
图 1.
TiBP或t-Bu-P4催化的甲基丙烯酸甲酯的基团转移聚合
Figure 1.
Group transfer polymerization of MMA mediated by TiBP or t-Bu-P4

有机化学家应用有机小分子化合物作为催化剂进行化学反应可追溯至100多年前.尽管该领域历史悠久, 但有机催化的概念直到2000年才被首次明确提出[1].此后, 有机催化领域快速发展, 并成为有机合成领域的研究热点.与金属催化剂比较, 有机催化剂只含有碳、氢、氧、氮、硫、磷等有机元素, 故具有更好的稳定性、更低的毒性及价格、更简便的分子设计等优点而被广泛应用.例如, 仅在2000年至2008年间就有超过2000篇论文报道了超过150种有机催化反应类型, 诸如丸善催化剂催化的烷基化反应、脯氨酸衍生物催化的曼尼希反应以及噁唑硼烷酮催化的向山羟醛缩合反应等[2].国内不少学者在有机催化不对称合成方面亦颇有成就[3-6].
在有机催化聚合方面, Hedrick等[7]于2001年首次报道了应用4-N, N-二甲基氨基吡啶催化L-丙交酯的开环聚合.此后, 大量的有机碱与酸相继被用于环状单体, 如取代的环氧烷、环酯、环酰氨、环碳酸酯等, 进行开环聚合的高效催化剂.其中, 较常见的有机酸有硫脲[8-10]、羧酸[11-13]、有机磷酸[14]、有机磺酸[15, 16]等; 较常见的有机碱有吡啶[7, 17, 18]、N-杂环卡宾[19-25]及磷腈碱[18, 26, 27]等.目前, 有机催化剂在高分子合成方面的应用主要集中在开环聚合与基团转移聚合两大聚合方法.有机催化开环聚合在近二十年被大量报道, 无论是在可聚单体、分子量及分子量分布控制、共聚、聚合机理、聚合物分子设计等各方面均有深入研究.相比之下, 采用有机化合物催化乙烯基单体的连锁聚合一直以来是有机催化聚合领域的一项极具挑战的工作.这也是这些年来本研究组所追求的一个重要目标, 并一直致力于丙烯酸衍生类单体的有机催化基团转移聚合.
基团转移聚合作为一种针对丙烯酸衍生类单体的有效可控聚合方法, 自1983年由杜邦公司开发以来[28], 已有36年的历史.其早期的开发是为了解决丙烯酸衍生类单体在阴离子聚合的实际生产中所需的低温问题.相较于阴离子聚合中的高活性烯醇负离子活性种, 基团转移聚合的活性种为以中性分子形式存在的硅烷基烯酮缩醛.活性末端的电中性一方面可降低其活性, 另一方面可避免该活性种由于极性过高而引起在溶剂中的聚集以及由于活性种的高活性所导致的链转移副反应.较之于其它聚合方法, 基团转移聚合所需的聚合条件温和、操作简便、副反应少, 特别适合于无结构缺陷的丙烯酸衍生类聚合物的制备.考虑到基团转移聚合的链引发与链增长在反应机理上均为向山-迈克尔这一基础有机化学反应, 原则上能催化该有机反应的催化剂均可催化基团转移聚合. 2007年以前, 亲核性的阴离子被作为基团转移聚合的路易斯碱型催化剂[29], 如Me3SiF2-[28, 30, 31]、HF2-[28, 30-32]、CN-[24, 30, 33, 34]、N3-[28, 33]、F-[31, 33, 34]、氧负离子[35-37]等.这些亲核性阴离子作为催化剂时通常配有一立体位阻较大的对阳离子, 以增进催化剂在溶剂中的溶解性及调节离子对结合的紧密程度.另一方面, 某些金属或过渡金属化合物被用于基团转移聚合的路易斯酸型催化剂, 如卤化锌[32, 34, 8-39]、有机铝化合物[32, 34, 40]、HgI2/R3SiI[29, 41-46]、CdI2/R3SiI[39]、Yb(OTf)3[47]及Sc(OTf)3[47]等.
作为20世纪80年代初出现的极少数的活性聚合方法, 基团转移聚合在1983~1995年间被广泛报道.在此期间, 潘容华等在这方面做了大量的工作.在1995~2007年间, 由于活性/可控自由基聚合的出现该聚合方法极少见诸于报道. 2007年以来, 有机小分子催化剂在该领域的应用极大地促进了基团转移聚合的发展.为区别传统法, 将应用有机小分子催化剂的基团转移聚合方法命名为“有机催化基团转移聚合”[48].在该方面, Waymouth组、Chen组以及Taton组分别做出了贡献. Waymouth等和Taton等分别报道了N-杂环卡宾催化的基团转移聚合[49-56], Taton等[57]还另外报道了有机膦化合物. Chen组[58-63]主要报道三苯基甲基阳离子盐及List磺酰胺类有机催化体系.本综述主要围绕作者近年来在该领域的工作, 从有机强碱催化的基团转移聚合、有机强酸催化的基团转移聚合、基于氢硅烷的新型有机催化基团转移聚合以及聚合机理4个方面对有机催化基团转移聚合的最新进展进行论述.
传统碱催化的基团转移聚合法所用的碱为亲核性阴离子, 该法对于甲基丙烯酸酯类单体的聚合比较有效, 但因亲核性阴离子易与单体、聚合物、溶剂等发生各种亲核取代反应而导致聚合物产物的分子量偏低、分子量分布变宽且化学结构上易产生缺陷等诸多问题.针对上述阴离子催化剂的过强亲核性, 于是弱亲核性的有机强碱被用于解决该问题.所用的有机强碱包括Verkade碱, 如[2, 5, 8, 9-四氮杂-1-磷杂双环[3.3.3]十一烷-2, 8, 9-三(1-甲基乙基)](TiBP), 及磷腈碱, 如1-叔丁基- 4, 4, 4-三(二甲氨基)-2, 2-二[三(二甲氨基)-正膦亚基氨基]-2Λ5, 4Λ5-连二(磷氮基化合物)(t-Bu-P4)[64].较之亲核性阴离子, 有机强碱的优势在于: (1)催化能力更强, (2)在有机溶剂中溶解性更好, (3)由于位阻大不与单体或聚合物中的极性官能团发生亲核取代反应, 可避免因催化剂的亲核性所导致的各种副反应.有机催化剂的这些特征, 使其在甲基丙烯酸酯类单体的聚合中具有更大优势.
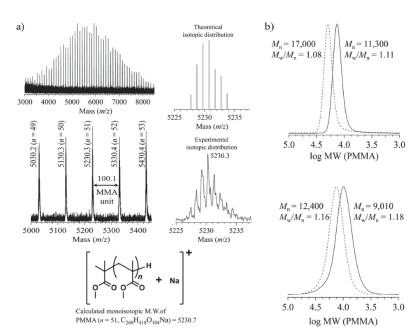
以TiBP或t-Bu-P4为催化剂、1-甲氧基-1-三甲基硅烷基-2-甲基-1-丙烯(SKAMe)为引发剂、在THF或甲苯中聚合甲基丙烯酸甲酯(MMA), 短时间内便定量得到分子量高, 分散度低的聚甲基丙烯酸酯(PMMA, 如图 1所示).以TiBP为催化剂时, PMMA的数均分子量可控制在6.5~55.9 kg•mol-1, 分散度在1.05~1.14范围内.以t-Bu-P4为催化剂时, 可控制分子量在6.5~109.6 kg• mol-1, 分散度在1.15~1.20范围内.亲核性阴离子催化的基团转移聚合法得到的PMMA的分子量未曾超过70 kg•mol-1, 且聚合物的化学结构常因副反应产生缺陷.从这一点来看, 有机强碱催化的基团转移聚合具有明显的优势.进一步对所得聚合物结构进行基质辅助激光解析电离飞行时间质谱(MALDI-TOF MS)解析以及扩链聚合结果(图 2)表明通过有机强碱催化的基团转移聚合法得到的聚合物与目标产物具有完全相同的化学结构, 聚合过程呈现无链转移与链终止的活性聚合特点.此外、研究还发现此方法对不含活泼质子的甲基丙烯酸酯具有普适性.此外、中等碱性的TiBP还适合某些丙烯酸酯, 如丙烯酸叔丁酯的聚合.
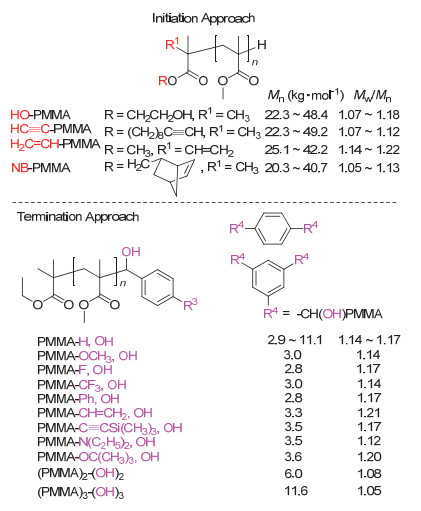
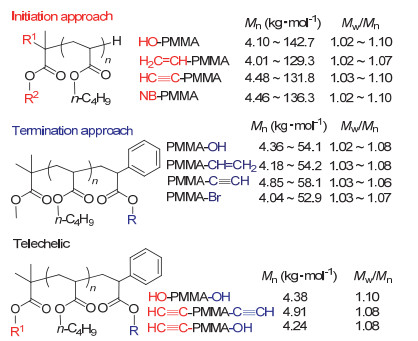
基于该方法的活性特点, 采取引发和终止法可精准制备末端官能化聚合物.引发法指在SKA引发剂中引入官能团, 经聚合得到起始末端官能化的聚合物.通过引发法, 合成了起始末端含羟基、乙烯基、乙炔基以及降冰片烯基的PMMA (Figure 3)[65].终止法是指在单体聚合结束后利用聚合物的活性SKA末端继续与过量的官能化终止剂定量地进行迈克尔加成或羟醛缩合反应而得到终止末端官能化的聚合物[66].例如, MMA完全聚合后向聚合体系中加入相对于活性末端物质的量10~20倍的苯甲醛或其衍生物, 经反应可得终止末端单、双、或多官能化的聚合物.如应用多醛基化合物, 则得到多羟基官能化的星型聚合物.以上末端官能化聚合物的结构由MALDI-TOF MS测定, 均与理论设计的聚合物具有完全一致的化学组成与结构.该方法特点在于可实现起始末端及终止末端的定量官能化, 同时所得聚合物具有极其窄的分子量分布, 这在除阴离子聚合之外的其他方法中难以实现.
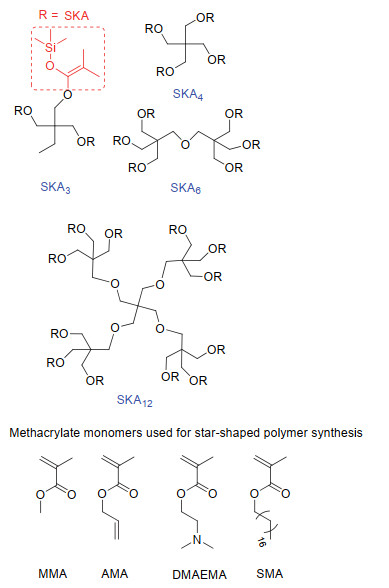
除聚甲基丙烯酸酯的末端官能化之外, 当所使用的引发剂为多官能的星型引发剂时, 可简便地制备无化学结构缺陷的星型聚合物[67-69].如图 4所示, 以t-Bu-P4为催化剂, 以SKA3、SKA4、SKA6或SKA12为引发剂, 进行甲基丙烯酸酯的聚合, 可分别得到具目标分子量且分布极窄的3、4、6或12臂星型聚甲基丙烯酸酯(表 1).例如, 所得星型PMMA的绝对分子量可高达256 kg•mol-1, 且分散度均低于1.17.聚合物臂链的切断实验表明聚合过程中星型聚合物的臂链进行了均匀的链增长.此方法对不含活泼氢的甲基丙烯酸酯单体, 如甲基丙烯酸2-(N, N-二甲基氨基)乙酯(DMAEMA)、甲基丙烯酸异丙酯(AMA)以及甲基丙烯酸十八烷酯(SMA)等, 均具有普适性.
 下载:
导出CSV
下载:
导出CSV
| Star-shaped polymer | Star-shaped initiator | Mw(expt.)/ (kg·mol-1)a |
Mw/Mnb |
| PMMA3 | SKA3 | 3.9~132.0 | 1.07~1.14 |
| PMMA4 | SKA 4 | 5.5~130.3 | 1.07~1.17 |
| PMMA6 | SKA 6 | 7.5~131.6 | 1.06~1.15 |
| PMMA12 | SKA 12 | 28.2~264.9 | 1.06~1.12 |
| PDMAEMA3 | SKA3 | 19.3~160c | 1.20~1.27d |
| PDMAEMA4 | SKA 4 | 24.0~254c | 1.16~1.32d |
| PDMAEMA6 | SKA 6 | 30.2~296c | 1.09~1.27d |
| PDMAEMA12 | SKA 12 | 34.3~419c | 1.13~1.39d |
| PAMA4 | SKA 4 | 13.6~21.2 | 1.11~1.16 |
| PSMA4 | SKA 4 | 34.5~65.2 | 1.06~1.08 |
| a Absolute molar masses determined in THF by SEC equipped with an multi-angle laser light scattering (MALS) detector. b Dispersity determined by SEC equipped with an RI detector in THF on the basis of PMMA standards. c Absolute molar masses determined by MALS-SEC in DMF containg 0.01 mol•L-1 LiCl. d Polydispersity determined by SEC equipped with an RI detector in DMF containg 0.01 mol•L-1 LiCl on the basis of poly(N, N-dimethy- lacrylamide) standards. | |||
传统酸催化的基团转移聚合法应用具有路易斯酸性的金属或过渡金属化合物活化单体, 但因其路易斯酸性过低导致在聚合中需使用单体物质的量10%~20%才能使聚合顺利进行.查阅资料可知, 传统路易斯酸催化的基团转移聚合法得到的聚合物分子量未曾超过4.0 kg•mol-1.鉴于传统酸的缺点, 我们将目光聚焦于有机强酸.有机强酸可以是有机强Brönsted酸, 如双(三氟甲烷)磺酰亚胺(HNTf2)及2, 3, 4, 5, 6-五氟苯基双(三氟甲磺酰基)甲烷(C6F5CHTf2)等, 也可以是有机路易斯酸, 如N-(三甲基硅基)双(三氟甲磺酰基)亚胺(Me3SiNTf2)及三(五氟苯基)硼烷(B(C6F5)3)等.单体为不含活泼氢的丙烯酸衍生类单体, 包括甲基丙烯酸酯、丙烯酸酯及丙烯酰胺.与传统金属或过渡金属催化剂相比, 有机强酸具有很强的酸性, 其催化的聚合优势明显.例如, 所需的有机酸用量仅为引发剂物质的量的1%~5%;线型聚合物的分子量可达100 kg•mol-1以上; 单体定量转化为聚合物所需的时间短等.
针对甲基丙烯酸酯的聚合, 有机强酸可以是有机Brönsted酸, 如HNTf2及C6F5CHTf2, 也可以是有机路易斯酸, 如Me3SiNTf2.当使用Brönsted酸时, 该酸先与引发剂SKA反应原位生成有机硅路易斯酸.聚合中, 硅路易斯酸与单体中的羰基配位并活化单体, 是聚合过程中真正的催化剂.不同于碱催化聚合中催化剂活化的是引发剂, 酸催化聚合中路易斯酸活化的是单体, 有关聚合机理的讨论将在后述内容中详细展开.采用有机酸催化的基团转移聚合法聚合MMA时, 聚合条件温和, 可很好地控制聚合物分子量及分子量分布(图 5所示).例如, 当MMA与SKAMe的摩尔比在150之内变化时, 催化剂的用量为引发剂物质的量的百分之几时即可保证单体在短时间内完全聚合得到目标分子量的聚合物, 且分散度低于1.07.另外, 降低聚合温度可提高聚合物的立构规整性.例如, 在常温下聚合得到的聚合物的间同结构的比例只有70%左右, 而在-55 ℃进行聚合时, 这一比例可提高到90%[70].
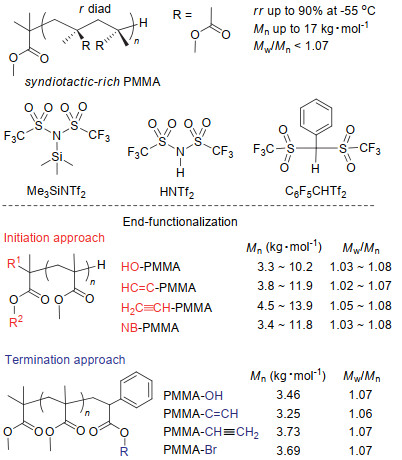
与碱催化方法一样, 通过有机酸催化的基团转移聚合同样可合成具目标结构的末端官能化聚甲基丙烯酸酯.聚合时使用官能化的引发剂便可在PMMA的起始末端定量地引入羟基、乙烯基、乙炔基或降冰片烯基, 而采用官能化的α-苯基丙烯酸酯作为终止剂则可在PMMA的终止末端分别引入不同的官能基团.比如, MMA完全聚合后继续向聚合体系中加入相对于SKA活性末端10倍物质的量的官能化α-苯基丙烯酸酯进行反应, 随后脱除保护基可得到终止末端含羟基、炔基、乙烯基或溴的PMMA[65].此外、采用Me3SiNTf2与星型引发剂在低温下聚合MMA时, 可得到间同比例高的星型PMMA[71].
因聚丙烯酸酯主链的活泼氢易发生转移反应, 所以其精准合成一直以来都是高分子合成领域的难点.可控自由基聚合及阴离子聚合都难以得到高转化率、高分子量、无化学缺陷的聚丙烯酸酯.与其它聚合方法相比, 有机路易斯酸催化的基团转移聚合法则在丙烯酸酯的聚合中表现出色[72, 73], 其具有: (ⅰ)聚合体系简单, (ⅱ)单体纯化简单且定量转化, (ⅲ)有机酸用量少, (iv)聚合速度快(<5 min), 及(ⅴ)可得到高分子量(>100 kg• mol-1)、无化学缺陷的聚合物等特点.与甲基丙烯酸酯不同, 丙烯酸酯聚合时的活性末端具有更高的反应性, 聚合中SKA活性末端需要用立体位阻大的硅烷基来稳定以避免链转移反应的发生.所用的引发剂可为1-甲氧基-1-三(异丙基)硅基-2-甲基-1-丙烯(SKAiPr)或1-甲氧基-1-三(苯基)硅基-2-甲基-1-丙烯(SKAPh)等.单体的纯化较为简单, 将购入的单体在分子筛中静置一段时间后经蒸馏便能满足纯化要求.所使用的单体不能含有活泼氢或含有能与催化剂发生反应的基团, 如酸性环境下易断键的酯基等.满足以上条件的丙烯酸酯包括疏水的丙烯酸甲、乙、正丁、2-乙基己酯、环己、桥式四氢双环戊二烯等, 亲水的丙烯酸甲氧基乙酯及2-(甲氧基乙氧基)乙酯等, 或官能化的丙烯酸烯丙酯、炔丙酯等均为可聚单体.该聚合方法特别适合于合成分子量高、分布窄且结构明确的聚丙烯酸酯.例如, 通过该方法合成得到的聚丙烯酸甲酯(PMA)、聚丙烯酸乙酯(PEA)、聚丙烯酸正丁酯(PnBA)、聚丙烯酸2-乙基己酯(PEHA)、聚丙烯酸环己酯(PCyHA)及聚丙烯酸2-甲氧基乙酯(PMEA)的数均分子量分别可达97.5、108.4、141.9、220.0、166.7及140.3 kg•mol-1, 而分子量分布大多低于1.10, 如图 6所示.
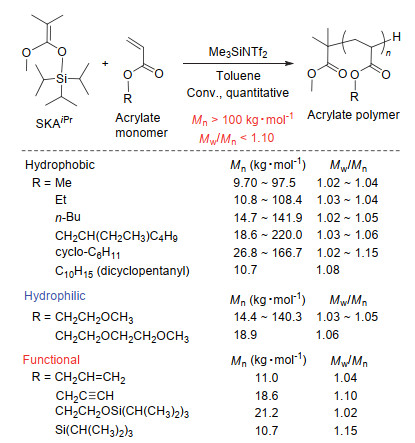
通过官能化引发剂以及终止剂的设计, 有机强酸催化的基团转移聚合同样可简便地实现末端官能化或遥爪型聚丙烯酸酯的制备[74](图 7).聚丙烯酸酯的末端官能化与上述通过有机强酸催化的基团转移聚合法合成末端官能化聚甲基丙烯酸酯的方式和反应原理一致, 在此不做赘述.
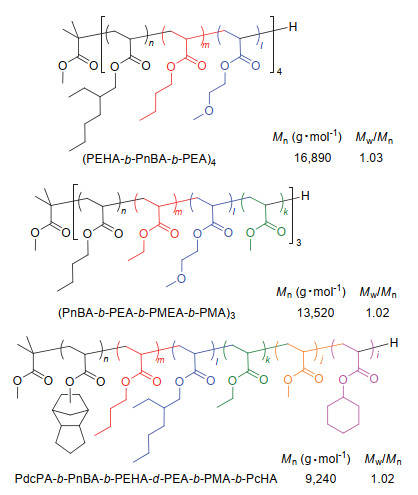
在有机酸催化的基团转移聚合过程中, 单体能够定量转化且短时间内聚合物活性末端不发生链转移反应, 这一特点使该方法特别适用于通过连续滴加单体的方法来精准制备不同丙烯酸酯的嵌段共聚物[69].在第一单体聚合完成后, 向反应体系续加第二单体至聚合结束则可得到双嵌段的共聚物.以此类推, 多次连续滴加单体则得到多嵌段的共聚物.例如, 向含SKAiPr和Me3SiNTf2的甲苯溶液中重复四次持续依次加入EHA, nBA及EA, 可得到十二嵌段(PEHA-b-PnBA-b-PEA)4共聚物(图 8).根据连续滴加单体的次数与不同组合, 则可得到各式各样的嵌段共聚物.
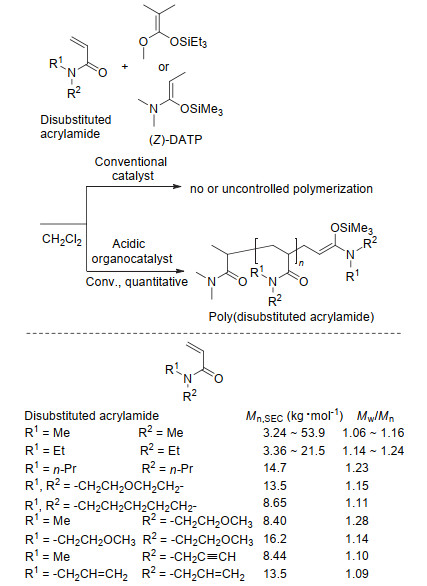
除甲基丙烯酸酯和丙烯酸酯之外, 有机酸催化的基团转移聚合也适用于单体活性不同的N, N-二取代丙烯酰胺的可控聚合(如图 9所示)[75-77].与甲基丙烯酸酯和丙烯酸酯的聚合采用三烷基硅烷基烯酮缩醛为引发剂不同, N, N-二取代丙烯酰胺的可控聚合需应用与单体具有相似结构的三烷基硅烷基烯酮缩醛胺(SKAm)引发剂.比如, 在N, N-二甲基丙烯酰胺(DMAA)的基团转移聚合中, 当应用过渡金属催化剂(如ZnX2, X=Cl、Br与I)时, 聚合不进行或不受控制.与之相比, 当采用SKAm为引发剂、有机Brönsted酸或路易斯酸为催化剂时, 聚合以可控的方式快速进行, 可得到分子量高且分散度低的聚(N, N-二甲基丙烯酰胺)(PDMAA).其分子量可被精准控制在54 kg•mol-1之内, 且分散度低于1.06.进一步研究表明应用与聚合物活性末端具有相同或相似结构的三烷基硅烷基烯酮缩醛胺为引发剂可以很好地平衡引发速率与链增长速率, 即使得引发速率不低于链增长速率从而使聚合可控.此外, 当采用三乙基硅烷基烯酮缩醛(SKAEt)为引发剂、有机酸或路易斯酸催化的二取代丙烯酰胺的聚合可得到与设计分子量一致且分子量分布在1.20附近的聚合物, 比上述采用SKAm时分布宽[73].除DMAA外, 有机强酸催化的基团转移聚合同样适用于其它不含活泼氢的二取代丙烯酰胺单体, 如N, N-二乙基丙烯酰胺(DEAA)、N, N-二丙基丙烯酰胺(nPAA)、4-丙烯酰吗啉(NAM)、N, N-二丙烯酰基哌(API)、N-(2-甲氧基乙基)-N-甲基丙烯酰胺(MMEAA)、N, N-双(2-甲氧基乙基)丙烯酰胺(BMEAA)、N-甲基-N-丙炔丙烯酰胺(MPAA)以及N, N-二烯丙基丙烯酰胺(DAlAA)等.
与前述方法一样, 在聚合中采用官能化引发剂和终止剂可以得到起始、终止或两末端官能化的聚(N, N-二取代丙烯酰胺)(如图 10所示)[78].例如, 在温敏性的聚(N, N-二乙基丙烯酰胺)(PDEAA)的起始和终止末端分别引入炔基及叠氮基后, 对得到的末端官能化PDEAA进一步通过分子内或分子间点击反应可合成线型、星型、环状等具各种拓扑结构的PDEAA, 进而可研究不同拓扑结构对温敏性的影响.
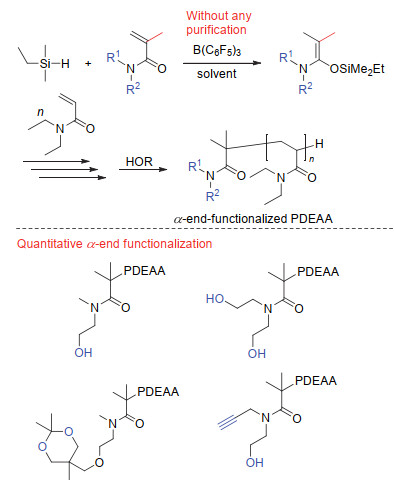
上述基团转移聚合法均涉及对水氧极其敏感的SKA或SKAm引发剂的直接使用, 引发剂的这一缺点给实际的实验操作或工业化生产带来极大挑战.此外, 绝大多数引发剂是非商品化的, 需经过繁杂的合成才能得到.鉴于此, 近年来我们开发了一种以三(五氟苯基)硼烷(B(C6F5)3)为催化剂, 以化学性质稳定的三取代氢硅烷为潜在性起始物的新型基团转移聚合法[79-82].此新型聚合方法中, B(C6F5)3被作为双功能型催化剂来促进(1)丙烯酸衍生类单体的原位1, 4-氢硅烷化生成实质的引发剂SKA或SKAm, 及(2)单体的基团转移聚合.此新型基团转移聚合法适合的单体与上述相同, 为不含活泼氢的甲基丙烯酸酯、丙烯酸酯及丙烯酰胺.其聚合反应式如图 11所示.

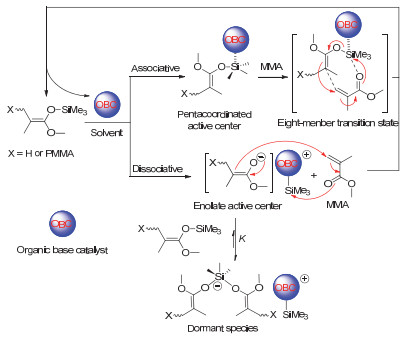
该聚合方法中, 三取代氢硅烷的选择至关重要.当三取代氢硅烷的取代基的立体位阻过大时, 聚合需长时间或根本不进行, 得到的分子量与理论分子量也会产生较大偏差.另一方面, 当三取代氢硅烷的取代基的立体位阻过低时, 由于三取代氢硅烷对活性末端的稳定化作用变差, 聚合过程中活性末端易发生分子内的链转移反应, 使得聚合的引发效率降低, 分子量分布出现双峰等结果.通过对三取代氢硅烷的筛选发现:针对甲基丙烯酸酯和丙烯酸酯的聚合, 二甲基苯基氢硅烷对分子量及分散度的控制最好; 而针对二取代丙烯酰胺的聚合, 二甲基乙基氢硅烷最优.聚合溶剂可为极性非质子溶剂, 如二氯甲烷、三氯甲烷、1, 2-二氯乙烷等.在该类极性溶剂中, 聚合反应具有较高的聚合速率.除此之外, 非极性的非质子溶剂, 如, 甲苯、乙苯、二甲苯等, 也可作为该聚合反应的溶剂使用.在该类溶剂中, 聚合具有较慢的速率.对于介电常数大、带有强配位能力孤电子对的极性溶剂及质子溶剂, 如THF, DMSO等, 则不可作为该聚合反应的溶剂使用. R1、R2及R3可为烷基, 也可为一些官能基团, 但不能含羟基、酚羟基、羧基、氨基等含有活泼氢的基团.如果含有该类基团, 需采用硅烷基、苄基等保护基团进行保护.另外, 一些容易与催化剂发生路易斯酸碱配位或反应的单体也不能直接用该方法聚合.例如, 当R1取代基为季碳取代基(如叔丁基)时, 由于此时的酯结构可被催化剂活化断裂, 得到的烷氧基负离子可使催化剂失活.再如, 当单体中含低位阻的杂原子基团(如N, N-二甲基氨基乙基)时, 由于该基团中杂原子的孤电子对的配位能力强于单体中的羰基, 使得催化剂与该基团中杂原子配位而失活.该方法中, 氢硅烷中的氢最终成为聚合物的起始末端.经醇等化合物终止后, 醇中的活泼氢被加成到聚合物的终止末端.聚合物的两末端均为氢原子, 这与其他聚合方法具有典型的不同之处.通过该方法, 聚合物的分子量可被很好地控制在数千至数万的范围之内, 而分子量分布低于1.20.聚合动力学的研究表明聚合过程具有很好的活性聚合的特点.关于这一点将在以下聚合机理中具体探讨.
另外, 采用巧妙的实验设计, 该聚合方法同样可用于聚合物起始及终止末端的官能化设计.例如, N, N-二取代丙烯酰胺的聚合反应中, 在1, 4-氢硅烷化反应阶段不使用可聚单体而使用基团转移聚合中不可自聚的N, N-二取代甲基丙烯酰胺, 则N, N-二取代甲基丙烯酰胺只与二甲基乙基氢硅烷发生1, 4-氢硅烷化反应原位生成SKAm, 而不会发生接下来的链引发及链增长反应.之后, 往反应体系中加入可聚的N, N-二取代丙烯酰胺继续反应, 则可得到起始末端含有一个N, N-二取代甲基丙烯酰胺残基的聚(N, N-二取代丙烯酰胺)(PDEAA).当反应中采用的是官能化的N, N-二取代甲基丙烯酰胺时, 则最终得到的聚合物便为起始末端官能化的聚合物(如图 12所示).通过该方法, 我们得到与目标结构一致的起始末端单羟基、双羟基及羟基/炔基双官能化的PDEAA.同理, 通过不可聚α, β-不饱和酯的选择, 同样可实现聚丙烯酸酯及聚甲基丙烯酸酯的起始末端的定量官能化.另一方面, 针对该方法中聚合物终止末端的官能化, 则可采取前述的终止剂进行.如聚合物活性末端与α-苯基丙烯酸酯进行向山-迈克尔反应或与苯甲醛的羟醛缩合反应等.在此不再赘述.
有机催化基团转移聚合可分为有机碱催化的聚合以及有机酸催化的聚合.有机碱为催化剂时, 聚合过程为活化引发剂的机理.有机酸为催化剂时, 聚合过程为单体活化机理[48, 83-85].在有机碱催化的聚合中, 根据有机碱的碱性强弱又可分为协同(Associative)及解离(Dissociative)机理.以SKAMe为引发剂、MMA聚合为例的协同聚合机理与解离聚合机理如图 13所示.当有机碱的碱性低(如TiBP)时聚合一般以协同机理进行, 反之碱性高(如t-Bu-P4)时则倾向以解离机理进行.在协同机理过程中, 引发剂/聚合物活性末端所含的亲电性三烷基硅烷基首先与催化剂作用形成五配位的硅中间体而被活化, 该中间体与单体相互作用形成一个八元环的过渡态.随后, 该引发剂与单体之间发生迈克尔加成反应而使链引发/链增长进行.与此同时, 三烷基硅烷基由聚合物活性末端转移至新加成单体的羰基氧上使被加成单体形成新的活性中心并伴随催化剂的释放.引发剂/活性中心的活化及迈克尔加成反应循环进行则活性聚合物末端持续进行链增长直至单体消耗完全.在该聚合机理中, 催化剂与引发剂/聚合物活性末端形成五配位硅中间体的平衡速度要比形成八元环过渡态和进行链增长反应的速率快得多, 从而使聚合可控.解离机理中, 有机强碱活化的也是引发剂或聚合物的活性末端, 但有机强碱的强亲核性使得引发剂/聚合物活性末端的硅氧键断裂而产生反应性极强的烯醇负离子.该烯醇负离子作为活性中心与单体进行迈克尔加成反应使链引发/链增长进行.此外, 高反应性烯醇负离子除与单体进行反应, 也与其它未被活化的引发剂或聚合物末端反应形成低反应活性的二烯醇化硅酸盐(休眠种).在该机理中, 高反应性烯醇负离子的活性种与低反应活性的二烯醇化硅酸盐休眠种所形成的平衡倾向于后者.由于达成该平衡的速率比链引发/链增长快, 从而导致该聚合反应可控.有机碱催化的基团转移聚合对甲基丙烯酸酯的聚合具有很好的可控性, 但对于丙烯酸酯或丙烯酰胺的聚合则表现不佳.
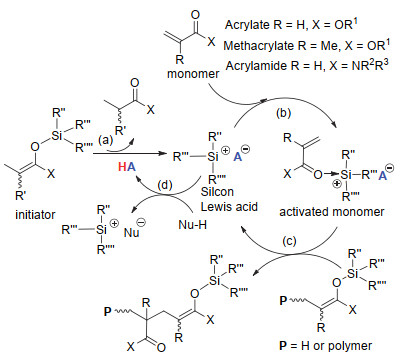
与有机碱催化的基团转移聚合不同, 有机酸催化的基团转移聚合为单体活化机理.以有机强Brönsted酸(HA)催化的聚合为例(图 14), 聚合过程主要由(a)硅路易斯酸的原位生成, (b)单体活化以及(c)链引发/链增长反应组成.首先, HA与等物质的量的引发剂反应生成硅路易斯酸, 接着该路易斯酸与单体中的羰基进行配位使单体被活化.之后, 被活化的单体与引发剂/聚合物活性末端进行迈克尔加成使链引发/链增长得以完成.伴随着链引发/链增长反应的进行, 聚合物活性末端的硅烷基转移至新加成单体的羰基氧上而形成新的活性中心.与此同时, 被活化单体上的硅阳离子与其对阴离子重新结合并脱离被活化单体使催化剂得以重生.单体活化与加成反应循环进行直至单体消耗完成.研究发现聚合物末端的硅烷基完全来自于引发剂, 在链引发/链增长过程中不与催化剂中的硅阳离子发生交换反应.如果聚合体系中含活泼氢化合物, 则生成的硅路易斯酸可与该化合物发生酸碱中和反应(d).在聚合中若直接使用有机强路易斯酸, 则基元反应(a)不存在, 而其它基元反应相同.当使用硅路易斯酸催化聚合时, 聚合过程一般为对单体浓度的一级反应, 但当使用的催化剂是B(C6F5)3时, 聚合过程往往表现为对单体浓度的零级反应.这与路易斯酸与单体的配位能力紧密相关.
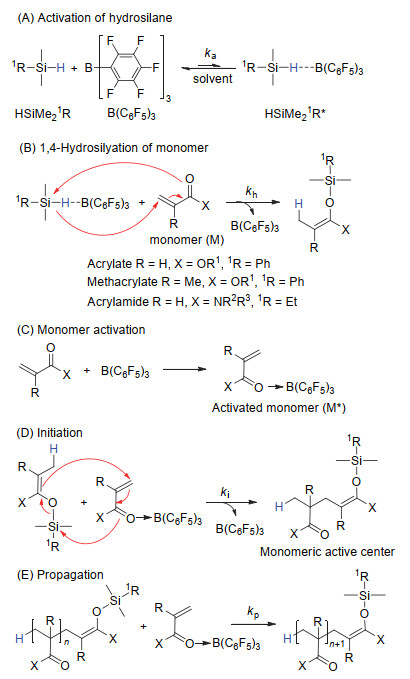
当使用B(C6F5)3/HSiMe21R这一催化/引发体系时, 除图 14中的(b)与(c)两步基元反应之外, 单体与HSiMe21R在B(C6F5)3催化下的1, 4-氢硅烷化反应也必须考虑在聚合的基元反应之中.因此, 该聚合方法的基元反应包括(A)HSiMe2R的活化(形成活化的氢化硅烷, HSiMe21R*), (B)单体与HSiMe21R*反应原位生成SKA或SKAm, (C)单体活化, (D)链引发及(E)链增长(图 15), 其中, (A)与(B)为1, 4-氢硅烷化反应的基元反应.在α, β-不饱和酮、酯及酰胺的1, 4-氢硅烷化反应的机理研究中已知HSiMe2R的活化为该反应的速率决定步骤.因此, 基元反应(A)除平衡常数需足够大之外, 正相反应的速率ka还必须满足ka≥ki≥kp方可使该聚合反应成为可控聚合.不管是丙烯酸酯、甲基丙烯酸酯还是丙烯酰胺, 当选择适当的HSiMe21R时, 聚合均能成为活性/可控聚合.因此, 选择合适的HSiMe21R是该聚合方法的关键.
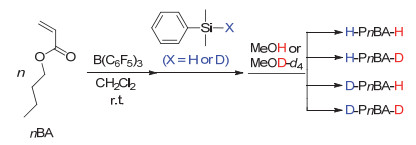
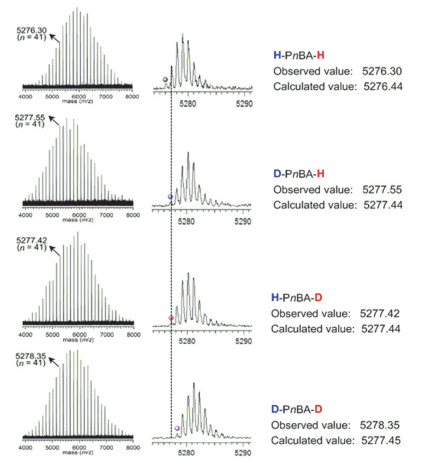
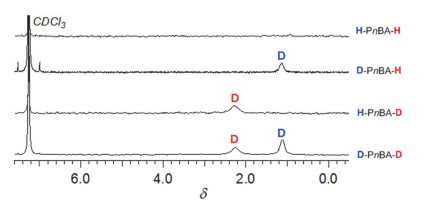
该聚合反应的末端氘代研究表明聚合物的起始末端全部来自于氢硅烷中的氢原子且在聚合过程中不存在任何副反应.采用普通/氘代二甲基苯基氢硅烷(HSiMe2Ph或DSiMe2Ph)为起始物、普通/氘代甲醇(CH3OH或CD3OD)为终止剂的不同组合方式于二氯甲烷中室温下聚合nBA(聚合条件为: [nBA]0/[HSiMe2Ph或DSiMe2Ph]0/[B(C6F5)3]0=50/1/0.05), 可得到四组具有不同起始及终止末端的聚合物, 分别为H-PnBA-H、D-PnBA-H、H-PnBA-D以及D-PnBA-D (理论Mn=6.4 kg•mol-1, 如图 16所示).对所得四组聚合物进行MALDI-TOF MS及2H NMR测定(图 17及图 18所示)追踪发现:二甲基苯基氢硅烷及甲醇中的H/D在引发及终止聚合反应后定量进入聚合物的起始及终止末端. MALDI-TOF MS的测试结果显示四组聚合物均只有一组聚合物离子峰, 而且每一聚合物离子峰的峰值均与设计的聚合物的理论离子峰值完全一致.这一结果表明:聚合发生后, 二甲基苯基氢硅烷中的H/D定量成为聚合物的起始末端; 而在使用甲醇终止后, 聚合物的终止末端全来自于甲醇羟基中的H/D. 2H NMR测定的结果同样可得以上结论.这些结果亦表明聚合过程中未有任何副反应发生, 聚合为活性聚合.利用末端氘代的方法, 甲基丙烯酸酯及丙烯酰胺的聚合也得到同样的结论.
针对该聚合反应的动力学研究进一步表明:利用该聚合反应针对可聚单体进行聚合时, 聚合过程表现为对单体浓度呈现零级反应的特点.这一特点决定于B(C6F5)3对单体的定量配位作用所导致的被活化单体浓度在除聚合晚期外的整个聚合过程中始终保持与B(C6F5)3的初始浓度相等, 即[M*]=[B(C6F5)3]0.根据聚合速率方程的定义:
|
$ R_{\mathrm{p}}=-\frac{\mathrm{d}[\mathrm{M}]}{\mathrm{d} t}=k_{\mathrm{p}}[\mathrm{SKA}]\left[\mathrm{M}^{*}\right] $ |
(ⅰ) |
此处, kp为聚合速率常数.由上述分析可知, 聚合过程中不存在副反应, 则
|
$ [\mathrm{SKA}]=\left[\mathrm{HSiMe}_{2}{ }^{1} \mathrm{R}\right]_{0} $ |
(ⅱ) |
由方程(ⅰ)与(ⅱ)可推导出聚合过程的动力学方程(ⅲ)
|
$ \text { Conv. }(\%)=\frac{k_{\mathrm{p}}\left[\mathrm{HSiMe}_{2}{^{1}} \mathrm{R}\right]_{0}\left[\mathrm{B}\left(\mathrm{C}_{6} \mathrm{F}_{5}\right)_{3}\right]_{0} t}{[\mathrm{M}]_{0}} \times 100 \% $ |
(ⅲ) |
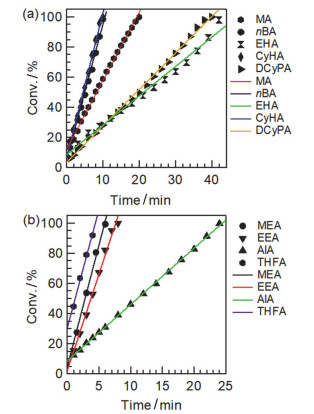
很明显, 除了在反应末期当单体消耗殆尽时引起[M*]≠[B(C6F5)3]0, 该聚合动力学方程表现为对单体浓度的零级关系, 即单体的转化率与反应时间成正比.而该理论推导与实验得到的动力学图示呈零级关系完全吻合.例如, 图 19所示的各类丙烯酸酯的聚合动力学图示表明该聚合反应为零级反应.同样的零级反应现象在甲基丙烯酸酯及丙烯酰胺的聚合过程中同样被观察到.另外, 动力学图中的直线斜率即为
有机催化基团转移聚合无论是在聚合物分子量及分子量分布方面, 还是在聚合物结构控制的精准度方面均胜于传统基团转移聚合方法.在甲基丙烯酸酯的聚合控制方面, 采用有机强碱催化的基团转移聚合可得到分子量高于100 kg•mol-1且分子量分布窄的聚甲基丙烯酸酯, 同时在聚合中可避免副反应的发生.另一方面, 采用有机强酸催化的基团转移聚合在低温可得到具有间同结构比例高的聚甲基丙烯酸酯.在丙烯酸酯的聚合控制方面, 利用有机酸催化剂可同时实现(1)高分子量、低分散度聚丙烯酸酯的合成, (2)定量的单体转化率, (3)少量的催化剂用量(相对于引发剂量的0.5~5 mol%), 以及(4)精准的聚合物结构控制, 而传统金属或过渡金属化合物催化的基团转移聚合即使使用相对于单体10~30 mol%的催化剂用量, 所得聚合物的分子量也不超过10 kg•mol-1.在丙烯酰胺的聚合控制方面, 采用有机酸催化的基团转移聚合首次实现了二取代丙烯酰胺的可控聚合, 是基团转移聚合领域一次重要突破.除甲基丙烯酸酯、丙烯酸酯以及丙烯酰胺外, 对其他具有类似结构的α, β-不饱和酯及酰胺等的聚合行为尚待深入研究与探讨.
其次, 根据基团转移聚合为活性聚合的特点以及基于官能性引发剂及终止剂在该领域的巧妙应用, 可实现聚甲基丙烯酸酯、聚丙烯酸酯以及聚丙烯酰胺的起始/终止末端的定量官能化或遥爪聚合物的制备.这些末端官能化聚合物可作为标准的聚合物素材为之后的聚合物末端的功能化修饰, 嵌段聚合物的合成以及其他功能性材料的制备提供完备的前驱体材料.该方面具有广阔的应用开发空间.
再次, 星型引发剂的应用为合成精准星型聚甲基丙烯酸酯、聚丙烯酸酯以及聚丙烯酰胺提供方向.除星型均聚物外, 通过同类单体的嵌段共聚也可得到嵌段型星聚合物.加之终止末端官能化的合成路线, 可制备各类由聚甲基丙烯酸酯、聚丙烯酸酯或聚丙烯酰胺以及聚醚、聚酯、聚氨基酸等组合而成的嵌段型星聚合物等.对研究该类聚合物的物理性能提供很好的合成手段以及良好的研究素材.当然, 应用有机催化基团转移聚合方法开发聚合物的其他拓扑结构有待进一步拓展.
B(C6F5)3/HSiMe21R这一新型催化/引发体系的应用为聚合物的分子设计及制备带来新的可能, 尤其是对水氧稳定的氢化硅烷的应用为实验操作带来巨大的便利, 并免除了高反应性硅烷基烯醇缩醛及硅烷基烯醇缩醛胺类引发剂的繁杂合成过程.鉴于当前B(C6F5)3的价格昂贵, 开发价廉催化剂或可回收交联型固载催化剂具有现实意义.
基团转移聚合已被杜邦公司以及德国BYK公司用于均聚物及嵌段聚合物的实际生产.作为一种实际被工业化应用的聚合方法, 有机催化基团转移聚合的新进展进一步为该聚合方法的实际应用奠定更为坚实的基础.总之, 我们坚信在不久的将来有机催化基团转移聚合方法的进一步完善无论在学术研究中, 还是在实际工业化生产中具有更为广阔的天地.
Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 4243. doi: 10.1021/ja000092s
MacMillan, D. W. C. Nature 2008, 455, 304. doi: 10.1038/nature07367
Xiao, Y.; Wang, Y.; Zhou, Z. Chinese J. Org. Chem. 2019, 39, 2203. doi: 10.6023/cjoc201703070
王黎明, 林赓, 赵美君, 刘东垚, 金瑛, 有机化学, 2018, 38, 642. http://www.cqvip.com/QK/93463X/201803/674815966.htmlWang, L.; Lin, G.; Zhao, M.; Liu, D.; Jin, Y. Chinese J. Org. Chem. 2018, 38, 642 (in Chinese). http://www.cqvip.com/QK/93463X/201803/674815966.html
马世雄, 钟源, 王守磊, 许兆青, 常民, 王锐, 化学学报, 2014, 72, 825. http://www.cqvip.com/QK/91047X/201407/72888866504849524855484857.htmlMa, S.; Zhong, Y.; Wang, S.; Xu, Z.; Chang, M.; Wang, R. Acta Chim. Sinica 2014, 72, 825 (in Chinese). http://www.cqvip.com/QK/91047X/201407/72888866504849524855484857.html
Zhao, H.; Meng, W.; Yang, Z.; Tian, T.; Sheng, Z.; Li, H.; Song, X.; Zhang, Y.; Yang, S.; Li, B. Chinese J. Chem. 2014, 32, 417. doi: 10.1002/cjoc.201400166
Nederberg, F.; Connor, E. F.; Möller, F.; Glauser, M. T.; Hedrick, J. L. Angew. Chem. Int. Ed. 2001, 40, 2712. doi: 10.1002/1521-3773(20010716)40:14<2712::AID-ANIE2712>3.0.CO;2-Z
Misaka, H.; Kakuchi, R.; Zhang, C. H.; Sakai, R.; Satoh, T.; Kakuchi, T. Macromolecules 2009, 42, 5091. doi: 10.1021/ma900712p
Dove, A. P.; Pratt, R. C.; Lohmeijer, B. G. G.; Waymouth, R. M.; Hedrick, J. L. J. Am. Chem. Soc. 2005, 127, 13798. doi: 10.1021/ja0543346
Miyake, G. M.; Chen, E. Y.-X. Macromolecules 2011, 44, 4116. doi: 10.1021/ma2007199
Sanda, F.; Sanada, H.; Shibasaki, Y.; Endo, T. Macromolecules 2002, 35, 680. doi: 10.1021/ma011341f
Persson, P. V.; Schroder, J.; Wickholm, K.; Hedenstrom, E.; Iversen, T. Macromolecules 2004, 37, 5889. doi: 10.1021/ma049562j
Oledzka, E.; Narine, S. S. J. Appl. Polym. Sci. 2011, 119, 1873. doi: 10.1002/app.32897
Makiguchi, K.; Satoh, T.; Kakuchi, T. Macromolecules 2011, 44, 1999. doi: 10.1021/ma200043x
Gazeau-Bureau, S.; Delcroix, D.; Martin-Vaca, B.; Bonrissou, D.; Navarro, C.; Magnet, S. Macromolecules 2008, 41, 3782. doi: 10.1021/ma800626q
Bourissou, D.; Martin-Vaca, B.; Dumitrescu, A.; Graullier, M.; Lacombe, F. Macromolecules 2005, 38, 9993. doi: 10.1021/ma051646k
Kadota, J.; Pavlović, D.; Desvergne, J.; Bibal, B.; Peruch, F.; Deffieux, A. Macromolecules 2010, 43, 8874. doi: 10.1021/ma101688d
Helou, M.; Miserque, O.; Brusson, J.; Carpentier, J.; Guillaume, S. M. Chem. Eur. J. 2010, 16, 13805. doi: 10.1002/chem.201001111
Connor, E. F.; Nyce, G. W.; Myers, M.; Mock, A.; Hedrick, J. L. J. Am. Chem. Soc. 2002, 124, 914. doi: 10.1021/ja0173324
Nyce, G. W.; Glauser, T.; Connor, E. F.; Mock, A.; Waymouth, R. M.; Hedrick. J. L. J. Am. Chem. Soc. 2003, 125, 3046. doi: 10.1021/ja021084+
Coulembier, O.; Dove, A. P.; Pratt, R. C.; Sentman, A. C.; Culkin, D. A.; Mespouille, L.; Dubois, P.; Waymouth, R. M.; Hedrick, J. L. Angew. Chem. Int. Ed. 2005, 44, 4964. doi: 10.1002/anie.200500723
Csihony, S.; Culkin, D. A.; Sentman, A. C.; Dove, A. P.; Waymouth, R. M.; Hedrick, J. L. J. Am. Chem. Soc. 2005, 127, 9079. doi: 10.1021/ja050909n
Zhang, L.; Nederberg, F.; Pratt, R. C.; Waymouth, R. M.; Hedrick, J. L.; Wade, C. G. Macromolecules 2007, 40, 4154. doi: 10.1021/ma070316s
Nederberg, F.; Lohmeijer, B. G. G.; Leibfarth, F.; Pratt, R. C.; Choi, J.; Dove, A. P.; Waymouth, R. M.; Hedrick, J. L. Biomacromolecules 2007, 8, 153. doi: 10.1021/bm060795n
Lohmeijer, B. G. G.; Dubois, G.; Leibfarth, F.; Pratt, R. C.; Nederberg, F.; Nelson, A.; Waymouth, R. M.; Wade, C. G.; Hedrick, J. L. Org. Lett. 2006, 8, 4683. doi: 10.1021/ol0614166
Zhang, L.; Nederberg, F.; Pratt, R. C.; Waymouth, R. M.; Hedrick, J. L.; Wade, C. G. Macromolecules 2007, 40, 4154. doi: 10.1021/ma070316s
Zhang, L.; Nederberg, F.; Messman, J. M.; Pratt, R. C.; Hedrick, J. L.; Wade, C. G. J. Am. Chem. Soc. 2007, 129, 12610. doi: 10.1021/ja074131c
Webster, O. W.; Hertler, W. R.; Sogah, D. Y.; Farnham, W. B.; RajanBabu, T. V. J. Am. Chem. Soc. 1983, 105, 5706. doi: 10.1021/ja00355a039
Webster, O. W. Adv. Polym. Sci. 2004, 167, 1.
Schubert, W.; Bandermann, F. Makromol. Chem. 1989, 190, 2161. doi: 10.1002/macp.1989.021900916
Schubert, W.; Sitz, H. D.; Bandermann, F. Makromol. Chem. 1989, 190, 2193. doi: 10.1002/macp.1989.021900919
Sogah, D. Y.; Hertler, W. R.; Webster, O. W.; Cohen, G. M. Macromolecules 1987, 20, 1473. doi: 10.1021/ma00173a006
Schubert, W.; Bandermann, F. Makromol. Chem. 1989, 190, 2721. doi: 10.1002/macp.1989.021901106
Hertler, W. R.; Sogah, D. Y.; Webster, O. W. Macromolecules 1984, 17, 1415. doi: 10.1021/ma00137a021
Hellstern, A. M.; DeSimone, J. M.; McGrath, J. E. Polym. Prepr. 1988, 29, 342.
Dicker, I. B.; Cohen, G. M.; Farnham, W. B.; Hertler, W. R.; Laganis, E. D.; Sogah, D. Y. Macromolecules 1990, 23, 4034. doi: 10.1021/ma00220a002
Patrickios, C. S.; Hertler, W. R.; Abbott, N. L.; Hatton, T. A. Macromolecules 1994, 27, 930. doi: 10.1021/ma00082a008
Eggert, M.; Freitag, R. J. Polym. Sci. Part A: Polym. Chem. 1994, 32, 803.
Ute, K.; Tarao, T.; Hongo, S.; Ohnuma, K.; Hatada, K.; Kitayama, T. Polym. J. 1999, 31, 177. doi: 10.1295/polymj.31.177
Ute, K.; Ohnuma, H.; Shimizu, I.; Kitayama, T. Polym. J. 2006, 38, 999. doi: 10.1295/polymj.PJ2006041
Dicker, I. B. Polym. Prepr. 1988, 29, 114.
Zhuang, R.; Müller, A. H. E. Macromol. Symp. 1994, 85, 379. doi: 10.1002/masy.19940850128
Zhuang, R.; Müller, A. H. E. Macromolecules 1995, 28, 8035. doi: 10.1021/ma00128a010
Zhuang, R.; Müller, A. H. E. Macromolecules 1995, 28, 8043. doi: 10.1021/ma00128a011
Ute, K.; Tarao, T.; Hatada, K. Polymer 1997, 44, 7869.
Ute, K.; Tarao, T.; Kitayama, T. Polym. J. 2005, 37, 578. doi: 10.1295/polymj.37.578
White, D.; Matyjaszewski, K. Polym. Prepr. 1995, 36, 286.
Fuchise, K.; Chen, Y.-G.; Satoh, T.; Kakuchi, T. Polym. Chem. 2013, 4, 4278. doi: 10.1039/c3py00278k
Raynaud, J.; Ciolino, A.; Baceiredo, A.; Destarac, M.; Bonnette, F.; Kato, T.; Gnanou, Y.; Taton, D. Angew. Chem. Int. Ed. 2008, 47, 5390. doi: 10.1002/anie.200800490
Scholten, M. D.; Hedrick, J. L.; Waymouth, R. M. Macromolecules 2008, 41, 7399. doi: 10.1021/ma801281q
Raynaud, J.; Liu, N.; Gnanou, Y.; Taton, D. Macromolecules 2010, 43, 8853. doi: 10.1021/ma101478p
Raynaud, J.; Liu, N.; Fèvre, M.; Gnanou, Y.; Taton, D. Polym. Chem. 2011, 2, 1706. doi: 10.1039/c1py00077b
Scholten, M. D.; Hedrick, J. L.; Waymouth, R. M. Polym. Prepr. 2007, 48, 167.
Raynaud, J.; Gnanou, Y.; Taton, D. Macromolecules 2009, 42, 5996. doi: 10.1021/ma900679p
Raynaud, J.; Gnanou, Y.; Taton, D. PMSE Prepr. 2009, 101, 1771.
Zhang, Y.; Chen, E. Y.-X. Angew. Chem. Int. Ed. 2012, 51, 2465. doi: 10.1002/anie.201108019
Fèvre, M.; Vignolle, J.; Heroguez, V.; Taton, D. Macromolecules 2012, 45, 7711. doi: 10.1021/ma301412z
Zhang, Y.; Chen, E. Y.-X. Macromolecules 2008, 41, 36. doi: 10.1021/ma702015w
Zhang, Y.; Chen, E. Y.-X. Macromolecules 2008, 41, 6353. doi: 10.1021/ma801125y
Miyake, G. M.; Zhang, Y.; Chen, E. Y.-X. Macromolecules 2010, 43, 4902. doi: 10.1021/ma100615t
Zhang, Y.; Gustafson, O.; Chen, E. Y-X. J. Am. Chem. Soc. 2011, 133, 13674. doi: 10.1021/ja2053573
Chen, E. Y.-X. Chem. Rev. 2009, 109, 5157. doi: 10.1021/cr9000258
Zhang, Y.; Lay, F.; García-García, P.; List, B. Chem. Eur. J. 2010, 16, 10462. doi: 10.1002/chem.201000961
Kakuchi, T.; Chen, Y.-G.; Kitakado, J.; Mori, K.; Fuchise, K.; Satoh, T. Macromolecules 2011, 44, 4641. doi: 10.1021/ma200720p
Chen, Y.-G.; Takada, K.; Kubota, N.; Eric, O.-T.; Ito, T.; Isono, T.; Satoh, T.; Kakuchi, T. Polym. Chem. 2015, 6, 1830. doi: 10.1039/C4PY01564A
Eric, O.-T.; Chen, Y.-G.; Takada, K.; Sato, S.; Satoh, T.; Kakuchi, T. Polym. Chem. 2015, 6, 7841. doi: 10.1039/C5PY01112D
Chen, Y.-G.; Fuchise, K.; Kawaguchi, S.; Satoh, T.; Kakuchi, T. Macromolecules 2011, 44, 9091. doi: 10.1021/ma202103d
Hsu, J.-C.; Chen, Y.-G.; Kakuchi, T.; Chen, W.-C. Macromolecules 2011, 44, 5168. doi: 10.1021/ma2006377
Kikuchi, S.; Chen, Y.-G.; Fuchise, K.; Takada, K.; Kitakado, J.; Sato, S.; Satoh, T.; Kakuchi, T. Polym. Chem. 2014, 5, 4701. doi: 10.1039/C4PY00290C
Kakuchi, R.; Chiba, K.; Fuchise, K.; Sakai, R.; Satoh, T.; Kakuchi, T. Macromolecules 2009, 42, 8747. doi: 10.1021/ma902006d
Chen, Y.-G.; Takada, K.; Fuchise, K.; Satoh, T.; Kakuchi, T. J. Polym. Sci. Part A: Polym. Chem. 2012, 50, 3277. doi: 10.1002/pola.26123
Takada, K.; Fuchise, K.; Chen, Y.-G.; Satoh, T.; Kakuchi, T. J. Polym. Sci. Part A: Polym. Chem. 2012, 50, 3560. doi: 10.1002/pola.26140
Takada, K.; Ito, T.; Kitano, K.; Tuschida, S.; Takagi, Y.; Chen, Y.-G.; Satoh, T.; Kakuchi, T. Macromolecules 2015, 48, 511. doi: 10.1021/ma502298v
Takada, K.; Fuchise, K.; Kubota, N.; Ito, T.; Chen, Y.-G.; Satoh, T.; Kakuchi, T. Macromolecules 2014, 47, 5514. doi: 10.1021/ma501106e
Fuchise, K.; Sakai, R.; Satoh, T.; Sato, S.; Narumi, A.; Kawaguchi, S.; Kakuchi, T. Macromolecules 2010, 43, 5589. doi: 10.1021/ma1005765
Fuchise, K.; Chen, Y.-G.; Takada, K.; Satoh, T.; Kakuchi, T. Macromol. Chem. Phys. 2012, 213, 1604. doi: 10.1002/macp.201200146
Kikuchi, S.; Chen, Y.-G.; Kitano, K.; Takada, K.; Satoh, T.; Kakuchi, T. Polym. Chem. 2015, 6, 6845. doi: 10.1039/C5PY01104C
Kikuchi, S.; Chen, Y.-G.; Ichinohe, E.; Sato, S.; Duan, Q.; Shen, X.-D.; Kakuchi, T. Macromolecules 2016, 49, 4828. doi: 10.1021/acs.macromol.6b01075
Fuchise, K.; Tsuchida, S.; Takada, K.; Chen, Y.-G.; Satoh, T.; Kakuchi, T. ACS Macro. Lett. 2014, 3, 1015. doi: 10.1021/mz5004689
Chen, Y.-G.; Kitano, K.; Tsuchida, S.; Kikuchi, S.; Takada, K.; Satoh, T.; Kakuchi, T. Polym. Chem. 2015, 6, 3502. doi: 10.1039/C5PY00294J
Kikuchi, S.; Chen, Y.-G.; Kitano, K.; Satoh, T.; Kakuchi, T. Macromolecules 2016, 49, 3049. doi: 10.1021/acs.macromol.6b00190
Chen, Y.-G.; Jia, Q.; Ding, Y., ; Sato, S.; Xu, L.; Zang, C.; , Shen, X.; Kakuchi, T. Macromolecules 2019, 52, 844. doi: 10.1021/acs.macromol.8b02245
Chen Y.-G.; Kakuchi, T. Chem. Rec. 2016, 16, 2161. doi: 10.1002/tcr.201600034
陳友根, 沈賢徳, 覚知豊次, 日本接着学会誌, 2017, 53, 432.Chen, Y.-G.; Shen, X.-D.; Kakuchi, T. Journal of The Adhesion Society of Japan, 2017, 53, 432.
Chen, Y.-G.; Fuchise, K.; Satoh, T.; Kakuchi, T. In Anionic Polymerization: Principles, Practice, Strength, Consequences, and Applications, Springer, Tokyo, 2015, pp. 451~494

图 1 TiBP或t-Bu-P4催化的甲基丙烯酸甲酯的基团转移聚合
Figure 1 Group transfer polymerization of MMA mediated by TiBP or t-Bu-P4

图 2 (a) 由TiBP催化的GTP获得的PMMA的MALDI-TOF MS图(反射模式)及(b)一次聚合(实线)和扩链聚合(虚线)获得的PMMA的SEC曲线: (上)TiBP催化, (下) t-Bu-P4催化
Figure 2 (a) MALDI-TOF MS spectra (reflector mode) of the PMMA obtained from TiBP-catalyzed GTP and (b) SEC traces of PMMA obtained from the first polymerization (solid line) and subsequent post-polymerization (dashed line) catalyzed by TiBP (above) and t-Bu-P4 (below)

图 3 通过t-Bu-P4催化的基团转移聚合制备末端官能化的PMMA
Figure 3 Synthesis of end-functionalized PMMA by t-Bu-P4-catalyzed GTP

图 4 t-Bu-P4催化的基团转移聚合中应用的多官能引发剂及甲基丙烯酸酯单体
Figure 4 Multifunctional initiators and methacrylate monomers used in t-Bu-P4-catalyzed GTP

图 5 有机酸催化的MMA的基团转移聚合及其末端官能化.
Figure 5 GTP of MMA using acidic organic catalysts and its end-functionalization.

图 6 有机酸催化的丙烯酸酯的基团转移聚合.
Figure 6 GTP of acrylaye monomers using acidic organic catalysts.

图 7 通过有机酸催化的基团转移聚合制备α-, ω-和α, ω-末端官能化的聚丙烯酸正丁酯
Figure 7 Preparation of α-, ω- and α, ω-end-functionalized poly(n-butyl acrylate) by GTP using acidic organic catalysts
图 8 采用Me3SiNTf2催化的基团转移聚合法制备丙烯酸酯的多嵌段共聚物
Figure 8 Multiblock acrylate polymers obtained from Me3SiNTf2- catalyzed GTP of acrylate monomers

图 9 有机酸催化的N, N-二取代丙烯酰胺的基团转移聚合
Figure 9 GTP of N, N-disubsitituted acrylamides using acidic organocatalyst

图 10 通过有机酸催化GTP设计α-, ω-和α, ω-末端官能化及环状聚(N, N-二取代丙烯酰胺)
Figure 10 Design of α-, ω- and α, ω-end-functionalized, and cyclic poly(N, N-dialkylacrylamide) by GTP using acidic organic catalysts

图 11 B(C6F5)3/氢硅烷组合为催化/引发体系的丙烯酸衍生类单体的原位基团转移聚合法
Figure 11 An in-situ GTP method for acrylic-derived monomers using B(C6F5)3/hrdrosilane combination as a catalysis/initiation system

图 12 基于Me2EtSiH和各种功能化甲基丙烯酰胺的基团转移聚合法制备α-单羟基、二羟基、炔基及羟基双官能化的PDEAA
Figure 12 Synthesis of defect-free α-monohydroxyl, dihydrooxyl, ethynyl, hydroxyl end-functionalized PDEAAs by GTP using Me2EtSiH and various functional methacrylamides

图 13 有机强碱催化的基团转移聚合中的associative和dissociative反应机理.
Figure 13 The associative and dissociative mechanisms in the group transfer polymerization using strong organic base catalyst.

图 14 有机强Brönsted酸催化的基团转移聚合的反应机理
Figure 14 Mechanism of the group transfer polymerization using strong organic Brönsted acid catalyst

图 15 基于B(C6F5)3/HSiMe21R催化体系的基团转移聚合的反应机理
Figure 15 Mechanism of group transfer polymerization using B(C6F5)3/HSiMe21R catalytic system

图 16 以Me2PhSiH或Me2PhSiD为一种起始成分, 并以CH3OH或CD3OD为终止剂, 通过B(C6F5)3催化的nBA的GTP合成H-PnBA-H, H-PnBA-D, D-PnBA-H和D-PnBA-D
Figure 16 Synthesis of H-PnBA-H, H-PnBA-D, D-PnBA-H and D-PnBA-D by B(C6F5)3-catalyzed GTP of nBA using Me2PhSiH or Me2PhSiD as one initiating component and CH3OH or CD3OD as the terminator

图 17 H-PnBA-H, H-PnBA-D, D-PnBA-H和D-PnBA-D的MALDI- TOF MS图谱
Figure 17 The MALDI-TOF MS spectra of the obtained (a) H-PnBA-H, (b) H-PnBA-D, (c) D-PnBA-H and (d) D-PnBA-D

图 18 (a) H-PnBA-H, (b) H-PnBA-D, (c) D-PnBA-H和(d) D-PnBA-D的2H NMR图谱
Figure 18 The 2H NMR (62 MHz) spectra of the obtained (a) H-PnBA-H, (b) H-PnBA-D, (c) D-PnBA-H, and (d) D-PnBA-D, in CHCl3 at room temperature, respectively

图 19 (a) MA, nBA, EHA, CyHA与DCyPA和(b) MEA, EEA, AlA和THFA聚合反应的零级动力学图
Figure 19 Zero-order kinetic plots for the polymerizations of (a) MA, nBA, EHA, CyHA and DCyPA and (b) MEA, EEA, AlA, and THFA
表 1 THF中通过t-Bu-P4催化的GTP法合成的甲基丙烯酸酯星型聚合物
Table 1. Star-shaped methacrylate polymers synthesized by t-Bu-P4- catalyzed GTP in THF
| Star-shaped polymer | Star-shaped initiator | Mw(expt.)/ (kg·mol-1)a |
Mw/Mnb |
| PMMA3 | SKA3 | 3.9~132.0 | 1.07~1.14 |
| PMMA4 | SKA 4 | 5.5~130.3 | 1.07~1.17 |
| PMMA6 | SKA 6 | 7.5~131.6 | 1.06~1.15 |
| PMMA12 | SKA 12 | 28.2~264.9 | 1.06~1.12 |
| PDMAEMA3 | SKA3 | 19.3~160c | 1.20~1.27d |
| PDMAEMA4 | SKA 4 | 24.0~254c | 1.16~1.32d |
| PDMAEMA6 | SKA 6 | 30.2~296c | 1.09~1.27d |
| PDMAEMA12 | SKA 12 | 34.3~419c | 1.13~1.39d |
| PAMA4 | SKA 4 | 13.6~21.2 | 1.11~1.16 |
| PSMA4 | SKA 4 | 34.5~65.2 | 1.06~1.08 |
| a Absolute molar masses determined in THF by SEC equipped with an multi-angle laser light scattering (MALS) detector. b Dispersity determined by SEC equipped with an RI detector in THF on the basis of PMMA standards. c Absolute molar masses determined by MALS-SEC in DMF containg 0.01 mol•L-1 LiCl. d Polydispersity determined by SEC equipped with an RI detector in DMF containg 0.01 mol•L-1 LiCl on the basis of poly(N, N-dimethy- lacrylamide) standards. | |||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们