表 1
反应条件的优化
Table 1.
Optimization of reaction conditions

硅卡宾(R2Si׃, silylene)是卡宾的同族类似物, 它具有空轨道和孤对电子而表现出Lewis酸性和碱性.与卡宾不同的是, 硅卡宾的基态为单线态, Lewis酸性很强导致其在常规条件下非常不稳定[1].近年来研究发现[2], 硅卡宾可以通过与Lewis碱配位形成相对稳定的化合物(R2Si←Donor), 并表现出很强的给电子能力及亲核性, 因此, 硅卡宾化学近年来得到了迅速发展.虽然硅卡宾的金属配合物已有不少报道[2, 3], 但与卡宾及膦配体配合物相比, 其类型及数目仍然非常有限, 特别是用于调控催化反应的报道非常少见[3-6].硅是地壳中最丰富的元素之一, 同时有机硅化合物通常具有很好的生物相容性, 因此, 发展有机硅的配合物化学对可持续化学及发展新颖的化学转化无疑具有重要的价值.
有机硼是重要的C—C偶联中间体, 烯烃及炔烃的硼氢化反应是制备有机硼最直接的方法[7].通过金属催化的方法可以很好地调控反应的区域及立体选择性, 因此, 催化硼氢化反应已成为选择性合成有机硼化合物最高效及原子经济性合成方法, 受到大家广泛关注.近年来, 铁催化的硼氢化反应也备受关注. 2013年, Enthaler等[8]报道了Fe2(CO)9(A)催化的炔烃硼氢化反应, 主要生成E式构型的产物.同年, Thomas团队[9]利用吡啶二亚胺配体及其衍生物与铁形成的络合物(B)在活化剂的存在下催化炔烃及烯烃的硼氢化反应. 2017年, Nishibayashi团队[10]报道了吡咯双膦螯合型PNP配体的铁氢络合物(C)催化的末端炔烃硼氢化反应, 同样生成E式构型的产物. 2020年, Findlater团队[11]利用含BIAN (bis(2, 6-diisopropylaniline)acenaphthene)配体的铁配合物在助催化剂的存在下, 可高效地催化炔烃及烯烃的硼氢化反应.
我们在前期研究发现, 有机硅卡宾配体可以稳定铁的氮气配合物D(Chart 1), 并可以很好调控铁的氧化态变化.进一步研究发现, D可以催化氮气的硅化反应, 表现出较高的催化活性[12].为了进一步研究该配合物的功能, 我们研究了D在催化炔烃硼氢化反应中的应用, 发现该铁配合物对硼氢化反应具有很好的调控能力.
|
|
我们以苯乙炔的硼氢化反应作为模板, 对催化反应的条件进行了优化.首先以配合物D为催化剂, 在nD/ n苯乙炔=5%的情况下, 对反应温度, 硼化试剂及溶剂等条件进行了优化(表 1), 反应进程可通过1H NMR及GC-MS进行监测.从表 1可以看出, 反应在常温条件下(Entries 5 and 6)收率很低, 当温度升到80 ℃时, 可以达到几乎定量的收率(Entries 1 and 7).溶剂对反应也有很大的影响, 反应在甲苯中进行明显优于在四氢呋喃及正己烷中(Entries 1~3).值得注意的是, 配合物D在氮气氛围中催化活性及选择性均比在氩气氛围中更好(Entries 1与4), 这很有可能是含有氮气配体的催化剂D在氮气气氛下相对比较稳定的缘故.当用儿茶酚硼烷(HBCat)代替频哪醇硼烷(HBPin)作硼氢化试剂时, 该体系的选择性有所提高(表 1, 比较Entries 1与7以及Entries 5与6).但催化剂用量降低时(表 1, 比较Entries 7与8)转化率明显降低.所有实验表明, 硼氢化产物以E式构型为主, 其E/Z比例为最高可达97:3.基于这些结果, 我们以D为催化剂, nD/n苯乙炔=5%, 80 ℃, 氮气气氛及甲苯溶剂中, 以HBCat为硼化试剂对反应底物进行了扩展.
 下载:
导出CSV
下载:
导出CSV
 |
|||||||
| Entrya | Borane | Solvent | T/℃ | Gas | Yield/% | E/Z | |
| 1 | HBPin | toluene | 80 | nitrogen | > 99 | 95:5 | |
| 2 | HBPin | THF | 80 | nitrogen | 64 | 94:6 | |
| 3 | HBPin | n-hexane | 80 | nitrogen | 60 | 93:7 | |
| 4 | HBPin | toluene | 80 | argon | 86 | 93:7 | |
| 5 | HBPin | toluene | 25 | nitrogen | 38 | 85:15 | |
| 6 | HBcat | toluene | 25 | argon | 36 | 95:5 | |
| 7 | HBCat | toluene | 80 | nitrogen | > 99 | 97:3 | |
| 8b | HBCat | toluene | 80 | nitrogen | 86 | 97:3 | |
| a 0.20 mmol of alkynes and borane, 1 mL of solvent. b 0.005 mmol of catalyst. | |||||||
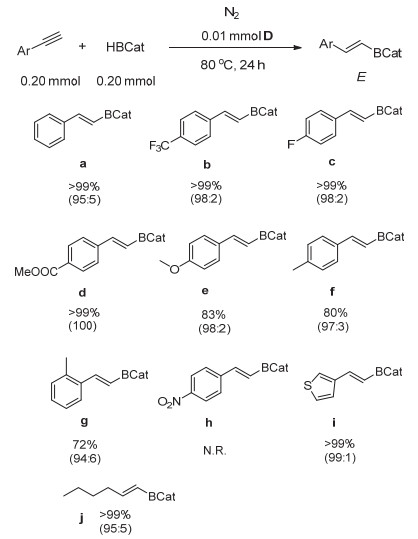
我们对不同取代基的末端炔烃进行了研究, 如图 1所示, 反应无论是对苯基和芳杂环取代还是烷基取代的炔烃都显示很好的区域及立体选择性(图 1, a、i、j和k).此外, 我们研究了苯环上取代基的电子效应对反应的影响:当苯基对位上带有吸电子基团如三氟甲基、氟及酯基时, 反应速率更快, 产物选择性更好(图 1, b、c、d); 当苯环带有给电子基团如甲氧基、甲基时反应转化率明显降低(图 1, e、f和g).但硝基苯取代的炔烃不反应, 这可能是由于硝基会使催化剂分解而造成的(图 1, h).另外, 底物位阻增加也对催化反应有明显的抑制(图 1, g).该催化体系对噻吩取代的炔烃显示更好的活性及选择性(图 1, i);对于烷基取代炔烃反应活性高, 但产物选择性稍有降低(图 1, k).
进一步研究发现, 该催化反应不适用于内炔烃, 这很有可能是由于内炔烃有较大的空间位阻的缘故.
尽管铁催化的硼氢化反应有一定数量的报道[8-11, 13], 但对其反应机理一直存在较大的分歧.相当一部分的铁催化的硼氢化反应需要额外的强碱来对铁配合物进行活化, 其目的是原位产生活性低价态铁或铁的氢化物[9, 11, 13a].已有证据表明:低价态铁及铁的氢化物都有可能是反应的关键中间体, 但反应途径不同.到目前为止, 推测的低价态铁对B—H键的氧化加成以及与炔烃形成配合物的初始机理都有报道.另一方面, 也有研究表明:炔烃对Fe—H及Fe—B键的插入反应都有可能是其催化循环的关键步骤[13].
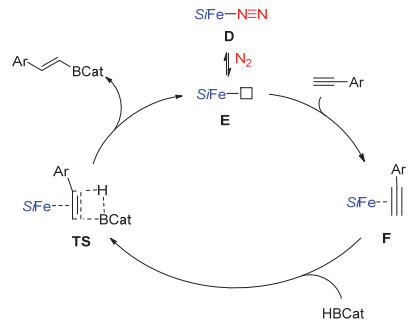
我们分别尝试了D与HBCat及炔烃的反应, 前者反应复杂, 没能分离和检测到明确的产物.但与炔烃反应时发现发生了炔烃的三聚, 形成了多取代苯.通过对铁的硅卡宾配合物的电子结构分析, 我们发现硅卡宾具有很强的接受电子能力, 与CO一样是典型的π-酸配体.因此, 配合物D与B—H键进行氧化加成反应会受到硅卡宾配体的抑制.基于以上分析, 我们认为D催化的硼氢化反应的初始步骤很有可能是通过铁中心对炔烃的活化引发的(图 2).催化剂D首先失去配位的氮气产生不饱和配合物E, 继而与炔烃进行配位形成中间体F.炔烃由于配位被活化进而与B—H键进行加成形成产物脱去,同时,再次生成催化剂E.由于炔烃配位, 该反应的区域及立体选择性主要通过空间位阻来进行调控, 硼原子主要加成在位阻小的末端.由于硅卡宾-亚胺配体有很大的位阻, 内炔烃很难与E进行配位, 因此反应很难发生.
以硅卡宾-亚胺双齿配体与铁形成的氮气配合物为催化体系, 实现了末端炔烃与儿茶酚硼烷的选择性硼氢化反应.反应产物同时具有较高的收率及E式选择性.与一些已有铁催化剂相比[9, 11, 13a], 该体系无需碱添加剂, 对芳基及烷基取代的末端炔烃显示相同的区域及立体选择性, 表明空间位阻起主要作用.下一步我们将进一步研究硅卡宾配合物与底物的作用本质, 进一步认识硅卡宾对金属反应性的调控的规律, 发展新的转化过程, 进一步拓展和发展硅卡宾化学.
一般反应过程如下:将配合物D (0.006 g, 0.01 mmol)和甲苯(1 mL)加到15 mL带磁子的封管中, 形成溶液.然后将炔烃(0.20 mmol)及儿茶酚硼烷(0.02 g, 0.20 mmol)依次加入体系中. 80 ℃下反应24 h后, 冷至室温, 抽干溶剂.将粗产品溶于CDCl3中进行1H NMR测试以确定反应转化率.将粗产品溶于乙酸乙酯经滤膜过滤后进行GC-MS分析来确定产物构型比例.
产物分离:待反应完成后, 旋干反应液, 通过柱层析分离得到产物.
(a) Trinquier, G. J. Am. Chem. Soc. 1990, 112, 2130. (b) Apeloig, Y.; Pauncz, R.; Miriam, K.; West, R. Steiner, W.; Chapman, D. Organometallics 2003, 22, 3250. (c) Sasamori, T.; Tokitoh, N. In Encyclopedia of Inorganic Chemistry II, Ed.: King, R. B., John Wiley & Sons: Chichester, U.K., 2005, p. 1698.
For selected papers: (a) Blom, B.; Stoelzel, M.; Driess. M. Chem. Eur. J. 2013, 19, 40. (b) Blom, B.; Gallego, D.; Driess, M. Inorg. Chem. Front. 2014, 1, 134. (c) Zhou, Y.-P.; Driess. M. Angew. Chem. Int. Ed. 2019, 58, 3715. (d) Raoufmoghaddam, S.; Zhou, Y. P.; Wang, Y.; Driess, M. J. Organomet. Chem. 2017, 829, 2. (e) Haaf, M.; Schmedake, T. A.; West, R. Acc. Chem. Res. 2000, 33, 704. (f) Yao, S.; Xiong, Y.; Driess, M. Organometallics 2011, 30, 1748. (g) Cui, H.; Teng, P.; Zhang, E.; Lu, J.; Zhang, F.; Wu, M. Chin. J. Chem. 2017, 35, 401. (h) Cui, H.; Cui, C. Chin. J. Org. Chem. 2016, 36, 626 (in Chinese). (崔海燕, 崔春明, 有机化学, 2016, 36, 626.)
(a) Jutzi, P.; Kanne, D.; Krfiger, C. Angew. Chem. Int. Ed. 1986, 25, 164. (b) Blom, B.; Stoelzel, M.; Driess. M. Chem. Eur. J. 2013, 19, 40. (c) Zhou, L.; Li, Y.; Lin, F.; Tian, D.; Lei, Q.; Fang, W.; Xie, H. Chin. J. Org. Chem. 2015, 35, 698 (in Chinese). (周莉, 李阳, 林芙蓉, 田迪英, 雷群芳, 方文军, 谢湖均, 有机化学, 2015, 35, 698.) (d) Wang, L.; Guo, J.; Li, Y.; Su, Y.; Liu, J.; Li, Y.; Wang, S.; Shimada, S.; Huang, W. Chin. J. Chem. 2017, 35, 507.
(a) Troadec, T.; Prades, A.; Rodriguez, R.; Mirgalet, R.; Baceiredo, A.; Saffon-Merceron, N.; Branchadell, V.; Kato, T. Inorg. Chem. 2016, 55, 8234. (b) Iimura, T.; Akasaka, N.; Iwamoto, T. Organometallics 2016, 35, 4071. (c) Iimura, T.; Akasaka, N.; Kosai, T.; Iwamoto, T. Dalton Trans. 2017, 46, 8868.
(a) Cabeza, J. A.; García-Á lvarez, P.; González-Á lvarez, L. Chem. Commun. 2017, 53, 10275. (b) Ren, H.; Zhou, Y.-P.; Bai, Y.; Cui, C.; Driess, M. Chem. Eur. J. 2017, 23, 5663. (c) Brück, A.; Gallego, D.; Wang, W.; Irran, E.; Driess, M.; Hartwig, J. F. Angew. Chem. Int. Ed. 2012, 51, 11478. (d) Zhou, Y.-P.; Raoufmoghaddam, S.; Szilvási, T.; Driess, M. Angew. Chem. Int. Ed. 2016, 55, 12868. (e) Wang, Y.; Kostenko, A.; Yao, S.; Driess, M. J. Am. Chem. Soc. 2017, 139, 13499.
(a) Fürstner, A.; Krause, H.; Lehmann, C. W. Chem. Commun. 2001, 2372. (b) Khoo, S.; Cao, J.; Yang, M.-C.; Shan, Y.-L.; Su, M.-D.; So, C.-W. Chem. Eur. J. 2018, 24, 14329. (c) Zhang, M.; Liu, X.; Shi, C.; Ren, C.; Ding, Y.; Roesky, H. W. Z. Anorg. Allg. Chem. 2008, 634, 1755. (d) Gallego, D.; Brgck, A.; Irran, E.; Meier, F.; Kaupp, M.; Driess, M.; Hartwig, J. F. J. Am. Chem. Soc. 2013, 135, 15617. (e) Tan, G.; Enthaler, S.; Inoue, S.; Blom, B.; Driess, M. Angew. Chem. Int. Ed. 2015, 54, 2214. (f) Qi, X.; Sun, H.; Li, X.; Fuhr, O.; Fenske, D. Dalton Trans. 2018, 47, 2581. (g) Mo, Z.; Kostenko, A.; Zhou, Y.-P.; Yao, S.; Driess, M. Chem. Eur. J. 2018, 24, 14608. (h) Schmidt, M.; Blom, B.; Szilvási, T.; Schomä cker, R.; Driess, M. Eur. J. Inorg. Chem. 2017, 1284. (i) Someya, C. I.; Haberberger, M.; Wang, W.; Enthaler, S.; Inoue, S. Chem. Lett. 2013, 42, 286.
(a) Bracher, F.; Litz, T.; J. Prakt. Chem./Chem.-Ztg. 1996, 338, 386. (b) Brown, H. C.; Chen, J. J. Org. Chem. 1981, 46, 3978. (c) Brown, H. C.; Rao, B. S. J. Am. Chem. Soc. 1959, 81, 6423. (d) Crockett, M. P.; Tyrol, C. C.; Wong, A. S.; Li, B.; Byers, J. A. Org. Lett. 2018, 20, 5233. (e) Hartwig, J. F. Acc. Chem. Res. 2011, 45, 864. (e) Martin, R.; Buchwald, S. L. Acc. Chem. Res. 2008, 41, 1461. (f) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457.
Haberberger, M.; Enthaler, S. Chem. Asian J. 2013, 8, 50. doi: 10.1002/asia.201200931
Greenhalgh, M. D.; Thomas, S. P. Chem. Commun. 2013, 49, 11230. doi: 10.1039/c3cc46727a
Nakajima, K.; Kato, T.; Nishibayashi, Y. Org. Lett. 2017, 19, 4323. doi: 10.1021/acs.orglett.7b01995
Singh, A.; Shafiei-Haghighi, S.; Smith, C. R.; Unruh, D. K.; Findlater, M. Asian J. Org. Chem. 2020, 9, 416. doi: 10.1002/ajoc.201900615
Bai, Y.; Zhang, J.; Cui, C. Chem. Commun. 2018, 54, 8124. doi: 10.1039/C8CC03734E
(a) Docherty, J. H.; Peng, J.; Dominey, A. P.; Thomas, S. P. Nat. Chem. 2017, 9, 595. (b) Gorgas, N.; Alves, L. G.; Stöger, B.; Martins, A. M.; Veiros, L. F.; Kirchner, K. J. Am. Chem. Soc. 2017, 139, 8130.
表 1 反应条件的优化
Table 1. Optimization of reaction conditions
|
|||||||
| Entrya | Borane | Solvent | T/℃ | Gas | Yield/% | E/Z | |
| 1 | HBPin | toluene | 80 | nitrogen | > 99 | 95:5 | |
| 2 | HBPin | THF | 80 | nitrogen | 64 | 94:6 | |
| 3 | HBPin | n-hexane | 80 | nitrogen | 60 | 93:7 | |
| 4 | HBPin | toluene | 80 | argon | 86 | 93:7 | |
| 5 | HBPin | toluene | 25 | nitrogen | 38 | 85:15 | |
| 6 | HBcat | toluene | 25 | argon | 36 | 95:5 | |
| 7 | HBCat | toluene | 80 | nitrogen | > 99 | 97:3 | |
| 8b | HBCat | toluene | 80 | nitrogen | 86 | 97:3 | |
| a 0.20 mmol of alkynes and borane, 1 mL of solvent. b 0.005 mmol of catalyst. | |||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们