Huang, W. Acc. Chem. Res. 2002, 35, 1096. doi: 10.1021/ar010160v
-
[118]
Iijima, S.; Ichihashi, T. Nature 1993, 363, 603. doi: 10.1038/363603a0
-
[119]
Dresselhaus, M. S.; Dresselhaus, G.; Eklund, P. C. Science of Fullerenes and Carbon Nanotubes:Their Properties and Applications, Acedemic Press, San Diego, 1996. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[120]
Sun, X.; Zaric, S.; Daranciang, D.; Welsher, K.; Lu, Y.; Li, X.; Dai, H. J. Am. Chem. Soc. 2008, 130, 6551. doi: 10.1021/ja8006929
-
[121]
Zandonella, C. Nature 2001, 410, 734. doi: 10.1038/35071183
-
[122]
Zhang, D.; Ryu, K.; Liu, X.; Polikarpov, E.; Ly, J.; Tompson, M. E.; Zhou, C. Nano Lett. 2006, 6, 1880. doi: 10.1021/nl0608543
-
[123]
Pushparaj, V. L.; Shaijumon, M. M.; Kumar, A.; Murugesan, S.; Ci, L.; Vajtai, R.; Linhardt, R. J.; Nalamasu, O.; Ajayan, P. M. Proc. Natl. Acad. Sci. 2007, 104, 13574. doi: 10.1073/pnas.0706508104
-
[124]
Odom, T. W.; Huang, J.-L.; Kim, P.; Lieber, C. M. Nature 1998, 391, 62. doi: 10.1038/34145
-
[125]
Ebbesen, T.; Takada, T. Carbon 1995, 33, 973. doi: 10.1016/0008-6223(95)00025-9
-
[126]
Kosaka, M.; Ebbesen, T. W.; Hiura, H.; Tanigaki, K. Chem. Phys. Lett. 1995, 233, 47. doi: 10.1016/0009-2614(94)01416-S
-
[127]
Dunlap, B. I. Phys. Rev. B 1992, 46, 1933. doi: 10.1103/PhysRevB.46.1933
-
[128]
Dunlap, B. I. Phys. Rev. B 1994, 49, 5643. doi: 10.1103/PhysRevB.49.5643
-
[129]
Wei, D.; Liu, Y. Adv. Mater 2008, 20, 2815. doi: 10.1002/adma.200800589
-
[130]
Bandaru, P. R.; Daraio, C.; Jin, S.; Rao, A. M. Nat. Mater. 2005, 4, 663. doi: 10.1038/nmat1450
-
[131]
Yao, Z.; Postma, H. W. C.; Balents, L.; Dekker, C. Nature 1999, 402, 273. doi: 10.1038/46241
-
[132]
Pan, B. C.; Yang, W. S.; Yang, J. Phys. Rev. B 2000, 62, 12652. doi: 10.1103/PhysRevB.62.12652
-
[133]
Charlier, J. C. Acc. Chem. Res. 2002, 35, 1063. doi: 10.1021/ar010166k
-
[134]
Feng, B.; Zhuang, X. Faraday Discuss. 2019, DOI: 10.1039/C9FD00115H.
-
[135]
Michl, J.; Thulstrup, E. W. Tetrahedron 1976, 32, 205. doi: 10.1016/0040-4020(76)87002-0
-
[136]
Sidman, J. W.; McClure, D. S. J. Chem. Phys. 1956, 24, 757. doi: 10.1063/1.1742604
-
[137]
Tétreault, N.; Muthyala, R. S.; Liu, R. S.; Steer, R. P. J. Phys. Chem. A 1999, 103, 2524. doi: 10.1021/jp984407q
-
[138]
Mitchell, D. R.; Gillispie, G. D. J. Phys. Chem. 1989, 93, 4390. doi: 10.1021/j100348a003
-
[139]
Murai, M.; Amir, E.; Amir, R. J.; Hawker, C. J. Chem. Sci. 2012, 3, 2721. doi: 10.1039/c2sc20615c
-
[140]
Yamaguchi, Y.; Ogawa, K.; Nakayama, K.-i.; Ohba, Y.; Katagiri, H. J. Am. Chem. Soc. 2013, 135, 19095. doi: 10.1021/ja410696j
-
[141]
Xin, H.; Ge, C.; Jiao, X.; Yang, X.; Rundel, K.; McNeill, C. R.; Gao, X. Angew. Chem., Int. Ed. 2018, 57, 1322. doi: 10.1002/anie.201711802
-
[142]
Nishimura, H.; Ishida, N.; Shimazaki, A.; Wakamiya, A.; Saeki, A.; Scott, L. T.; Murata, Y. J. Am. Chem. Soc. 2015, 137, 15656. doi: 10.1021/jacs.5b11008
-
[143]
Amir, E.; Murai, M.; Amir, R. J.; Cowart, J. S.; Chabinyc, M. L.; Hawker, C. J. Chem. Sci. 2014, 5, 4483. doi: 10.1039/C4SC02210F
-
[144]
Dias, J. R. J. Phys. Org. Chem.2007, 20, 395. doi: 10.1002/poc.1159
-
[145]
Yamaguchi, Y.; Takubo, M.; Ogawa, K.; Nakayama, K.-i.; Koganezawa, T.; Katagiri, H. J. Am. Chem. Soc. 2016, 138, 11335. doi: 10.1021/jacs.6b06877
-
[146]
Hou, I. C.-Y.; Shetti, V.; Huang, S.-L.; Liu, K.-L.; Chao, C.-Y.; Lin, S.-C.; Lin, Y.-J.; Chen, L.-Y.; Luh, T.-Y. Org. Chem. Front. 2017, 4, 773.
-
[147]
Sun, Q.; Hou, I. C.-Y.; Eimre, K.; Pignedoli, C. A.; Ruffieux, P.; Narita, A.; Fasel, R. Chem. Commun. 2019, 55, 13466.
-
[148]
Jessop, P. G. Green Chem. 2011, 13, 1391. doi: 10.1039/c0gc00797h
-
[149]
Ghasimi, S.; Landfester, K.; Zhang, K. A. ChemCatChem 2016, 8, 694. doi: 10.1002/cctc.201501102
-
[150]
Narayanam, J. M.; Stephenson, C. R. Chem. Soc. Rev. 2011, 40, 102. doi: 10.1039/B913880N
-
[151]
Ghasimi, S.; Bretschneider, S. A.; Huang, W.; Landfester, K.; Zhang, K. A. Adv. Sci. 2017, 4, 1700101. doi: 10.1002/advs.201700101
-
[152]
Kishida, K.; Horike, S.; Nakagawa, K.; Kitagawa, S. Chem. Lett. 2012, 41, 425. doi: 10.1246/cl.2012.425
-
[153]
Anderson Jr., A. G.; Steckler, B. M. J. Am. Chem. Soc. 1959, 81, 4941. doi: 10.1021/ja01527a046
-
[154]
Barman, S.; Khutia, A.; Koitz, R.; Blacque, O.; Furukawa, H.; Iannuzzi, M.; Yaghi, O. M.; Janiak, C.; Hutter, J.; Berke, H. J. Mater. Chem. A 2014, 2, 18823. doi: 10.1039/C4TA04393F
-
[155]
Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; Keeffe, M.; Yaghi, O. M. Science 2002, 295, 469. doi: 10.1126/science.1067208
-
[156]
Rowsell, J. L. C.; Spencer, E. C.; Eckert, J.; Howard, J. A. K.; Yaghi, O. M. Science 2005, 309, 1350. doi: 10.1126/science.1113247
-
[157]
Rowsell, J. L. C.; Eckert, J.; Yaghi, O. M. J. Am. Chem. Soc. 2005, 127, 14904. doi: 10.1021/ja0542690
-
[158]
Nakagawa, K.; Tanaka, D.; Horike, S.; Shimomura, S.; Higuchi, M.; Kitagawa, S. Chem. Commun. 2010, 46, 4258. doi: 10.1039/c0cc00027b
-
[159]
Holovics, T. C.; Robinson, R. E.; Weintrob, E. C.; Toriyama, M.; Lushington, G. H.; Barybin, M. V. J. Am. Chem. Soc. 2006, 128, 2300. doi: 10.1021/ja053933+
-
[160]
Sun, S.; Zhuang, X.; Wang, L.; Zhang, B.; Ding, J.; Zhang, F.; Chen, Y. J. Mater. Chem. C 2017, 5, 2223. doi: 10.1039/C6TC05362A
-
[161]
Yang, C.; Schellhammer, K. S.; Ortmann, F.; Sun, S.; Dong, R.; Karakus, M.; Mics, Z.; Lffler, M.; Zhang, F.; Zhuang, X.; Cánovas, E.; Cuniberti, G.; Bonn, M.; Feng, X. Angew. Chem., Int. Ed. 2017, 56, 3920. doi: 10.1002/anie.201700679
-
[1]
-

图 4 石墨烯中的拓扑缺陷: (a) 5-7位错; (b) 5-8-5位错; (c) 55-77簇状拓扑缺陷; (d) 555-777簇状拓扑缺陷; (e) 闭环的花形拓扑缺陷; (f) 非周期晶界; (g) θ=32.2°周期晶界; (h) θ=0°周期平动晶界[35]
Figure 4 Topological defects in graphene: (a) 5-7 dislocation; (b) 5-8-5 dislocation; (c) 55-77 clustered topological defect; (d) 555-777 clustered topological defect; (e) closed loop flower topological defect; (f) aperiodic grain boundary; (g) θ=32.2° periodic grain boundary; (h) θ=0° periodic translational grain boundary[35]
图 7 (a)构象熵的理论模型; (b)含有晶格空位的单原子晶体的构象熵
Figure 7 (a) Theoretical model of conformational entropy; (b) conformational entropy of monatomic crystal with lattice vacancies
图 10 (a), (b)分别为(1, 0)和(1, 1)位错的原子结构; (c), (d)分别为θ=21.8° (LAGB I)和θ=32.2° (LAGB II)对称的大角度晶界的原子结构; (e)各种平面(实心符号)和弯曲(空心符号)晶界结构的单位长度γ的晶界能量与取向角θ的关系[32]
Figure 10 (a), (b) Atomic structures of (1, 0) and (1, 1) dislocations, respectively; (c), (d) atomic structures of the θ=21.8° (LAGB I) and the θ=32.2° (LAGB II) symmetric large-angle grain boundaries, respectively; (e) grain-boundary energy per unit length γ as a function of misorientation angle θ for various flat (filled symbols) and buckled (open symbols) grain-boundary structures[32]
图 11 (a)~(d)由一系列Stone-Wales键旋转形成的石墨烯中的位错; (e), (f)分别为一个和四个Stone-Wales键旋转得到的DFT原子模型(步骤1和步骤4); (g)每一步的形成能[96]
Figure 11 (a)~(d) Dislocations in graphene formed by rotations of a series of Stone-Wales bond; (e), (f) atomistic models obtained within DFT for one and four Stone-Wales bond rotations (steps 1 and 4); (g) Formation energy at each step[96]

图 13 (a)~(c)分别为d5d7, 585和t5t7拓展缺陷的模拟隧道扫描显微镜图像; (d)含有5/7元环的Phagraphene和Phagraphene的恒高原子力显微镜图像[115, 116]
Figure 13 Simulated STM images for the (a) d5d7, (b) 585, and (c) t5t7 extended defects; (d) phagraphene with seVeral 5/7-membered rings and constant-height atomic force microscopy image of phagraphene[115, 116]

图 14 (a) 二维石墨烯包裹成管状的示意图; (b) 5-77-5缺陷; (c)图(b)中所示四种5-77-5 缺陷的形成能和半径之间的关系; (d)管中离轴5-77-5缺陷的形成能与不同手性的关系[132, 133]
Figure 14 (a) Schematic diagram of graphene undergoing curling to form carbon nanotubes; (b) 5-77-5 defect; (c) The formation energy of 5-77-5 defect vs. radius for the four groups as shown in (b); (d) Formation energy of the off-axis 5-77-5 defect in tubes vs. different chiralities[132, 133]

图 16 (a)线性的聚薁聚合物; (b) 2, 6-聚薁的非接触原子力显微镜图像; (c)薁分子的前线分子轨道; (d) 2, 6-聚薁的能带结构; (e)具有不同连接模式的聚薁的能带结构[144, 147]
Figure 16 (a) Polyazulene polymers in a linear conformation; (b) nc-AFM image of a 2, 6-polyazulene chain; (c) the shape of frontier orbitals of an azulene molecule; (d) band structure of 2, 6-polyazulene; (e) Band structures of polyazulenes with different connectivity patterns[144, 147]
图 17 (a) P-Az-1和P-Az-2的结构; (b)价带和导带的位置; (c)通过在级联催化循环中使用P-Az-1h或P-Az-2h以及FeIII或CuII作为助催化剂, 并使用甲酸作为水中的额外电子供体, 将CrVI光催化还原为CrIII; (d) P-Az-B的结构以及HOMO和LUMO谱带位置[149, 151]
Figure 17 (a) Structures of P-Az-1 and P-Az-2; (b) positions of the valence bands and conduction bands; (c) photocatalytic reduction of CrVI to CrIII by using P-Az-1h or P-Az-2h, FeIII or CuII as co-catalyst in a cascade catalytic cycle, and formic acid as an extra electron donor in water; (d) structure, HOMO and LUMO band positions of P-Az-B[149, 151]

图 18 (a) CID-13: [Zn(azdc)(bpy)]n, azdc=1, 6-薁二甲酸酯; bpy=4, 4'-联吡啶; (b) CID-3: [Zn(ndc)(bpy)]n, ndc=2, 7-萘二甲酸酯; (c) MOF650: Zn4O(2, 6-azd)3; (d) IRMOF-8: Zn4O(ndc)3[152, 154, 158]
Figure 18 (a) CID-13: [Zn(azdc)(bpy)]n, azdc=1, 6-azulenedicarbo- xylate; bpy=4, 4'-bipyridyl; (b) CID-3: [Zn(ndc)(bpy)]n, ndc=2, 7-naphthalenedicarboxylate; (c) MOF650: Zn4O(2, 6-azd)3; (d) IRMOF-8: Zn4O(ndc)3[152, 154, 158]
图 19 (a) 制造基于BCCF分子的二极管的示意图; (b) PDIBA-5和(c) DIBA的I-V曲线的Fowler-Nordheim图; (d)不同扫描速率下微超级电容器的叠层电容;(e)模拟的能带结构和状态密度; (f)越过费米能级的较低能带的电荷密度[160, 161]
Figure 19 (a) Schematic fabrication of BCCF-based molecular diode. Fowler-Nordheim plots for I-V curves of junctions based on (b) PDIBA-5 and (c) DIBA; (d) stack capacitance of the MSCs at different scan rates; (e) simulated band structure and density of states (DOS); (f) charge density of the lower band crossing the Fermi level[160, 161]
表 1 部分已知碳材料的标准摩尔熵[97, 98, 100-102]
Table 1. The standard molar entropy of some typical carbon materials[97, 98, 100-102]
Standard
Enthalpy of
Formation/
(J•mol-1•K-1)Standard
Gibbs Energy
of Formation/
(J•mol-1•K-1)Standard Molar
Entropy/
(J•mol-1•K-1)Diamond +1.895 +2.900 2.377 Impurity-free
nanocrystalline
diamond— — 3.08 Nanocrystalline
diamond— — 3.83 diamond — — 3.83 Graphite 0 0 5.740 Multi-walled
carbon
nanotube— — 6.439 Fullerene-C60 — — 27.3 Gaseous carbon +716.68 +671.26 158.10  下载: 导出CSV
下载: 导出CSV
-



















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 36
- 文章访问数: 10566
- HTML全文浏览量: 1534
