图 1.
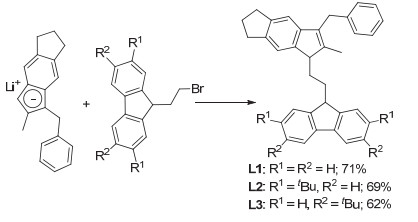
亚乙基桥联茚-芴类茂金属配合物
Figure 1.
Ethylene-bridged indenyl-fluorenyl metallocene complexes

丙烯配位聚合研究的核心在于高效(高活性、高选择性)金属催化剂的开发.目前, 丙烯配位聚合催化剂的研究主要导向于获得高分子量聚合物.然而, 随着丙烯齐聚物应用的广泛开发, 高性能丙烯齐聚催化剂的研究受到了越来越多的关注.
丙烯配位齐聚反应过程中, 通过对链终止反应的控制可以形成不同的端基, 如亚乙烯基、烯丙基和饱和端基等[1-6].其中, 含有烯丙基端基的丙烯齐聚物具有优异的可聚性及端基可修饰性, 其潜在用途广泛[2, 3, 7-12].从丙烯配位聚合的反应机理看, 烯丙基端基可由丙烯1, 2-插入后发生β-Me消除形成.然而, 由于C—C键活化断裂所需的能量较大, 而且C原子sp3杂化轨道的取向性较强, β-H消除反应往往比β-Me消除反应具有绝对的优势.
20世纪80年代, Watson等[13]在研究二(五甲基环戊二烯基)稀土金属配合物催化丙烯聚合反应时, 首次发现了金属中心β-位C—Me键的活化现象, 吸引了相关学者的深入研究.随后, Teuben等[14, 15]和Resconi等[16]指出, 茂金属催化剂配体结构上特定位置的位阻环境对β-H消除反应过渡态的形成具有一定的抑制作用, 迫使β-Me取向于金属的最低未占分子轨道(LUMO)轨道, 从而有效促进选择性β-Me消除反应的发生.近年来, 人们为了获得不同链长的烯丙基封端聚合产物, 导向选择性β-Me消除的茂金属催化剂成了研究热点[1, 9, 17-24], 然而至今为止兼具β-Me消除选择性和丙烯齐聚性能的催化剂报道仍然非常罕见. 1992年, Resconi等[16]对(CpMe5)2MCl2 (M=Zr, Hf)/MAO催化丙烯聚合反应的性能做了详细的研究, 该催化剂体系代表了首例具有较高β-Me消除选择性的丙烯齐聚茂金属催化剂.在0~50 ℃条件下, 锆催化剂体系具有82%~91%的β-Me消除选择性, 产物聚合度在4.5~95范围内, 铪催化剂体系的选择性为63%~98%, 产物聚合度较低(Pn=3.4~27.4). 2006年, Okuda课题组[20]在此基础上发展了一系列全烷基取代二(环戊二烯基)铪配合物, 其中(C5Me4iBu)2HfCl2/MAO体系可以催化丙烯以最高61.6 wt%的选择性二聚获得烯丙基端基C6产物4-甲基-1-戊烯, 但催化活性很低.由于β-Me消除选择性对此类配体结构的依赖性强, 除了以上两例外未见其它取代二(环戊二烯基)茂金属配合物催化丙烯选择性聚合获得烯丙基封端齐聚物的报道.
最近, 我们[17]报道了一类基于茚环3-位苄基取代的亚乙基桥联茚-芴锆、铪配合物(图 1), 其3-位苄基对丙烯齐聚反应过程中的β-Me消除选择性具有显著的促进作用, 其中锆催化剂体系在高温条件下(100~160 ℃)以较高的活性催化丙烯齐聚获得聚合度较低的产物.与其他类型的茂金属催化剂相比, 基于桥联茚-芴配体结构的茂金属配合物具有突出的可修饰性, 为进一步调控聚合活性和选择性提供了有利条件.
基于以上研究基础, 我们合成了一系列亚乙基桥联多取代茚-芴锆、铪配合物, 在保留茚环3-位苄基的同时, 在配体的其他位置引入了特定的取代基, 希望进一步调控金属中心的空间和电子环境, 以发展一系列新型高性能催化剂用于丙烯选择性齐聚获得烯丙基封端产物.
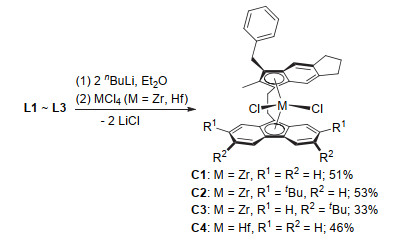
配体L1~L3通过5-苄基-6-甲基-1, 2, 3, 5-四氢-s-引达省的锂盐与相应的取代9-(2-溴乙基)芴发生亲核取代反应得到(Scheme 1).以L1~L3为原料, 将其制成双锂盐后直接与等物质的量的ZrCl4或HfCl4在乙醚中发生转金属化反应生成目标配合物(Scheme 2).通过重结晶纯化, 最终所得锆配合物为橙红色晶体, 铪配合物为黄色固体粉末.由于此类配合物在溶液中的稳定性较差, 导致了部分配合物在纯化过程中发生变质, 最终收率为33%~53%.
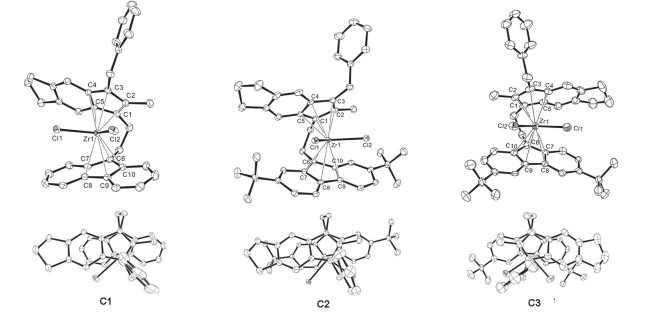
如图 2所示, 配合物C1~C3中金属中心的配位环境均为扭曲的准四面体构型.茚环3-位苄基朝向茚环上方远离金属中心的方向.此外, 从分子结构俯视图中可以看出, 配合物C1、C2和C3中的茚环相对于芴环均表现出向后方扭转的取向(远离配位氯原子方向), 说明金属中心前方空间环境较为拥挤.
在表 1中列出了配合物C1~C3的几个重要结构参数, 即金属中心与配体五元环中心的距离(M—CentInd、M—CentFlu)、茚环平面和芴环平面的二面角(α)、配体五元环中心与金属中心构成的夹角CentInd—M—CentFlu (β)、茚环和芴环的相对位置扭转角(RA).由于配体取代方式不同, 该系列配合物的各结构参数表现出一定的差异, 但总体与文献报道的亚乙基桥联茚-芴类配合物的参数值接近[17, 25-29].对比发现, 芴环叔丁基取代配合物C2和C3的α、Zr—CentInd、Zr—CentFlu均略微大于芴环不含取代基的配合物C1, 可能是由芴环上叔丁基对茚环的空间排斥作用所引起.该系列配合物中的茚环相对于芴环向后方扭转的角度RA在-11.30°到-17.07°之间.芴环叔丁基取代的配合物C2和C3中茚环向后扭转的角度大于C1, 其中芴环2, 7-位叔丁基取代的配合物C3的扭转角度最大.上述不同的茚环扭转角可能与金属中心前方空间的拥挤程度有关.
 下载:
导出CSV
下载:
导出CSV
 |
|||||
| Complexes | α/(°) | β/(°) | M—CentInd/nm | M—CentFlu/nm | RA/(°) |
| C1 | 60.48 | 128.76 | 0.2213 | 0.2267 | –11.30 |
| C2 | 61.25 | 128.07 | 0.2215 | 0.2268 | –17.07 |
| C3 | 61.70 | 128.89 | 0.2219 | 0.2285 | –14.77 |
以配合物C1~C4为主催化剂, 改性甲基铝氧烷(MMAO)或AliBu3/[Ph3C][B(C6F5)4](TIBA/TrB)为助催化剂, 催化丙烯齐聚结果见表 2.配合物ansa-C2H4-(3-Bn-Ind)(Flu)ZrCl2 (1)[30]为已报道的结构, 但尚未被用于催化丙烯齐聚研究, 故对其也进行了催化评价用于对比研究.
下载:
导出CSV
| Entry | Cat. | n(Al)/n(M) | Tp/℃ | Yield/g | Activity/(106 g"molM-1"h-1) | Allyl contentb/% | Vinylidene contentb/% | Mnc/(g"mol-1) |
| 1 | 1 | 4000 | 60 | 3.85 | 6.64 | 85 | 15 | 2800 |
| 2 | 4000 | 80 | 1.81 | 3.06 | 83 | 17 | 1700 | |
| 3 | 4000 | 100 | 1.32 | 2.11 | 71 | 29 | 900 | |
| 4 | C1 | 4000 | 40 | 3.76 | 6.01 | 40 | 61 | 1700 |
| 5 | 4000 | 60 | 2.19 | 3.51 | 41 | 59 | 1000 | |
| 6 | 4000 | 80 | 0.80 | 1.28 | 47 | 53 | 700 | |
| 7 | 4000 | 100 | 0.40 | 0.64 | 52 | 48 | 600 | |
| 8 | 2000 | 60 | 1.72 | 2.75 | 46 | 54 | 1500 | |
| 9 | 1000 | 60 | 1.42 | 2.27 | 51 | 49 | 2100 | |
| 10 | 400 | 60 | 1.19 | 1.91 | 46 | 54 | 1600 | |
| 11 | C2 | 4000 | 40 | 1.60 | 2.56 | 81 | 19 | 4500 |
| 12 | 4000 | 60 | 3.07 | 4.91 | 83 | 17 | 1400 | |
| 13 | 4000 | 80 | 2.15 | 3.44 | 85 | 15 | 800 | |
| 14 | 4000 | 100 | 1.67 | 2.67 | 86 | 14 | 600 | |
| 15 | 2000 | 60 | 1.80 | 2.88 | 83 | 17 | 1700 | |
| 16 | C3 | 4000 | 40 | 0.460 | 0.74 | 77 | 23 | 1800 |
| 17 | 4000 | 60 | 1.26 | 2.02 | 72 | 28 | 900 | |
| 18 | 4000 | 80 | 0.326 | 0.52 | 68 | 32 | 500 | |
| 19 | 4000 | 100 | 0.276 | 0.44 | 69 | 31 | 400 | |
| 20 | 2000 | 60 | 0.051 | 0.08 | 74 | 26 | 900 | |
| 21 d | C4 | — | 40 | 2.00 | 3.20 | 86 | 14 | 1000 |
| 22 d | — | 60 | 1.29 | 2.06 | 84 | 16 | 600 | |
| 23 d | — | 80 | 0.99 | 1.91 | 78 | 22 | 400 | |
| 24 d | — | 100 | 1.19 | 1.58 | — | — | dimers and trimerse | |
| a Conditions: MMAO as cocatalyst, toluene as solvent; V=25 mL; [Cat.]=5×10-5 mol/L; 30 min; 0.62 MPa of propylene. b Percentage of allyl or vinylidene ends in total unsaturated end groups, measured by 1H NMR spectroscopy. c Determined by 1H NMR spectroscopy. d Triisobutyl aluminum/[Ph3C][B(C6F5)4] (TIBA/TrB) as cocatalyst, [Hf]:[TIBA]:[TrB]=1:100:1.2. e Determined by GC. | ||||||||
齐聚产物的1H NMR分析表明产物中仅含有烯丙基和亚乙烯基两种不饱和端基, 根据文献报道[4, 17, 19, 22]分别由丙烯单体经1, 2-插入后发生β-Me消除反应和β-H消除反应形成.因此, 讨论中烯丙基端基的百分含量即等于β-Me消除选择性.
该系列配合物C1~C4催化丙烯聚合所得产物的分子量普遍较低, 在温和条件下(40~100 ℃)实现了丙烯的选择性齐聚, 获得了分子量在400到4500 g•mol-1范围内的齐聚产物(表 2).对比配合物1 (Entries 1~3)和C1 (Entries 5~7)的催化结果发现, 配合物C1中茚环2-位甲基以及5, 6-位亚丙基的引入对产物分子量具有明显的抑制作用, 这一结果由茚环2, 5, 6-位取代基对链增长反应速率以及链终止反应速率的综合影响所造成.
分析配合物催化丙烯齐聚活性发现, 茚环含有2-位甲基、5, 6-位亚丙基的锆配合物C1其催化活性明显低于2, 5, 6-位不含取代基的配合物1.然而, Rieger等[27]曾报道, 在配合物C2H4(Ind)(Flu)ZrCl2中进一步引入上述相同取代基后, 相应配合物C2H4 {2-Me-5, 6-[1, 3-(CH2)3]- Ind}(Flu)ZrCl2具有明显更高的催化丙烯聚合活性, 这一取代基效应与我们的结果相反.进一步文献调研发现, 在亚乙基桥联(茚)-(芴)类茂金属催化剂中引入2-位甲基后催化活性升高的现象普遍存在[27, 31, 32], 但其具体原因未见讨论.与这些文献所报道的催化剂结构相比较, 配合物C1和1的茚环3-位含有苄基取代, 金属中心的配位空间环境相对更为拥挤.因此, 我们认为对于该系列基于茚环3-位苄基取代的配合物, 茚环2-位甲基、5, 6-位亚丙基的进一步引入造成了金属中心周围的空间环境过于拥挤, 对丙烯单体的配位插入表现出了一定的抑制作用.
分析配合物C1和1催化丙烯齐聚所得产物的端基成分发现(表 2), 茚环的取代情况对烯丙基端基选择性(β-Me消除选择性)也有显著的影响.与我们的预期相反, 茚环进一步引入2-位甲基和5, 6-位亚丙基使得配合物C1在40~100 ℃条件下仅表现出40%~52%的β-Me消除选择性, 明显低于配合物1 (71%~85%).由于茚环2-位甲基距离活性中心较近, 我们认为β-Me消除选择性的降低可能主要由2-位甲基的引入所导致.通过之前的研究, 认为该类催化剂茚环3-位苄基与聚合活性链之间的非键空间作用是推动发生β-Me消除反应的主要动力[17].因此, 配合物茚环3-位苄基的具体空间取向会对β-Me消除选择性产生明显的影响, 苄基靠近金属中心的概率越大, 其与聚合链之间的非键空间作用越有效, 相应的β-Me消除选择性也越高.配合物C1中茚环2-位甲基处于3-位苄基的邻位, 其空间位阻作用一定程度上抑制了3-位苄基靠近金属中心的取向, 从而降低了茚环3-位苄基与聚合物链的非键作用倾向, 导致β-Me消除选择性也相应地有所降低(图 3).
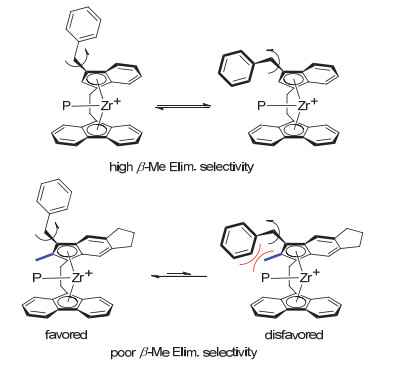
基于以上结果, 我们在配合物C1的芴环2, 7-位或3, 6-位进一步引入叔丁基, 期望通过增大配位中心的位阻环境来进一步促进β-Me消除反应, 并利用叔丁基的供电子性来提高配合物的催化活性.
比较配合物C1~C3催化丙烯齐聚所得产物的端基成分(表 2), 发现芴环2, 7-位或3, 6-位叔丁基的引入对烯丙基端基选择性(β-Me消除选择性)的提高均具有显著的作用.在40~100 ℃条件下, 催化剂C1仅表现出40%~52%的烯丙基端基选择性, 而芴环2, 7-位或3, 6-位引入叔丁基的C2和C3在相同条件下烯丙基端基选择性分别提高到81%~86%以及68%~77%.我们认为, 芴环2, 7-位或3, 6-位叔丁基与活性聚合链之间的非键空间作用有效地抑制了β-H消除反应过渡态的形成, 从而相对提高了β-Me消除反应的竞争性.与芴环3, 6-位叔丁基取代的C3相比, 芴环含有2, 7-位叔丁基的C2表现出更高的β-Me消除选择性, 这一结果与我们之前研究C2H4(3-Bn-Ind)(Flu')ZrCl2 (Flu'=2, 7-二叔丁基芴基、3, 6-二叔丁基芴基)系列配合物时的发现有所不同, 即之前是芴环3, 6-位叔丁基取代的络合物具有更高的β-Me消除选择性.对此, 我们认为本工作中C2和C3配体结构中茚环2-位甲基的存在也对金属中心造成一定的位阻效应, 这种情况下, 芴环2, 7-位叔丁基取代模式与C2、C3中的多取代茚基所构造出的配位环境更有利于β-Me消除过渡态的形成.相比之下, 芴环3, 6-位叔丁基与该多取代茚基的组合使得两个大π体系的开口有所增大, 故而对β-Me消除的选择性略低.
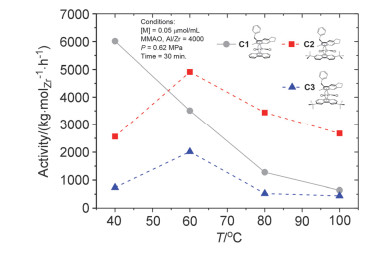
配合物中芴环的取代情况对催化丙烯齐聚活性也有显著的影响.从空间效应的角度, 芴环大位阻取代基的引入对烯烃的配位插入反应具有一定的抑制作用; 从电子因素的角度, 芴环2, 7-位或3, 6-位引入叔丁基后, 金属中心电子云密度有所上升, 有利于催化活性的提高[33, 34].其中, 空间位阻作用的有效程度与催化剂结构和反应温度有关.如图 4所示, 芴环2, 7-位叔丁基取代的配合物C2在较高温度条件下(60~100 ℃), 由于空间位阻作用不十分明显, 其催化丙烯齐聚活性主要受电子因素的影响, 明显高于相应芴环不含取代基的配合物C1.然而, 在较低温度条件下(<40 ℃), 空间位阻作用的有效性增强, 其催化活性反而低于配合物C1.对于配合物C3, 芴环3, 6-位叔丁基距离活性中心较近, 对单体配位插入的抑制作用也更为明显, 其电子因素方面对催化活性的促进作用被位阻效应所抵消, 因此C3在40~100 ℃条件下的催化活性普遍低于C1和C2.
分析配合物C1~C3催化丙烯齐聚所得产物的分子量(表 2), 发现芴环2, 7-位叔丁基取代的配合物C2催化所得产物的分子量较高; 然而, 3, 6-位叔丁基的引入对产物分子量的影响不大.这一结果主要由链增长反应速率(表现为活性)及链终止反应速率的综合影响所造成.
由于此类铪配合物在MMAO作用下, 其催化丙烯齐聚的活性物种会发生明显的失活现象[17, 35-38], 本文采用TIBA/TrB为助催化剂对铪配合物C4进行丙烯齐聚研究.结果显示, 催化剂体系C4/TIBA/TrB对丙烯齐聚表现出中等到高的催化活性(表 2, Entries 21~24).在较高温度条件下(80~100 ℃), C4/TIBA/TrB的催化活性甚至高于相应的锆催化剂体系C1/MMAO.与锆催化剂体系C1~C3/MMAO相比, 铪体系C4/TIBA/TrB催化所得齐聚物的分子量明显较低(40~80 ℃, 400~1000 g•mol-1), 100 ℃条件下甚至实现了丙烯的选择性二聚及三聚(经气相色谱分析以二聚物为主).由于Hf—C键略短于Zr—C键[16, 17], 铪配合物中配体取代基的空间效应更为有效, 其β-Me消除选择性也明显高于相应的锆体系C1/MMAO.由上可见, 铪体系在催化丙烯低聚获得低分子量烯丙基端基齐聚物(<1000 g•mol-1)方面具有一定的优势和潜力.
气相色谱分析结果表明, 配合物C4在100 ℃催化丙烯二聚所得产物中包含1-戊烯、4-甲基-1-戊烯、2-甲基-1-戊烯以及2, 4-二甲基-1-戊烯四种组分, 说明同时存在β-Me消除和β-H消除链终止反应[20, 39].此外, 我们还发现二聚产物中的烯丙基端基含量仅为49%, 明显低于在40~80 ℃条件下催化所得产物(Mn=400~1000 g•mol-1)中的烯丙基端基含量(78%~86%).这一现象进一步说明了活性齐聚物链与配体取代基之间的空间位阻效应是推动β-Me消除反应发生的驱动力.由于二聚过程中的活性聚合链较短, 其与配体位阻基团之间的位阻作用也较弱, 从而导致了β-Me消除反应相对于β-H消除反应的优势有所降低.
由于链终止反应的活化能远远大于链增长反应, 随着反应温度的升高, 配合物C1~C4催化丙烯聚合所得产物的分子量均呈现明显的下降趋势(表 2).然而, 在恒定丙烯压力的条件下, 温度变化对链转移反应的影响需要综合考虑热力学条件变化、单体浓度变化及聚合链长短等因素.因此, 对于不同的催化剂结构, 反应温度对端基选择性的影响表现出不同的变化规律.随温度的升高, 配合物C1和C2催化丙烯齐聚的β-Me消除选择性有所上升, 而C3和C4却表现出相反的趋势.总体来说, 该系列配合物催化丙烯齐聚的端基选择性受反应温度的影响较小; 配合物C2~C4在40~100 ℃以普遍较高的选择性催化丙烯齐聚获得不同链长的烯丙基封端产物.
关于Al/Zr物质的量之比对茂金属配合物催化烯烃聚合反应活性的影响, 人们普遍认为高Al/Zr物质的量之比条件下, 活性物种阴阳离子间的作用较弱, 其催化活性也相应较高.随着Al/Zr物质的量之比逐渐增大, 配合物C1~C3对丙烯齐聚的催化活性也普遍表现出上升趋势(表 2).其中催化剂体系C1/MMAO在Al/Zr物质的量之比低至400时, 仍然能够高效地催化丙烯齐聚(1.91×106 g•molZr-1•h-1).然而, 对于催化剂体系C3/MMAO, 当Al/Zr物质的量之比从4000降低至2000时, 其催化活性急剧下降.
此外, Al/Zr物质的量之比对齐聚产物的分子量及端基成分也有一定的影响.对于配合物C1, 产物分子量随Al/Zr物质的量之比的升高表现出一定的下降趋势, 说明聚合过程的链终止反应中可能存在一小部分聚合物链向Al中心转移的情况.对于配合物C2和C3, Al/Zr物质的量之比对产物分子量的影响较小, 说明链终止过程基本不涉及聚合物链向Al的转移.分析不同Al/Zr物质的量之比条件下催化所得产物的端基成分发现, 烯丙基端基含量受Al/Zr物质的量之比的影响较小, 对于不同结构的催化剂, 其影响规律也有所不同.这一现象主要由不同Al/Zr物质的量之比条件下活性物种阴阳离子对的空间及电子环境的差异所引起.
丙烯浓度对产物分子量以及端基选择性的影响如图 5和图 6所示(聚合体系中丙烯浓度根据丙烯压力换算获得[40]).配合物C1~C4与MMAO或TIBA/TrB组成的催化剂体系催化丙烯齐聚时, 丙烯浓度变化对产物分子量及链终止反应选择性的影响均较小.由于链增长反应速率通常对丙烯浓度呈一级动力学关系, 聚合产物分子量又几乎不受单体浓度的影响, 说明聚合过程中的总体链转移反应也与丙烯浓度呈一级动力学关系, 是涉及向丙烯单体转移的双分子过程, 且β-Me消除和β-H消除反应均如此[17, 41].
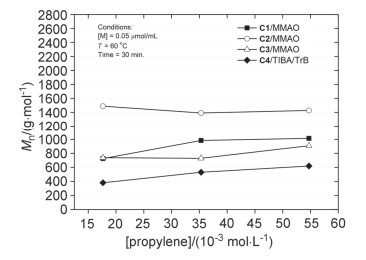
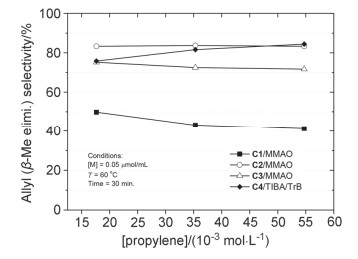
与文献报道的结果相比[16, 20], 本文发展的丙烯选择性齐聚催化剂C2~C4对反应条件的依赖性较小, 在较宽温度范围内、不同Al/Zr物质的量之比以及不同单体压力条件下, 均能保持较高的β-Me消除选择性以及中等到高的催化活性.
合成了数个亚乙基桥联多取代茚-芴锆、铪配合物C1~C4.该系列配合物与MMAO或TIBA/TrB组成的催化剂体系在40~100 ℃条件下以中等到高的活性、高达86%的β-Me消除选择性催化丙烯齐聚获得烯丙基封端产物, 产物分子量Mn为400~4500 g•mol-1.研究结果表明, 该系列配合物中茚环2-位甲基以及5, 6-位亚丙基的引入是控制产物分子量较低的关键, 但其对催化活性和β-Me消除选择性均表现出不利的影响.然而, 进一步在芴环引入2, 7-位或3, 6-位叔丁基使得锆配合物C2和C3的β-Me消除选择性得到显著提高.此外, 与相应的锆催化剂体系相比, 铪体系C4/TIBA/TrB具有明显较高的β-Me消除选择性, 产物分子量也较低.
该类催化剂的桥联茚-芴配体结构具有高度的可修饰性, 突破了丙烯齐聚反应中β-Me消除难以普遍化的现状.通过对这类催化剂结构的取代修饰, 有望进一步提高其催化丙烯聚合反应的活性及β-Me消除选择性.本研究为进一步发展高选择性丙烯齐聚催化剂、探索β-Me消除反应在丙烯转化反应中的策略性应用提供了重要参考.
涉及对水汽或氧气敏感的实验均在氩气保护下进行, 并采用标准Schlenk操作.乙醚、石油醚、正己烷、四氢呋喃、甲苯均采用在氩气保护下与钠丝一起加热回流的方法进行干燥处理, 并以二苯甲酮为指示剂, 回流至深紫色或深蓝色后收集使用.二氯甲烷采用加入CaH2加热回流的方法进行干燥处理.
中间体5-苄基-6-甲基-1, 2, 3, 5-四氢-s-引达省[42]、9-(2-溴乙基)芴[43]、1-[9-(2, 7-二叔丁基)芴基]-2-溴乙烷[43]、1-[9-(3, 6-二叔丁基)芴基]-2-溴乙烷[43]采用类似文献报道的方法合成.
取一100 mL Schlenk瓶, 抽烤并置换氩气三次以除水除氧.称取2.15 g (8.26 mmol) 5-苄基-6-甲基-1, 2, 3, 5-四氢-s-引达省于其中, 用30 mL乙醚溶解.液氮-乙醇浴冷却下用注射器缓慢滴加3.50 mL (2.36 mol/L, 8.26 mmol)正丁基锂的正己烷溶液.滴毕后自然升至室温搅拌反应. 48 h后, 冰-盐浴冷却下加入2.14 g (7.83 mmol) 9-(2-溴乙基)芴, 自然升至室温搅拌反应. 24 h后, 薄层色谱(TLC)跟踪显示未反应完全, 加入20 mL四氢呋喃, 继续搅拌反应2 h后, TLC跟踪显示反应基本完全.加入饱和氯化铵水溶液终止反应, 旋蒸除去溶剂后用二氯甲烷萃取(100 mL×3), 合并有机相后用无水硫酸镁干燥.滤除干燥剂, 旋蒸除去溶剂, 得白色固体粗产品, 用二氯甲烷重结晶得到2.50 g白色晶体, 产率71%. 1H NMR (CDCl3, 400 MHz) δ: 1.84~1.44 (m, 4H, CH2CH2CH2, Ind-CH2CH2-Flu), 1.90 (s, 3H, 2-Ind-CH3), 2.20~2.00 (m, 2H, Ind-CH2CH2-Flu), 3.00~2.77 (m, 4H, CH2CH2CH2), 3.18 (brs, 1H, 1-Ind-H), 3.97~3.81 (m, 3H, PhCH2 and 9-Flu-H), 6.97 (s, 1H, ArH), 7.02 (s, 1H, ArH), 7.49~7.15 (m, 11H, ArH), 7.78 (d, J=10.0 Hz, 2H, ArH); 13C NMR (CDCl3, 100 MHz) δ: 12.6, 24.3, 26.0, 26.1, 31.5, 32.9, 33.0, 47.3, 51.0, 114.8, 118.9, 119.9, 120.0, 124.3, 124.6, 126.0, 127.0, 127.08, 127.11, 127.12, 128.5, 128.6, 135.5, 140.1, 140.4, 141.5, 141.6, 142.2, 142.3, 144.7, 144.8, 147.2, 147.4. Anal. calcd for C35H32•0.11CH2Cl2: C 91.28, H 7.03; found C 91.56, H 7.01.
取一100 mL Schlenk瓶, 抽烤并置换氩气三次以除水除氧.称取2.20 g (8.45 mmol) 5-苄基-6-甲基-1, 2, 3, 5-四氢-s-引达省于其中, 用30 mL乙醚溶解.液氮-乙醇浴冷却下用注射器缓慢滴加3.58 mL (2.36 mol/L, 8.45 mmol)正丁基锂的正己烷溶液.滴毕后自然升至室温搅拌反应. 48 h后, 冰-盐浴冷却下加入3.10 g (8.05 mmol) 2, 7-二叔丁基-9-(2-溴乙基)芴, 自然升至室温搅拌反应. 24 h后, TLC跟踪显示未反应完全, 加入20 mL四氢呋喃, 继续搅拌反应2 h后, TLC跟踪显示反应基本完全.加入饱和氯化铵水溶液终止反应, 旋蒸除去溶剂后用二氯甲烷萃取(100 mL×3), 合并有机相后用无水硫酸镁干燥.滤除干燥剂, 旋蒸除去溶剂, 得白色固体粗产品.以硅胶为固定相, 石油醚/二氯甲烷(V:V=10:1)为洗脱剂, 柱层析分离获得白色固体3.15 g, 产率69%. 1H NMR (CDCl3, 400 MHz) δ: 1.38 [s, 9H, C(CH3)3], 1.40 [s, 9H, C(CH3)3], 1.82~1.20 (m, 4H, CH2CH2CH2, Ind-CH2CH2-Flu), 1.88 (s, 3H, 2-Ind-CH3), 2.16~1.98 (m, 2H, Ind-CH2CH2-Flu), 2.95~2.77 (m, 4H, CH2CH2CH2), 3.12 (brs, 1H, 1-Ind-H), 3.97~3.81 (m, 3H, PhCH2, 9-Flu-H), 6.96 (s, 1H, ArH), 7.00 (s, 1H, ArH), 7.21~7.13 (m, 1H, ArH), 7.32~7.22 (m, 4H, ArH), 7.43~7.32 (m, 3H, ArH), 7.46 (s, 1H, ArH), 7.64 (d, J=8.0 Hz, 2H, ArH); 13C NMR (CDCl3, 100 MHz) δ: 12.4, 23.0, 25.4, 26.0, 31.5, 31.87, 31.91, 32.9, 33.0, 35.1, 47.0, 50.8, 114.9, 118.5, 119.1, 119.2, 120.9, 121.2, 123.9, 124.0, 126.0, 128.5, 128.6, 135.3, 139.0, 139.1, 140.2, 140.33, 142.27, 142.29, 144.8, 144.9, 147.1, 147.3, 149.86, 149.91. Anal. calcd for C43H48: C 91.43, H 8.57; found C 91.17, H 8.63.
取一100 mL Schlenk瓶, 抽烤并置换氩气三次以除水除氧.称取2.40 g (9.22 mmol) 5-苄基-6-甲基-1, 2, 3, 5-四氢-s-引达省于其中, 用30 mL乙醚溶解.液氮-乙醇浴冷却下用注射器缓慢滴加3.91 mL (2.36 mol/L, 9.22 mmol)正丁基锂的正己烷溶液.滴毕后自然升至室温搅拌反应. 48 h后, 冰-盐浴冷却下加入3.40 g (8.83 mmol) 3, 6-二叔丁基-9-(2-溴乙基)芴, 自然升至室温搅拌反应. 24 h后, TLC跟踪显示未反应完全, 加入20 mL四氢呋喃, 继续搅拌反应2 h后, TLC跟踪显示反应基本完全.加入饱和氯化铵水溶液终止反应, 旋蒸除去溶剂后用二氯甲烷萃取(120 mL×3), 合并有机相后用无水硫酸镁干燥.滤除干燥剂, 旋蒸除去溶剂, 得白色固体粗产品.以硅胶为固定相, 石油醚/二氯甲烷(V:V=10:1)为洗脱剂, 柱层析分离获得白色固体3.08 g, 产率62%. 1H NMR (CDCl3, 400 MHz) δ: 1.43 [s, 9H, C(CH3)3], 1.44 [s, 9H, C(CH3)3], 1.86~1.49 (m, 4H, Ind-CH2CH2-Flu, CH2CH2CH2), 1.91 (s, 3H, 2-Ind-CH3), 2.16~1.99 (m, 2H, Ind-CH2CH2-Flu), 2.96~2.78 (m, 4H, CH2CH2CH2), 3.20 (br s, 1H, 1-Ind-H), 3.95~3.73 (m, 3H, PhCH2, 9-Flu-H), 6.94 (s, 1H, ArH), 7.04 (s, 1H, ArH), 7.20~7.12 (m, 1H, ArH), 7.28~7.20 (m, 5H, ArH), 7.37~7.28 (m, 3H, ArH), 7.79 (s, 2H, ArH); 13C NMR (CDCl3, 100 MHz) δ: 12.7, 25.1, 26.0, 26.7, 31.5, 31.9, 32.9, 33.0, 35.1, 46.6, 51.2, 114.8, 116.4, 116.5, 119.0, 123.9, 124.06, 124.10, 124.2, 126.0, 128.5, 128.6, 135.4, 140.0, 140.4, 141.5, 141.6, 142.2, 142.4, 144.8, 145.0, 150.13, 150.2. Anal. calcd for C43H48: C 91.43, H 8.57; found C 91.38, H 8.42.
取一50 mL Schlenk瓶, 抽烤并置换氩气三次以除水除氧.称取1.10 g (2.43 mmol)配体L1于其中, 加入30 mL乙醚, 充分搅拌.冰-盐浴冷却下用注射器抽缓慢滴加2.10 mL (2.36 mol/L, 4.9 mmol)正丁基锂的正己烷溶液.滴毕后自然升至室温搅拌反应, 生成大量黄色沉淀.反应48 h后, 冰-盐浴冷却下加入0.566 g (2.43 mmol) ZrCl4, 自然升至室温搅拌反应, 体系变为红色浊液.反应48 h后, 真空除去溶剂, 加入50 mL二氯甲烷, 搅拌2 h后离心分离得到红色清液, 浓缩至饱和后加入20 mL正己烷, 于-20 ℃冰箱重结晶, 分离获得橙红色细小晶体0.76 g, 产率51%. 1H NMR (CDCl3, 400 MHz) δ: 1.99 (s, 3H, 2-Ind-CH3), 2.13~1.86 (m, 2H, CH2CH2CH2), 3.02~2.74 (m, 4H, CH2CH2CH2), 4.01 (s, 2H, PhCH2), 3.11~3.98 (m, 1H, Ind-CH2CH2-Flu), 4.24~4.10 (m, 1H, Ind-CH2CH2-Flu), 4.78~4.63 (m, 1H, Ind-CH2CH2-Flu), 6.89 (d, J=8.4 Hz, 2H, ArH), 7.17~7.03 (m, 5H, ArH), 7.28 (t, J=8.0 Hz, 1H, ArH), 7.38 (t, J=8.0 Hz, 1H, ArH), 7.61~7.51 (m, 2H, ArH), 7.82~7.75 (m, 2H, ArH), 7.84 (d, J=8.4 Hz, 1H, ArH), 7.94 (d, J=8.4 Hz, 1H, ArH); 13C NMR (CDCl3, 100 MHz) δ: 13.2, 26.3, 29.2, 30.0, 32.0, 32.5, 32.8, 103.5, 116.5, 116.9, 118.1, 119.4, 122.3, 122.9, 123.7, 123.9, 124.3, 124.5, 125.4, 125.7, 126.0, 126.3, 127.6, 128.1, 128.3, 128.5, 129.6, 130.9, 140.6, 143.5, 144.7. Anal. calcd for C35H30Cl2Zr•0.5CH2Cl2: C 65.08, H 4.77; found C 64.84, H 5.12.
取一50 mL Schlenk瓶, 抽烤并置换氩气三次以除水除氧.称取1.00 g (1.77 mmol)配体L2于其中, 加入30 mL乙醚, 充分搅拌.冰-盐浴冷却下用注射器缓慢滴加1.50 mL (2.36 mol/L, 3.54 mmol)正丁基锂的正己烷溶液.滴毕后自然升至室温搅拌反应, 生成大量黄色沉淀.反应48 h后, 在冰-盐浴冷却下加入0.412 g (1.77 mmol) ZrCl4, 自然升至室温搅拌反应, 体系变为红色浊液.反应48 h后, 真空除去溶剂, 加入50 mL二氯甲烷, 搅拌2 h后离心分离得到红色清液, 浓缩至饱和后加入15 mL正己烷, 于-20 ℃冰箱重结晶, 分离获得橙红色晶体0.68 g, 产率53%. 1H NMR (CDCl3, 400 MHz) δ: 1.29 [s, 9H, C(CH3)3], 1.42 [s, 9H, C(CH3)3], 1.97 (s, 3H, 2-Ind-CH3), 2.02~1.91 (m, 2H, CH2CH2CH2), 3.02~2.70 (m, 4H, CH2CH2CH2), 3.93~3.81 (m, 1H, Ind-CH2CH2-Flu), 4.00 (s, 2H, PhCH2), 4.08~3.97 (m, 1H, Ind-CH2CH2-Flu), 4.21~4.09 (m, 1H, Ind-CH2CH2-Flu), 4.71~4.59 (m, 1H, Ind-CH2CH2-Flu), 6.88 (d, J=6.8 Hz, 2H, ArH), 7.16~7.03 (m, 4H, ArH), 7.34 (dd, J=8.8 Hz, 1.6 Hz, 1H, ArH), 7.43 (s, 1H, ArH), 7.73~7.61 (m, 3H, ArH), 7.77 (s, 1H, ArH), 7.82 (d, J=8.8 Hz, 1H, ArH); 13C NMR (CDCl3, 100 MHz) δ: 13.4, 26.5, 29.2, 30.0, 31.1, 31.2, 32.1, 32.5, 33.1, 35.3, 35.5, 103.2, 116.3, 116.9, 117.1, 117.9, 119.2, 120.4, 122.4, 123.7, 123.8, 124.1, 124.7, 125.3, 125.9, 126.7, 128.2, 128.4, 128.6, 129.9, 130.1, 140.7, 142.9, 144.6, 150.5, 150.7. Anal. calcd for C43H46Cl2Zr•0.4CH2Cl2: C 68.69, H 6.22; found C 68.55, H 6.51.
取一50 mL Schlenk瓶, 抽烤并置换氩气三次以除水除氧.称取1.10 g (1.95 mmol)配体L3于其中, 加入30 mL乙醚, 充分搅拌.冰-盐浴冷却下用注射器缓慢滴加1.65 mL (2.36 mol/L, 3.90 mmol)正丁基锂的正己烷溶液.滴毕后自然升至室温搅拌反应, 生成大量黄色沉淀.反应48 h后, 冰-盐浴冷却下加入0.454 g (1.95 mmol) ZrCl4, 自然升至室温搅拌反应, 体系变为红色浊液.反应48 h后, 真空去除溶剂, 加入50 mL二氯甲烷, 搅拌2 h后离心分离得到红色清液.真空抽除溶剂后, 用甲苯/正己烷溶解, 于-20 ℃冰箱重结晶, 分离获得橙红色晶体0.47 g, 产率33%. 1H NMR (CDCl3, 400 MHz) δ: 1.34 [m, 9H, C(CH3)3], 1.48 [m, 9H, C(CH3)3], 2.13~1.89 (m, 2H, CH2CH2CH2), 2.02 (s, 3H, 2-Ind-CH3), 3.02~2.72 (m, 4H, CH2CH2CH2), 4.19~3.82 (m, 5H, Ind-CH2CH2-Flu, PhCH2), 4.68~4.54 (m, 1H, Ind-CH2CH2-Flu), 6.91 (d, J=7.6 Hz, 2H, ArH), 7.23~7.02 (m, 5H, ArH), 7.52~7.42 (m, 2H, ArH), 7.80~7.67 (m, 3H, ArH), 7.87 (s, 1H, ArH); 13C NMR (CDCl3, 100 MHz) δ: 13.2, 26.7, 29.3, 30.0, 31.9, 32.1, 32.6, 32.9, 35.2, 35.5, 102.7, 116.5, 116.8, 117.9, 118.9, 119.1, 120.0, 121.7, 122.7, 123.2, 123.6, 123.7, 125.0, 125.5, 125.9, 126.3, 126.7, 127.7, 128.2, 128.4, 129.3, 130.3, 140.9, 142.9, 144.3, 148.8, 150.4. Anal. calcd for C43H46Cl2Zr: C 71.24, H 6.40; found C 71.01, H 6.57.
取一50 mL Schlenk瓶, 抽烤并置换氩气三次以除水除氧.称取0.90 g (1.60 mmol)配体L1于其中, 加入30 mL乙醚, 充分搅拌.冰-盐浴冷却下用注射器缓慢滴加1.35 mL (2.36 mol/L, 3.20 mmol)正丁基锂的正己烷溶液.滴毕后自然升至室温搅拌反应, 生成大量黄色沉淀.反应48 h后, 冰-盐浴冷却下加入0.512 g (1.60 mmol) HfCl4, 自然升至室温搅拌反应, 体系变为黄色浊液.反应48 h后, 真空除去溶剂, 加入50 mL二氯甲烷, 搅拌2 h后离心分离得到黄色清液.真空抽除溶剂后, 用甲苯/正己烷溶解, 于-20 ℃冰箱重结晶, 分离获得黄色固体粉末0.51 g, 产率46%. 1H NMR (CDCl3, 400 MHz) δ: 2.04~1.90 (m, 2H, CH2CH2CH2), 2.06 (s, 3H, 2-Ind- CH3), 3.04~2.77 (m, 4H, CH2CH2CH2), 4.01 (s, 2H, PhCH2), 4.06~3.95 (m, 1H, Ind-CH2CH2-Flu), 4.29~4.07 (m, 2H, Ind-CH2CH2-Flu), 4.72~4.59 (m, 1H, Ind- CH2CH2-Flu), 6.88 (d, J=7.2 Hz, 2H, ArH), 7.16~7.00 (m, 5H, ArH), 7.25 (t, J=8.0 Hz, 1H, ArH), 7.33 (t, J=7.6 Hz, 1H, ArH), 7.49 (d, J=8.4 Hz, 1H, ArH), 7.55 (t, J=7.2 Hz, 1H, ArH), 7.85~7.73 (m, 3H, ArH), 7.94 (d, J=8.4 Hz, 1H, ArH); 13C NMR (CDCl3, 100 MHz) δ: 13.0, 26.4, 28.5, 29.3, 32.0, 32.4, 32.7, 99.2, 113.9, 116.5, 116.7, 117.9, 122.1, 122.2, 123.59, 123.63, 124.2, 124.4, 125.3, 125.5, 125.8, 126.0, 127.3, 127.4, 128.09, 128.14, 128.5, 129.1, 129.3, 141.0, 143.0, 144.5. Anal. calcd for C35H30Cl2Hf•0.6CH2Cl2: C 56.94, H 4.19; found C 56.48, H 4.49.
于Schlenk操作线上抽烤安培瓶并置换氩气三次以除水除氧, 称取一定量的配合物于安培瓶, 用注射器加入所需的甲苯, 配置成一定浓度的催化剂溶液, 使用时间不超过24 h.
将50 mL玻璃内衬置于高压釜体内, 放入搅拌子, 密闭体系后抽真空, 同时将釜体用油浴加热至180 ℃, 保持该状态30 min, 期间置换丙烯三次以除水除氧.关闭真空阀, 保持0.05 MPa丙烯压力, 调节温度至聚合反应所需温度.在丙烯保护下, 用注射器加入所需量的甲苯, 关闭加料口后搅拌恒温.
恒温后, 加入助催化剂及主催化剂.对于MMAO体系, 其加料方式为:在丙烯保护下, 加入所需量的MMAO溶液后搅拌10 min; 然后, 加入所需量的催化剂溶液, 调节丙烯至所需压力, 齐聚反应开始.对于TIBA/TrB体系, 其加料方式为:在丙烯保护下, 加入所需量的TIBA溶液, 搅拌10 min; 然后, 加入所需量的催化剂溶液, 搅拌反应10 min; 最后, 加入所需量的TrB溶液, 调节丙烯至所需压力, 齐聚反应开始.
齐聚反应结束时, 关闭丙烯进气阀, 停止搅拌, 快速用冰-盐浴冷却后缓慢卸去丙烯压力; 打开釜体取出玻璃内衬, 在冰-盐浴冷却下加入200 μL内标正庚烷, 然后再缓慢加入2.0 mL甲醇, 充分搅拌后取少量液体经过滤后封装于密闭玻璃管内, 用于易挥发齐聚物组份的分析.在剩下的反应液中滴加质量分数为3%的盐酸-甲醇溶液, 充分搅拌1 h以上.过滤得到体系中的不溶齐聚物.分出滤液中的有机相, 水洗三次后合并有机相, 用无水硫酸镁干燥后过滤, 旋蒸除去溶剂后获得可溶产物, 进一步真空干燥至恒重后称重.
O'Reilly, M. E.; Dutta, S.; Veige, A. S. Chem. Rev. 2016, 116, 8105. doi: 10.1021/acs.chemrev.6b00054
Janiak, C. Coord. Chem. Rev. 2006, 250, 66. doi: 10.1016/j.ccr.2005.02.016
Janiak, C.; Blank, F. Macromol. Symp. 2006, 236, 14. doi: 10.1002/masy.200690047
Janiak, C.; Lange, K. C. H.; Marquardt, P.; Krüger, R.-P.; Hanselmann, R. Macromol. Chem. Phys. 2002, 203, 129. doi: 10.1002/1521-3935(20020101)203:1<129::AID-MACP129>3.0.CO;2-C
Resconi, L.; Camurati, I.; Sudmeijer, O. Top. Catal. 1999, 7, 145. doi: 10.1023/A:1019115801193
陈志康, 毛远洪, 曹育才, 梁胜彪, 宋莎, 倪晨, 刘振宇, 叶晓峰, 沈安, 朱红平, 有机化学, 2018, 38, 2937. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346644.shtmlChen, Z.; Mao, Y.; Cao, Y.; Liang, S.; Song, S.; Ni, C.; Liu, Z.; Ye, X.; Shen, A.; Zhu, H. Chin. J. Org. Chem. 2018, 38, 2937. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346644.shtml
Tsou, A. H.; López-Barrón, C. R.; Jiang, P.; Crowther, D. J.; Zeng, Y. Polymer 2016, 104, 72. doi: 10.1016/j.polymer.2016.09.088
Ohtaki, H.; Deplace, F.; Vo, G. D.; LaPointe, A. M.; Shimizu, F.; Sugano, T.; Kramer, E. J.; Fredrickson, G. H.; Coates, G. W. Macromolecules 2015, 48, 7489. doi: 10.1021/acs.macromol.5b01975
Schöbel, A.; Lanzinger, D.; Rieger, B. Organometallics 2013, 32, 427. doi: 10.1021/om300781a
Rose, J. M.; Mourey, T. H.; Slater, L. A.; Keresztes, I.; Fetters, L. J.; Coates, G. W. Macromolecules 2008, 41, 559. doi: 10.1021/ma702190c
Weng, W.; Markel, E. J.; Peacock, A. J.; Dekmezian, A. H. Macromol. Rapid Commun. 2001, 22, 1488. doi: 10.1002/1521-3927(20011201)22:18<1488::AID-MARC1488>3.0.CO;2-I
Markel, E. Macromolecules 2000, 33, 8541. doi: 10.1021/ma001087b
Watson, P. L.; Roe, D. C. J. Am. Chem. Soc. 1982, 104, 6471. doi: 10.1021/ja00387a064
Eshuis, J. J. W.; Tan, Y. Y.; Teuben, J. H.; Renkema, J. J. Mol. Catal. 1990, 62, 277. doi: 10.1016/0304-5102(90)85223-5
Eshuis, J. J. W.; Tan, Y. Y.; Meetsma, A.; Teuben, J. H.; Renkema, J.; Evens, G. G. Organometallics 1992, 11, 362. doi: 10.1021/om00037a061
Resconi, L.; Piemontesi, F.; Franciscono, G.; Abis, L.; Fiorani, T. J. Am. Chem. Soc. 1992, 114, 1025. doi: 10.1021/ja00029a035
Zhang, L.; Ma, H. Chin. J. Polym. Sci. 2019, 37, 578. doi: 10.1007/s10118-019-2224-1
Machat, M. R.; Lanzinger, D.; Pöthig, A.; Rieger, B. Organometallics 2017, 36, 399. doi: 10.1021/acs.organomet.6b00814
Bader, M.; Marquet, N.; Kirillov, E.; Roisnel, T.; Razavi, A.; Lhost, O.; Carpentier, J.-F. Organometallics 2012, 31, 8375. doi: 10.1021/om300957k
Suzuki, Y.; Yasumoyo, T.; Mashima, K.; Okuda, J. J. Am. Chem. Soc. 2006, 128, 13017. doi: 10.1021/ja063717g
Moscardi, G.; Resconi, L.; Cavallo, L. Organometallics 2001, 20, 1918. doi: 10.1021/om000680e
Weng, W.; Markel, E. J.; Dekmezian, A. H. Macromol. Rapid Commun. 2000, 21, 1103. doi: 10.1002/1521-3927(20001101)21:16<1103::AID-MARC1103>3.0.CO;2-F
Resconi, L.; Piemontesi, F.; Camurati, I.; Sudmeijer, O.; Nifant'ev, I. E.; Ivchenko, P. V.; Kuz'mina, L. G. J. Am. Chem. Soc. 1998, 120, 2308. doi: 10.1021/ja973160s
Resconi, L.; Jones, R. L.; Rheingold, A. L.; Yap, G. P. A. Organometallics 1996, 15, 998. doi: 10.1021/om950197h
Wang, Y.; Huang, W.; Ma, H.; Huang, J. Polyhedron 2014, 76, 81. doi: 10.1016/j.poly.2014.03.019
Deisenhofer, S.; Feifel, T.; Kukral, J.; Klinga, M.; Leskela, M.; Rieger, B. Organometallics 2003, 22, 3495. doi: 10.1021/om030212f
Dietrich, U.; Hackmann, M.; Rieger, B.; Klinga, M.; Leskela, M. J. Am. Chem. Soc. 1999, 121, 4348. doi: 10.1021/ja9833220
Cobzaru, C.; Deisenhofer, S.; Harley, A.; Troll, C.; Hild, S.; Rieger, B. Macromol. Chem. Phys. 2005, 206, 1231. doi: 10.1002/macp.200400551
Rieger, B.; Jany, G.; Fawzi, R.; Steimann, M. Organometallics 1994, 13, 647. doi: 10.1021/om00014a041
Alt, H. G.; Jung, M. J. Organomet. Chem. 1999, 580, 1. doi: 10.1016/S0022-328X(98)00736-0
Thomas, E. J.; Rausch, M. D.; Chien, J. C. W. Organometallics 2000, 19, 4077. doi: 10.1021/om000256d
Thomas, E. J.; Chien, J. C. W.; Rausch, M. D. Macromolecules 2000, 33, 1546. doi: 10.1021/ma991463w
Schmid, M. A.; Alt, H. G.; Milius, W. J. Organomet. Chem. 1995, 501, 101. doi: 10.1016/0022-328X(95)05640-B
Spaleck, W.; Antberg, M.; Rohrmann, J.; Winter, A.; Bachmann, B.; Kiprof, P.; Behm, J.; Herrmann, W. A. Angew. Chem., Int. Ed. Engl. 1992, 31, 1347. doi: 10.1002/anie.199213471
Chen, E. Y.-X.; Marks, T. J. Chem. Rev. 2000, 100, 1391. doi: 10.1021/cr980462j
Busico, V.; Cipullo, R.; Cutillo, F.; Friederichs, N.; Ronca, S.; Wang, B. J. Am. Chem. Soc. 2003, 125, 12402. doi: 10.1021/ja0372412
Stapleton, R. A.; Galan, B. R.; Collins, S.; Simons, R. S.; Garrison, J. C.; Youngs, W. J. J. Am. Chem. Soc. 2003, 125, 9246. doi: 10.1021/ja030121+
Busico, V.; Cipullo, R.; Pellecchia, R.; Talarico, G.; Razavi, A. Macromolecules 2009, 42, 1789. doi: 10.1021/ma900066n
Mise, T.; Kageyama, A.; Miya, S.; Yamazaki, H. Chem. Lett. 1991, 1525.
Busico, V.; Brita, D.; Caporaso, L.; Cipullo, R.; Vacatello, M. Macromolecules 1997, 30, 3971. doi: 10.1021/ma970042g
Stehling, U.; Diebold, J.; Kirsten, R.; Roell, W.; Brintzinger, H. H.; Juengling, S.; Muelhaupt, R.; Langhauser, F. Organometallics 1994, 13, 964. doi: 10.1021/om00015a033
Aitola, E.; Surakka, M.; Repo, T.; Linnolahti, M.; Lappalainen, K.; Kervinen, K.; Klinga, M.; Pakkanen, T.; Leskela, M. J. Organomet. Chem. 2005, 690, 773. doi: 10.1016/j.jorganchem.2004.09.089
Perumattam, J.; Shao, C.; Confer, W. L. Synthesis 1994, 1181.

图 3 茚环3-位苄基的取向对β-Me消除反应的影响
Figure 3 Influence of 3-benzyl orientation on β-Me elimination

图 5 丙烯浓度对产物分子量的影响
Figure 5 Influence of propylene concentration on product molecular weight

图 6 丙烯浓度对β-Me消除选择性的影响
Figure 6 Influence of propylene concentration on β-methyl elimination selectivity
表 1 配合物C1~C3的部分结构参数
Table 1. Selected geometric parameters of complexes C1~C3
|
|||||
| Complexes | α/(°) | β/(°) | M—CentInd/nm | M—CentFlu/nm | RA/(°) |
| C1 | 60.48 | 128.76 | 0.2213 | 0.2267 | –11.30 |
| C2 | 61.25 | 128.07 | 0.2215 | 0.2268 | –17.07 |
| C3 | 61.70 | 128.89 | 0.2219 | 0.2285 | –14.77 |
 下载: 导出CSV
下载: 导出CSV
表 2 配合物1和C1~C4催化丙烯齐聚反应结果a
Table 2. Propylene oligomerization catalyzed by complexes 1 and C1~C4
| Entry | Cat. | n(Al)/n(M) | Tp/℃ | Yield/g | Activity/(106 g"molM-1"h-1) | Allyl contentb/% | Vinylidene contentb/% | Mnc/(g"mol-1) |
| 1 | 1 | 4000 | 60 | 3.85 | 6.64 | 85 | 15 | 2800 |
| 2 | 4000 | 80 | 1.81 | 3.06 | 83 | 17 | 1700 | |
| 3 | 4000 | 100 | 1.32 | 2.11 | 71 | 29 | 900 | |
| 4 | C1 | 4000 | 40 | 3.76 | 6.01 | 40 | 61 | 1700 |
| 5 | 4000 | 60 | 2.19 | 3.51 | 41 | 59 | 1000 | |
| 6 | 4000 | 80 | 0.80 | 1.28 | 47 | 53 | 700 | |
| 7 | 4000 | 100 | 0.40 | 0.64 | 52 | 48 | 600 | |
| 8 | 2000 | 60 | 1.72 | 2.75 | 46 | 54 | 1500 | |
| 9 | 1000 | 60 | 1.42 | 2.27 | 51 | 49 | 2100 | |
| 10 | 400 | 60 | 1.19 | 1.91 | 46 | 54 | 1600 | |
| 11 | C2 | 4000 | 40 | 1.60 | 2.56 | 81 | 19 | 4500 |
| 12 | 4000 | 60 | 3.07 | 4.91 | 83 | 17 | 1400 | |
| 13 | 4000 | 80 | 2.15 | 3.44 | 85 | 15 | 800 | |
| 14 | 4000 | 100 | 1.67 | 2.67 | 86 | 14 | 600 | |
| 15 | 2000 | 60 | 1.80 | 2.88 | 83 | 17 | 1700 | |
| 16 | C3 | 4000 | 40 | 0.460 | 0.74 | 77 | 23 | 1800 |
| 17 | 4000 | 60 | 1.26 | 2.02 | 72 | 28 | 900 | |
| 18 | 4000 | 80 | 0.326 | 0.52 | 68 | 32 | 500 | |
| 19 | 4000 | 100 | 0.276 | 0.44 | 69 | 31 | 400 | |
| 20 | 2000 | 60 | 0.051 | 0.08 | 74 | 26 | 900 | |
| 21 d | C4 | — | 40 | 2.00 | 3.20 | 86 | 14 | 1000 |
| 22 d | — | 60 | 1.29 | 2.06 | 84 | 16 | 600 | |
| 23 d | — | 80 | 0.99 | 1.91 | 78 | 22 | 400 | |
| 24 d | — | 100 | 1.19 | 1.58 | — | — | dimers and trimerse | |
| a Conditions: MMAO as cocatalyst, toluene as solvent; V=25 mL; [Cat.]=5×10-5 mol/L; 30 min; 0.62 MPa of propylene. b Percentage of allyl or vinylidene ends in total unsaturated end groups, measured by 1H NMR spectroscopy. c Determined by 1H NMR spectroscopy. d Triisobutyl aluminum/[Ph3C][B(C6F5)4] (TIBA/TrB) as cocatalyst, [Hf]:[TIBA]:[TrB]=1:100:1.2. e Determined by GC. | ||||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们