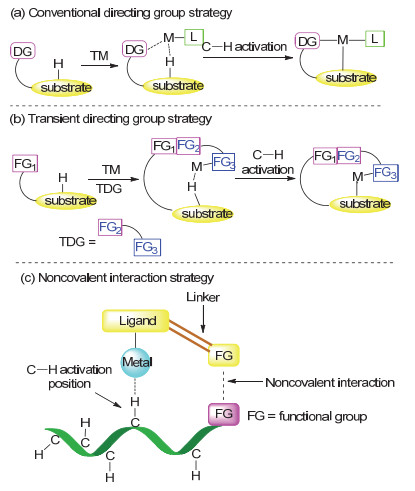
图式 1.
位点选择性碳氢键官能团化策略
Scheme 1.
Diverse strategy for site-selective C—H functionalization

近年来, 过渡金属催化的碳氢键官能团化反应成为有机合成领域的研究热点之一[1].相对于传统偶联反应, 其无需对起始原料进行预官能团化, 能够将惰性碳氢键切断, 进行直接官能团化, 从而具有原子经济性和步骤经济性等优点, 受到化学家们的广泛关注.然而, 有机分子中常常具有多个化学环境相似的碳氢键, 要实现特定碳氢键的官能团化是极具挑战性的.为了对碳氢键活化位点进行精准控制, 目前最常用的策略是利用底物本身配位官能团或者共价键连接的配位基团, 即导向基策略.导向基团与过渡金属进行配位后, 通常通过动力学有利的五元环金属中间体, 选择性活化邻近位置的碳氢键, 实现特定位点选择性(Scheme 1a)[2].然而, 引入的导向基团通常需要额外的步骤进行连接和脱除, 这就使得合成步骤变长, 成本增加, 也大大降低了反应的效率.为了更好地解决这一问题, Yu课题组开创性地发展了基于可逆共价键的“瞬态导向策略”[3], 在实现位点选择性精准控制的同时, 避免了传统共价键连接的导向基团的连接和脱除(Scheme 1b)[4].除了以上策略, 非共价键策略近年来也初现端倪, 为惰性碳氢键官能团化提供了一种全新的思路[5].在本文中, 我们将从非共价键在选择性碳氢键官能团化的应用角度进行阐述, 希望对该领域的研究提供参考和展望.
一般而言, 在导向碳氢键活化过程中, 强的导向基团和过渡金属作用使得反应具有更高的选择性, 然而, 由于形成的环金属中间体具有相对更强的稳定性, 因此在一定程度上, 这种强作用力也可能对反应活性有一定的影响.作为互补, 非共价键在选择性碳氢健活化中的作用日益凸显出来.非共价键一般包括氢键、静电作用、离子对作用、π-π堆积效应、范德华力和疏水作用力等.非共价键作用常见于生物系统和有机合成之中, 例如, 非共价键在有机小分子催化、酶催化、超分子化学和药物开发中都起着重要的作用[6, 7].在这里主要介绍过渡金属与非共价键共同作用的选择性碳氢键官能团化反应.在该策略中, 配体的官能团与底物分子的官能团通过非共价键作用, 在过渡金属催化下对特定位置的碳氢键进行活化(Scheme 1c).
对于非共价键参与的位点选择性碳氢键活化, 常常受到如下三个因素的影响: (1)底物的官能团需要和配体的官能团发生非共价作用, 使得过渡金属能够对特定位点的碳氢键进行活化; (2)过渡态的稳定性; (3)反应体系对非共价作用的兼容性.接下来将按照非共价键的作用类型来分, 主要有氢键作用、离子对作用、路易斯酸碱对作用、静电作用等.
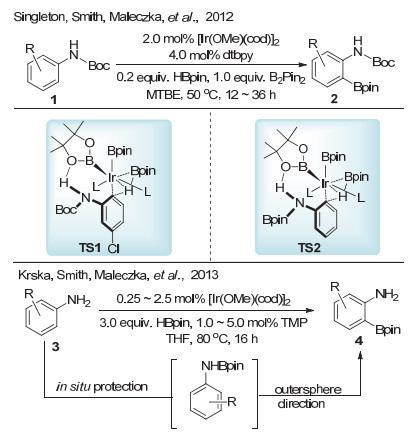
2012年, Singleton等[8]报道了铱催化叔丁氧羰基(Boc)保护的苯胺邻位碳氢键硼化反应(Scheme 2).在该反应中, 硼化选择性地发生在苯胺的邻位.通过实验和理论计算研究表明, 这种选择性主要是由于底物分子中氮上的氢原子与硼酸频哪醇酯中的氧原子形成了氢键(TS1).在铱催化剂的作用下, 对苯胺邻位碳氢键进行活化, 实现高选择性硼化.该反应能够兼容间位和对位具有取代基的叔丁氧羰基保护的苯胺, 而对于邻位具有取代基的底物作者未给出例子.之后, 该硼化反应很顺利地拓展到无保护的苯胺底物上[9].相对于之前的硼酸频哪醇酯作为硼基的主要来源, 作者采用频哪醇硼烷作为硼基来源, 与苯胺原位形成中间体(TS2), 之后通过之前类似的机理实现苯胺的邻位硼化.
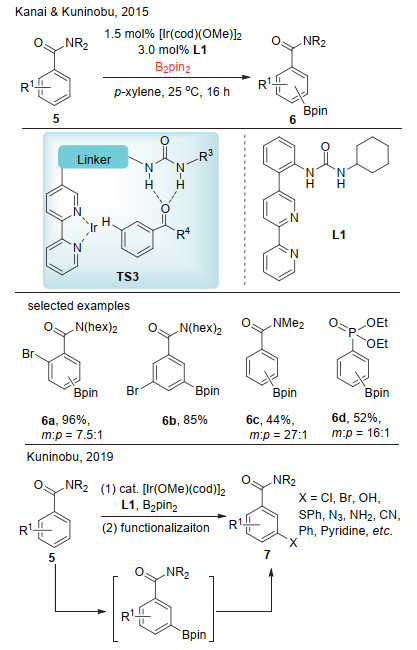
2015年, Kanai和Kuninobu课题组[10]报道了铱催化的苯甲酰胺间位选择性碳氢键硼化反应(Scheme 3).他们的间位选择性硼化策略也是基于氢键, 对于配体的设计有着较高的要求.他们认为, 反应基于TS3反应模型.首先, 苯甲酰胺的羰基氧与配体中的脲形成氢键, 而后过渡金属铱与配体分子中的联吡啶结构进行配位, 进而对底物分子的间位或者对位碳氢键进行活化.通过对配体分子结构的有效调节, 最终配体L1更有利于间位碳氢键活化.该反应底物适用范围广, 各种取代的苯甲酰氨、杂环和苯基磷酸酯等都能很好兼容, 能够得到选择性较高的间位硼化产物.在这之后, Kuninobu课题组[11]进一步利用这一策略, 将底物进行间位硼化后, 未经分离而进一步将其转化成其他官能团, 包括溴、氯、羟基、苯硫基、叠氮、氰基、芳基、烯基、烷基和烯丙基等.
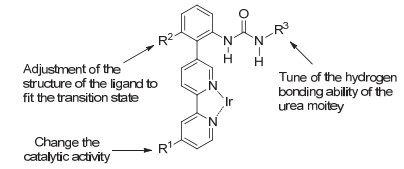
Kuninobu和Kanai课题组[12]对基于氢键策略的芳环化合物间位选择性碳氢键硼化反应中含脲结构的联吡啶配体进行了深入的探究(Scheme 4).他们发现, 改变R1位点能够改变配体的催化活性; 调节R2位点能够调整反应的过渡态, 而调节R3位点则能够改变脲结构与羰基形成氢键的能力.这一系统性的研究有望为未来配体设计提供初步的结构-活性关系. Sunoj课题组[13]通过密度泛函理论(DFT)对该反应的机理进行了研究, 他们发现硫脲结构的酰胺氢对反应选择性有很大影响, 该氢原子能够与底物中的羰基形成氢键作用, 在能量上更加趋向于活化底物间位.并且底物6c与含有配体L1的催化剂铱之间的非共价作用使得间位和对位活化时能量有所差异, 间位碳氢键活化的过渡态在能量上比对位要低27.6 kJ•mol-1, 而这个能量差值可以使间位和对位的选择性达到27:1.
2017年, Phipps等[14]报道了苄胺衍生物的间位碳氢键硼化反应(Scheme 5).他们利用联吡啶磺酸盐中的磺酸根与底物分子的酰胺氢形成氢键作用, 实现了间位碳氢键的选择性硼化.该反应能够兼容一系列三氟乙酰基保护的苄胺、苯乙胺和苯丙胺, 对于吡啶杂环也有较好的兼容性.为了验证底物与配体之间存在氢键作用, 他们进行了一系列对照实验.首先, 在不同溶剂中, 核磁滴定表明底物分子和配体间存在相互作用, 即使在极性溶剂乙腈中其作用也非常明显.其次, 当底物的酰胺氢被取代后, 其间位硼化和对位硼化产物的比例显著降低, 说明配体与底物之间的氢键作用能够影响反应的选择性.值得一提的是, 该类磺酸根配体最初由Phipps课题组[15]设计用于离子对作用的碳氢键活化(见2.2节).
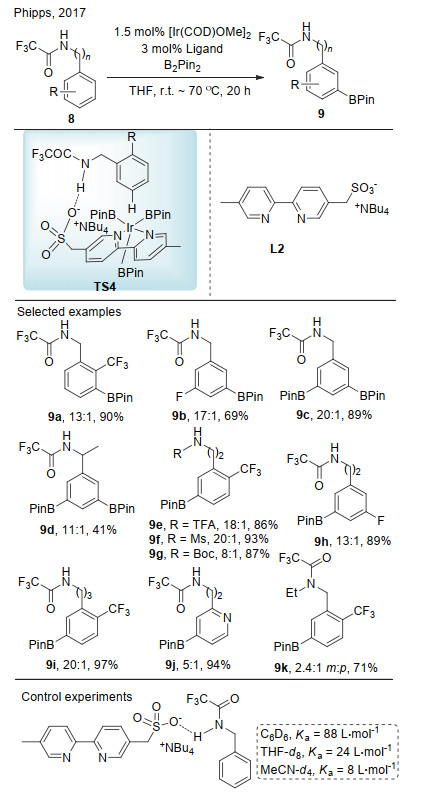
铱催化芳香酰胺的碳氢键硼化反应, 由于电子效应的影响, 其邻位碳氢健的活化是不利的.为克服这一困难, Reek课题组[16]报道了氢键控制的铱催化芳香酰胺邻位选择性碳氢键硼化反应(Scheme 6).该反应采用二级酰胺作为底物, 设计了含有吲哚结构的联吡啶骨架配体L3, 该配体的吲哚氮氢和酰胺氮氢能够与底物酰胺的氧形成氢键, 从而通过过渡态TS5实现铱催化酰胺的邻位碳氢键的活化, 最终高选择性地得到邻位硼化产物.
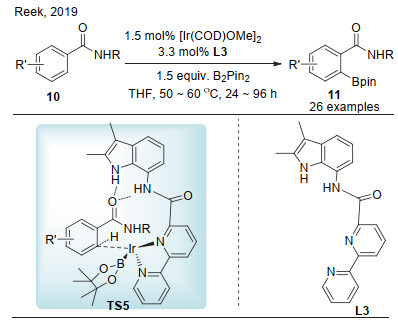
2016年, Phipps课题组[15]报道了一种全新的非共价键策略用于铱催化间位碳氢键硼化反应(Scheme 7).该反应中底物为季铵盐, 配体为具有磺酸根的联吡啶.通过离子交换, 底物的阳离子与配体的磺酸根离子产生离子对作用, 配体中联吡啶与铱催化剂配位, 进而选择性活化底物分子中的间位碳氢键.一系列的苄胺、苯胺和含杂环的季铵盐均能够高选择性地发生间位碳氢键硼化.为体现该反应的应用价值, 作者通过偶联反应将季铵盐进一步转化为联芳化合物.在这之后, 该课题组进一步利用该策略, 将底物范围拓展到苯乙胺和苯丙胺的季铵盐衍生物之中[17].在这些工作的基础上, Phipps课题组[18]继续将该方法用于芳基膦盐的硼化之中.
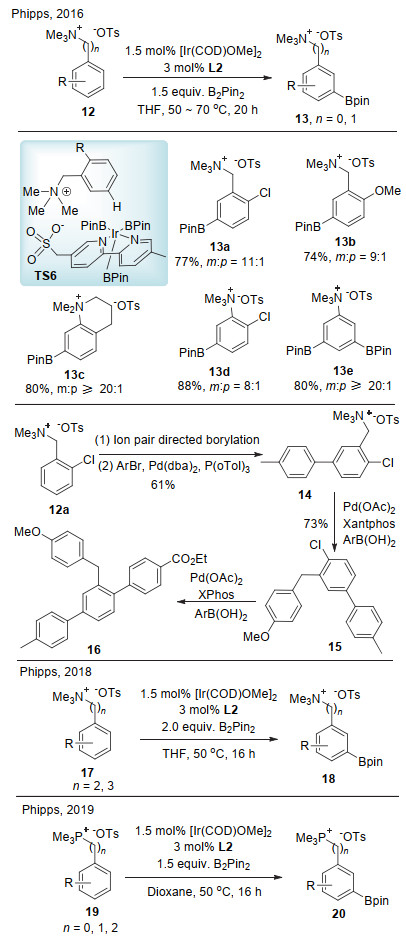
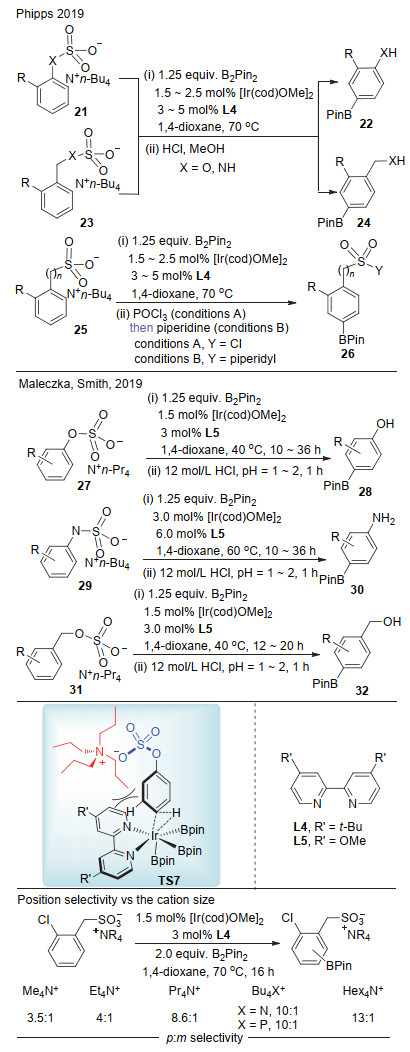
最近, Phipps课题组[19a], Maleczka和Smith课题组[19b]分别报道了基于离子对策略的铱催化对位选择性碳氢键硼化(Scheme 8).在该反应中, 底物(苯酚、苄醇、苯胺或苄胺)先转化成相应的硫酸盐或磺酰盐, 其阳离子部分为四烷基(丙基或者丁基)季铵盐, 利用该季铵盐的大位阻效应阻止底物间位碳氢键金属化, 从而高选择性地实现了对位碳氢键硼化.硼化之后的铵盐在浓盐酸作用下水解得到对位硼化的苯酚、苯胺或苄醇产物. Phipps等以2-氯苄基磺酸盐为底物, 对阳离子大小与对/间位选择性进行了系统研究.实验表明, 对位选择性随着阳离子的增大而明显增加.
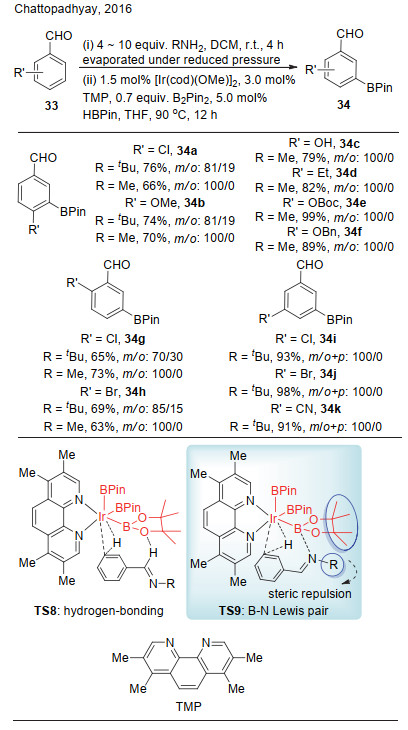
2016年, Chattopadhyay课题组[20]报道了非共价作用促进的铱催化芳香醛间位选择性碳氢键硼化(Scheme 9).该反应通过芳基醛与烷基胺反应现场生成亚胺, 亚胺与铱催化剂上的频哪醇硼通过非共价作用, 诱导实现铱催化的间位碳氢键硼化, 3, 4, 7, 8-四甲基-1, 10-邻菲咯啉(TMP)作为配体调控催化中心金属的电性.他们认为, 反应的选择性可能是来源于TS8中的氢键作用, 或者是TS9中氮-硼的路易斯酸碱对作用.作者认为, 在TS8中, 亚胺的取代基远离反应中心, 因此选择性应该不受烷基胺上取代烷基(R)的大小影响; 而与此相反, 在TS9中, 由于烷基胺的取代基(R)与频哪醇的位阻排斥, 位阻更小的R基团更加有利于反应.基于以上设想以及反应中甲胺给出比叔丁胺更好的反应选择性, 作者倾向于认为该反应的选择性是基于路易斯酸碱对作用TS9.这一工作虽然不是通过设计的配体与底物的路易斯酸碱对作用, 但也为后续工作提供了有益的参考.
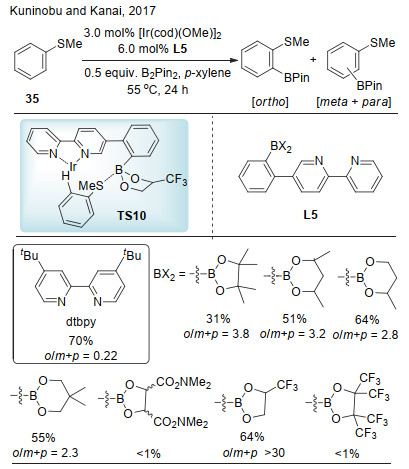
2017年, Kuninobu和Kanai课题组[21]报道了芳香硫醚的邻位硼化反应.该反应中底物的硫原子与配体的硼原子之间形成路易斯酸碱对作用(Scheme 10, TS10).他们通过研究发现, 通过配体结构改变能够调控反应的选择性, 当4, 4-二-叔丁基联吡啶(dtbpy)作为配体时其邻位选择性较差.对联吡啶进一步改造, 当采用配体L4类型配体时, 邻位选择性显著增加.配体上的硼保护基的电性对反应的活性和邻位的选择性也有非常大的影响.在硼的保护基团中引入三氟甲基, 其产率和邻位选择性均显著提高(64%, o/m+p>30).一系列的3位或者4位取代的芳基硫醚均能够以较高的邻位选择性得到间位硼化产物, 含有吡啶、呋喃和吡咯等的杂芳环硫醚则难以适用于该体系, 可能是杂芳环本身杂原子与硼原子的路易斯酸碱对作用的影响.
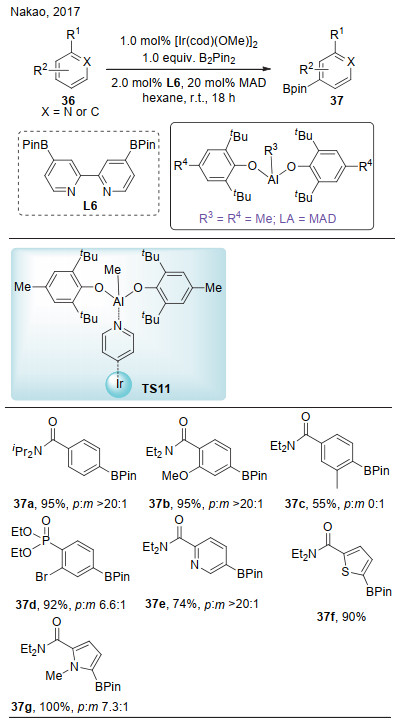
2017年, Nakao课题组[22]报道了芳香化合物的对位选择性碳氢键硼化反应(Scheme 11).该反应中, 底物上的路易斯碱官能团和路易斯酸形成酸碱对, 通过电性传递使得底物的芳环更加缺电子, 从而提高反应活性(TS11).同时, 配位于铱上的配体与大位阻的铝之间产生排斥作用, 在空间上阻碍了金属铱对邻位和间位碳氢键的活化, 从而实现了对位碳氢键的高选择性活化.该反应的底物范围广, 对于苯甲酰胺、芳基磷酸酯以及吡啶等杂环都有着良好的兼容性.
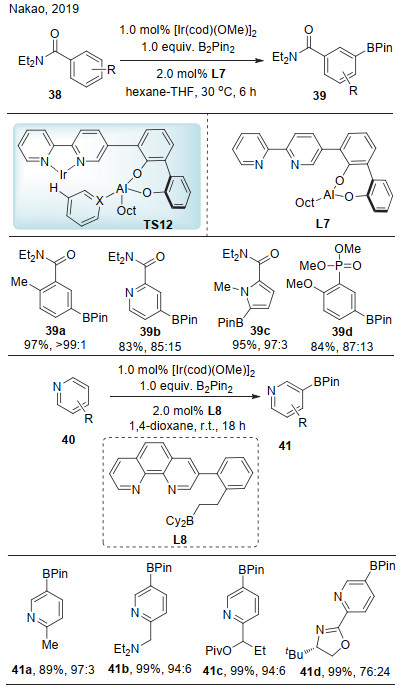
2019年, Nakao课题组[23]将该铱铝双功能催化剂应用于芳香化合物间位碳氢键的选择性硼化(Scheme 12).其催化剂结构主要包括可用于与铱配位的联吡啶或菲咯啉结构和联吡啶与铝的连接部分.具有路易斯酸性的烷基铝能够识别酰胺的羰基或者sp2杂化的氮.通过系统的研究, 他们发现具有烷基铝联苯氧化物的2, 2'-联吡啶配体L7, 在铱催化下能实现苯甲酰胺的高选择性间位碳氢键硼化反应.当使用1, 10-菲咯啉配体(L8)时, 可以实现吡啶C5 (C3)位选择性碳氢键硼化.
2017年, Chattopadhyay课题组[24]报道了铱催化芳香酯选择性对位碳氢键硼化反应(Scheme 13).该反应采用8位连接2, 2-联吡啶的喹诺酮配体模板L9.作者认为, L9的喹诺酮单元在反应中可以互变异构为2-羟基喹啉, 与反应体系中的叔丁醇钾反应, 形成L9的钾盐, 钾离子与底物的酯羰基形成路易斯酸碱对, 通过如Scheme 13所示过渡态TS13, 实现对位碳氢键硼化.该反应能够选择性活化芳香酯的对位, 对于杂环底物也具有较好的位置选择性.为验证该模型的可能性, 作者进行了几组对照实验.首先, 当采用L10作为配体时, 其对位与间位的选择性达到33/1.将配体用甲基保护后得到L11, 其对位与间位选择性仅为1.9/1.再次, 作者以L9为配体, 在体系中加入18-冠-6醚, 当体系中没有叔丁醇钾存在时, 其对位与间位选择性为5.6/1;当体系中具有叔丁醇钾时, 由于18-冠-6醚捕获钾离子, 其对位与间位选择性由33/1降低为1.8/1.0, 该结果进一步验证了钾离子对反应的选择性有着较大的影响, 证明体系中可能存在钾离子与酯羰基的弱相互作用.
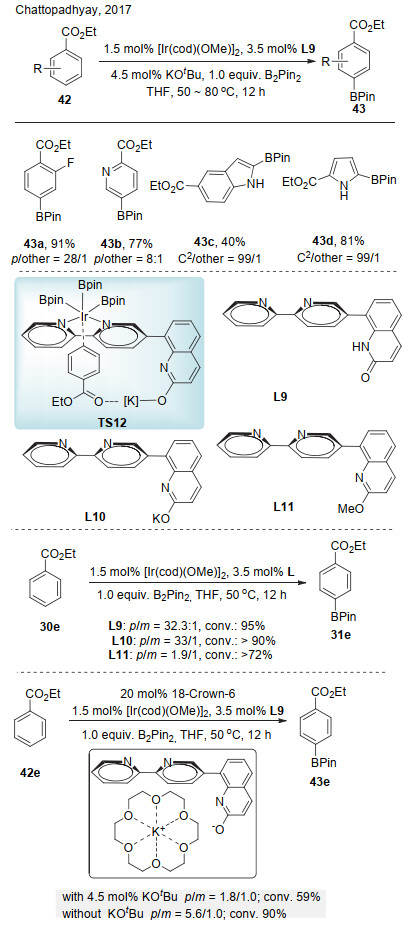
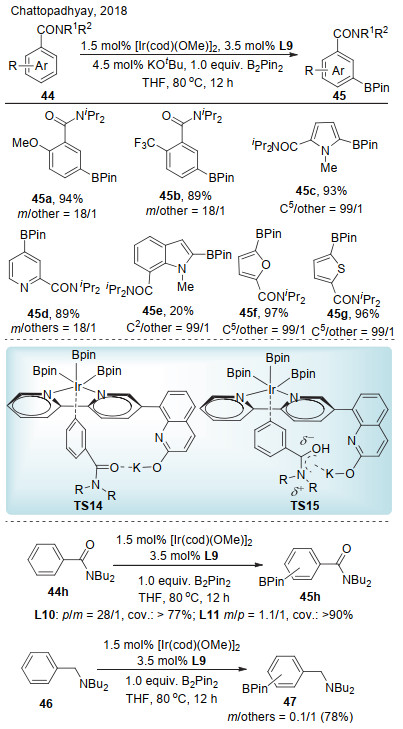
Chattopadhyay课题组[25]随后将配体L9应用于铱催化的芳香酰胺类化合物间位碳氢键的选择性硼化(Scheme 14).该反应能够兼容吡啶、呋喃、噻吩和吲哚等杂环化合物.当用L10作为配体时, 其对位与间位的选择性为28/1;当用L11作为配体时, 其间位与对位的选择性为1.1/1;将底物变为苄胺, 其选择性急剧下降.以上对照实验表明, 与之前反应类似, 体系中存在钾离子与底物酰胺的非共价键作用.他们推测可能有两种作用方式:酰胺羰基与钾离子的路易斯酸碱作用(TS14)或者离子-π作用(TS15).作者认为, 经由TS15的离子-π作用将显著受到底物电性和反应溶剂极性的影响.对照实验表明, 无论是给电子(44a, MeO)还是缺电子底物(44b, CF3)都给出相当的反应活性和选择性; 另外, 反应溶剂换成非极性溶剂正己烷, 产率略有下降, 但选择性几乎相同.因此, 虽然无法完全排除离子-π作用, 但作者更倾向于反应是经由路易斯酸碱对作用.
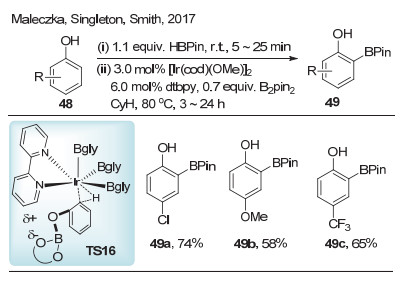
2017年, Maleczka等[26]报道了铱催化苯酚邻位的硼化反应(Scheme 15).在该反应中, 底物苯酚和频哪醇硼烷形成保护的苯酚, 其实验和计算研究表明, 在过渡态中, 底物中的OBPin官能团带有部分负电荷, 而联吡啶配体相对而言具有正电性, 通过过渡态TS16实现邻位碳氢键硼化.值得注意的是, 要实现苯酚邻位的选择性硼化, 苯酚对位通常需要引入大于氟原子的官能团.在实验的基础上, 作者通过密度泛函理论(DFT)推测出电性对反应活性与选择性影响, 得到与实验相一致的结果, 进一步证实该过渡态模型的可能性.
近年来, 设计和发展基于非共价键作用的配体模板为选择性碳氢键活化提供了一种全新的模式, 通过不同的非共价键作用, 使得催化体系的中心金属、配体模板及底物分子共同作用, 实现精准可控的选择性活化.目前的研究主要集中在氢键作用、离子对作用、路易斯酸碱对作用和静电作用等.显而易见的是, 该领域尽管已经有所发展, 并初步显示出其潜在的应用性, 但该领域仍然存在着一些关键问题亟待解决. (1)目前的报道局限于铱(Ⅰ)催化的碳氢键硼化反应, 使用的催化金属为铱(Ⅰ), 其他金属尚没有报道, 反应类则局限于硼化反应.这类催化反应的特点是, 整个反应过程为氧化还原中性体系, 反应体系比较简洁, 无需额外的氧化还原剂和添加剂.而众所周知, 绝大多数过渡金属催化的碳氢键活化对配体、溶剂和添加剂等体系的微调具有极大的依赖性, 铱(Ⅰ)催化的非共价键作用的碳氢键硼化得益于联砒啶类优势骨架配体的应用, 而对于其他过渡金属, 目前尚无具有广泛适用性的配体体系得到应用.另外, 非共价键作用通常依赖烷烃、甲苯、四氢呋喃和1, 4-二氧六环等非极性溶剂, 对反应的适用性也提出了更高的要求.其次, 反应体系添加剂, 如酸、碱、无机盐和氧化剂等, 对非共价键作用体系不可避免地产生一定的影响.因此, 未来在该领域的成功有赖于发展具有促进作用的普适性配体和温和的反应体系.最近, Yu[27]及Maiti[28]课题组发展了基于钯-氮强配位作用的双功能的模板, 实现钯催化的杂芳环的选择性碳氢键官能团化, 有望为该领域的进一步发展提供借鉴. (2)底物类型、选择性、底物范围和反应类型仍有极大的提升空间[29]. (3)能否发展手性非共价键的配体模板实现具有前手性化合物的去对称化和不对称碳氢键活化是该领域的极大挑战[30].虽然目前该领域尚处于初级阶段, 随着研究的进一步深入, 我们相信会有更多的非共价作用模式被开发出来, 为该领域的发展注入蓬勃的生命.最后, 利用非共价键作用进行碳氢键的精准官能团化也将为有机合成、药物开发、超分子化学等领域的发展开启新的篇章.
For recent reviews on C-H activation, see: (a) Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094. (b) Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147. (c) Wencel-Delord, J.; Droge, T.; Liu, F.; Glorius, F. Chem. Soc. Rev. 2011, 40, 4740. (d) Pan, F.; Shi, Z. Acta Chim. Sinica 2012, 70, 1679. (潘菲, 施章杰, 化学学报, 2012, 70, 1679.) (e) Yuan, Y.; Song, S.; Jiao, N. Acta Chim. Sinica 2015, 73, 1231. (袁逸之, 宋颂, 焦宁, 化学学报, 2015, 73, 1231.) (f) Zhang, M.; Zhang, Y.; Jie, X.; Zhao, H.; Li, G.; Su, W. Org. Chem. Front. 2014, 1, 843. (g) Xu, J.; Lu, P.; Ye, J.; Liu, G. Acta Chim. Sinica 2015, 73, 1294. (徐佳斌, 陈品红, 叶金星, 刘国生, 化学学报, 2015, 73, 1294.) (h) Daugulis, O.; Roane, J.; Tran, L. D. Acc. Chem. Res. 2015, 48, 1053. (i) He, G.; Wang, B.; Nack, W. A.; Chen, G. Acc. Chem. Res. 2016, 49, 635. (j) Rao, W.-H.; Shi, B.-F. Org. Chem. Front. 2016, 3, 1028. (k) Yang, Y.; Lan, J.; You, J. Chem. Rev. 2017, 117, 8787. (l) He, J.; Wasa, M.; Chan, K. S. L.; Shao, Q.; Yu, J.-Q. Chem. Rev., 2017, 117, 8754. (m) Huang, J.; Gu, Q.; You, S.-L. Chin. J. Org. Chem. 2018, 38, 51. (黄家翩, 顾庆, 游书力, 有机化学, 2018, 38, 51.) (n) Ren, Q.; Nie, B.; Zhang, Y.; Zhang, J. Chin. J. Org. Chem. 2018, 38, 2465. (任青云, 聂飚, 张英俊, 张霁, 有机化学, 2018, 38, 2465). (o) Zhao, K.; Yang, L.; Liu, J.; Xia, C. R. Chin. J. Org. Chem. 2018, 38, 2833. (赵康, 杨磊, 刘建华, 夏春谷, 有机化学, 2018, 38, 2833.) (p) Wang, S.; Yan, F.; Wang, L.; Zhu, L. Chin. J. Org. Chem. 2018, 38, 291. (汪珊, 严沣, 汪连生, 朱磊, 有机化学, 2018, 38, 291.) (q) Xu, L.; Xu, H.; Lin, H.; Dai, H. Chin. J. Org. Chem. 2018, 38, 1940. (徐琳琳, 徐辉, 林海霞, 戴辉雄, 有机化学, 2018, 38, 1940) (r) Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. Chem. Rev. 2019, 119, 2192. (s) Zhang, S.; Liao, G.; Shi, B. Chin. J. Org. Chem. 2019, 39, 1522. (张硕, 廖港, 史炳锋, 有机化学, 2019, 39, 1522). (t) Wu, M.; Huang, X.; Zhang, H.; Li, P. Chin. J. Org. Chem. 2019, 39, 3114. (吴梅, 黄新平, 张海兵, 李鹏飞, 有机化学, 2019, 39, 3114). (u) Zhan, B.; Shi, B.-F. Chin. J. Org. Chem. 2019, 39, 3602. (占贝贝, 史炳锋, 有机化学, 2019, 39, 3602.
(a) Chen, Z.; Wang, B.; Zhang, J.; Yu, W.; Liu, Z.; Zhang, Y. Org. Chem. Front. 2015, 2, 1107. (b) Sambiagio, C.; Schö nbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig G.; Schaaf, P.; Wiesinger, T.; Farooq Zia, M.; Wencel-Delord, J.; Besset, T.; Maes, B. U. W.; Schnürch, M. Chem. Soc. Rev. 2018, 47, 6603. (c) Zhang, Q.; Shi, B.-F. Chin. J. Chem. 2019, 37, 647. (d) Rej, S.; Ano, Y.; Chatani, N. Chem. Rev. 2020, 120, 1788.
Zhang, F.-L.; Hong, K.; Li, T.-J.; Park, H.; Yu, J.-Q. Science 2016, 351, 252. doi: 10.1126/science.aad7893
For reviews and representative examples, see: (a) Gong, L.-Z. Acta Chim. Sinica 2018, 76, 817. (龚流柱, 化学学报, Acta Chim. Sinica 2018, 76, 817.) (b) Kim, D.-S.; Park, W.-J.; Jun, C.-H. Chem. Rev. 2017, 117, 8977. (c) Gandeepan, P.; Ackermann, L. Chem 2018, 4, 199. (d) John-Campbell, S. S.; Bull, J. A. Org. Biomol. Chem. 2018, 16, 4582. (e) Bhattacharya, T.; Pimparkar, S.; Maiti, D. RSC Adv. 2018, 8, 19456. (f) Qin, Y.; Zhu, L.; Luo, S. Chem. Rev. 2017, 117, 9433. (g) Sun, H.; Guimond, N.; Huang, Y. Org. Biomol. Chem. 2016, 14, 8389. (h) Xu, Y.; Su, T.; Huang, Z.; Dong, G. Angew. Chem., Int. Ed. 2016, 55, 2559. (i) Yao, Q.-J.; Zhang, S.; Zhan, B.-B.; Shi, B.-F. Angew. Chem., Int. Ed. 2017, 56, 6617. (j) Liu, Y.; Ge, H.; Liu, X.-H.; Park, H.; Hu, J.-H.; Hu, Y.; Zhang, Q.-L.; Wang, B.-L.; Sun, B.; Yeung, K.; Zhang, F.-L.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 888. (k) Chen, X. Y.; Ozturk, S.; Sorensen, E. J. Org. Lett. 2017, 19, 1140. (l) Liao, G.; Yao, Q.-J.; Zhang, Z.-Z.; Wu, Y.-J.; Huang, D.-Y.; Shi, B.-F. Angew. Chem., Int. Ed. 2018, 57, 3661. (m) Liao, G.; Li, B.; Chen, H.-M.; Yao, Q.-J.; Xia, Y.-N.; Luo, J.; Shi, B.-F. Angew. Chem. Int. Ed. 2018, 57, 17151. (n) Zhang, S.; Yao, Q.-J.; Liao, G.; Li, X.; Li, H.; Chen, H.-M.; Hong, X.; Shi, B.-F. ACS Catal. 2019, 9, 1956. (o) Song, H.; Li, Y.; Yao, Q.-J.; Jin, L.; Liu, L.; Liu, Y.-H.; Shi, B.-F. Angew. Chem., Int. Ed. 2020, DOI: 10.1002/anie.201915949.(p)Wu,Y.-J.;Yao,Q.-J.;Chen,H.-M.;Liao,G.;Shi,B.-F.Sci.China,Chem.2020,DOI:10.1007/s11426-020-9694-3.
Davis, H. J.; Phipps, R. J. Chem. Sci. 2017, 8, 864. doi: 10.1039/C6SC04157D
(a) For selected reviews on noncovalent interactions, see: Neel, A. J.; Hilton, M. J.; Sigman, M. S.; Toste, F. D. Nature 2017, 543, 637. (b) Müller-Dethlefs, K.; Hobza, P. Chem Rev. 2000, 100, 143. (c) Breugst, M.; von der Heiden, D.; Schmauck, J. Synthesis 2017, 49, 3224. (d) Hobza P, Müller-Dethlefs K. Non-Covalent Interactions, The Royal Society of Chemistry, Cambridge, 2009. (e) Scheiner, S. Noncovalent Forces, Heidelberg, Springer, 2015; (f) Schreiner, P. R. Chem. Soc. Rev. 2003, 32, 289. (g) Doyle, A. G.; Jacobsen, E. N. Chem Rev. 2007, 107, 5713;
(a) You, C.-C.; Zhang, M.; Liu, Y. Acta Chim. Sinica 2000, 58, 338. (尤长城, 张旻, 刘育, 化学学报, 2000, 58, 338.) (b) Xu, J.; Wang, Z.; Zhang, X. Acta Chim. Sinica 2016, 74, 467. (徐俊, 王治强, 张希, 化学学报, 2016, 74, 467.) (c) Zhu, J.; Lü, J.-G.; Zhou, Y.-J.; Li, Y.-W.; Chen, J.; Zhen, C.-H. Acta Chim. Sinica 2007, 65, 37. (朱驹, 吕加国, 周有骏, 李耀武, 陈军, 郑灿辉, 化学学报, 2007, 65, 37.) (d) Wheeler, S. E.; Seguin, T. J.; Guan, Y.; Doney, A. C. Acc. Chem. Res. 2016, 49, 1061. (e) Persch, E.; Dumele, O.; Diederich, F. Angew. Chem., Int. Ed. 2015, 54, 3290. (f) Jiang, H.; Li, Q.; Wang, G. Chin. J. Org. Chem. 2018, 38, 1065. (江华, 李巧连, 王光霞, 有机化学, 2018, 38, 1065.) (g) Jiao, Y.; Zhang, X. Acta Chim. Sinica 2018, 76, 659. (焦阳, 张希, 化学学报, 2018, 76, 659.) (h) Liu, C.-Z.; Wang, H.; Zhang, D.-W.; Zhao, X.; Li, Z.-T. Chin. J. Org. Chem. 2019, 39, 28. (刘传志, 王辉, 张丹维, 赵新, 黎占亭, 有机化学, 2019, 39, 28.)
Roosen, P. C.; Kallepalli, V. A.; Chattopadhyay, B.; Singleton, D. A.; Maleczka, R. E.; Smith, M. R. J. Am. Chem. Soc. 2012, 134, 11350. doi: 10.1021/ja303443m
Preshlock, S. M.; Plattner, D. L.; Maligres, P. E.; Krska, S. W.; Maleczka, R. E.; Smith, M. R. Angew. Chem., Int. Ed. 2013, 52, 12915. doi: 10.1002/anie.201306511
Kuninobu, Y.; Ida, H.; Nishi, M.; Kanai, M. Nat. Chem. 2015, 7, 712. doi: 10.1038/nchem.2322
Wang, J.; Torigoe, T.; Kuninobu, Y. Org. Lett. 2019, 21, 1342. doi: 10.1021/acs.orglett.9b00030
Lu, X.; Yoshigoe, Y.; Ida, H.; Nishi, M.; Kanai, M.; Kuninobu, Y. ACS Catal. 2019, 9, 1705. doi: 10.1021/acscatal.8b05005
Unnikrishnan, A.; Sunoj, R. B. Chem. Sci. 2019, 10, 3826. doi: 10.1039/C8SC05335A
Davis, H. J.; Genov, G. R.; Phipps, R. J. Angew. Chem., Int. Ed. 2017, 56, 13351. doi: 10.1002/anie.201708967
Davis, H. J.; Mihai, M. T.; Phipps, R. J. J. Am. Chem. Soc. 2016, 138, 12759. doi: 10.1021/jacs.6b08164
Bai, S.-T.; Bheeter, C. B.; Reek, J. N. H. Angew. Chem., Int. Ed. 2019, 58, 13039. doi: 10.1002/anie.201907366
Mihai, M. T.; Davis, H. J.; Genov, G. R.; Phipps, R. J. ACS Catal. 2018, 8, 3764. doi: 10.1021/acscatal.8b00423
Lee, B.; Mihai, M. T.; Stojalnikova, V.; Phipps, R. J. J. Org. Chem. 2019, 84, 13124. doi: 10.1021/acs.joc.9b00878
(a) Mihai, M.; Williams, B. D.; Phipps, R. J. J. Am. Chem. Soc. 2019, 141, 15477. (b) Montero Bastidas, J. R.; Oleskey, T. J.; Miller, S. L.; Smith, M. R.; Maleczka, R. E. J. Am. Chem. Soc. 2019, 141, 15483.
Bisht, R.; Chattopadhyay, B. J. Am. Chem. Soc. 2016, 138, 84. doi: 10.1021/jacs.5b11683
Li, H. L.; Kuninobu, Y.; Kanai, M. Angew. Chem., Int. Ed. 2017, 56, 1495. doi: 10.1002/anie.201610041
Yang, L.; Semba, K.; Nakao, Y. Angew. Chem., Int. Ed. 2017, 56, 4853. doi: 10.1002/anie.201701238
Yang, L.; Uemura, N.; Nakao, Y. J. Am. Chem. Soc. 2019, 141, 7972. doi: 10.1021/jacs.9b03138
Hoque, M. E.; Bisht, R.; Haldar, C.; Chattopadhyay, B. J. Am. Chem. Soc. 2017, 139, 7745. doi: 10.1021/jacs.7b04490
Bisht, R.; Hoque, M. E.; Chattopadhyay, B. Angew. Chem., Int. Ed. 2018, 57, 15762. doi: 10.1002/anie.201809929
Chattopadhyay, B.; Dannatt, J. E.; Andujar-De Sanctis, I. L.; Gore, K. A.; Maleczka, R. E.; Singleton, D. A.; Smith, M. R. J. Am. Chem. Soc. 2017, 139, 7864. doi: 10.1021/jacs.7b02232
Zhang, Z.; Tanaka, K.; Yu, J.-Q. Nature 2017, 543, 538. doi: 10.1038/nature21418
(a) Achar, T. K.; Ramakrishna, K.; Porey, S.; Pal, T.; Dolui, P.; Biswas, J. P.; Maiti, D. Chem.-Eur. J. 2018, 24, 17906. (b) Ramakrishna, K.; Biswas, J. P.; Jana, S.; Achar, T. K.; Porey, S.; Maiti, D. Angew. Chem., Int. Ed. 2019, 58, 13808.
Haldar, C.; Hoque, M. E.; Bisht, R.; Chattopadhyay, B. Tetrahedron Lett. 2018, 59, 1269. doi: 10.1016/j.tetlet.2018.01.098
(a) Giri, R.; Shi, B.-F.; Engle, K. M.; Maugel, N.; Yu, J.-Q. Chem. Soc. Rev. 2009, 38, 3242. (b) Wencel-Delord, J.; Colobert, F. Chem.-Eur. J. 2013, 19, 14010. (c) Zheng, C.; You, S.-L. RSC Adv. 2014, 4, 6173. (d) Gao, D.-W.; Gu, Q.; Zheng, C.; You, S.-L. Acc. Chem. Res. 2017, 50, 351. (e) Newton, C. G.; Wang, S.-G.; Oliveira, C. C.; Cramer, N. Chem. Rev. 2017, 117, 8908. (f) Yan, S.-Y.; Han, Y.-Q.; Yao, Q.-J.; Nie, X.-L.; Liu, L.; Shi, B.-F. Angew. Chem., Int. Ed. 2018, 57, 9093. (g) Saint-Denis, T. G.; Zhu, R.-Y.; Chen, G.; Wu, Q.-F.; Yu, J.-Q. Science 2018, 359, 759. (h) Liao, G.; Zhou, T.; Yao, Q.-J.; Shi, B.-F. Chem. Commun. 2019, 55, 8514. (i) Han, Y.-Q.; Ding, Y.; Zhou, T.; Yan, S.-Y.; Song, H.; Shi, B.-F. J. Am. Chem. Soc. 2019, 141, 4558. (j) Luo, J.; Zhang, T.; Wang, L.; Liao, G.; Yao, Q.-J.; Wu, Y.-J.; Zhan, B.-B.; Lan, Y.; Lin, X.-F.; Shi, B.-F. Angew. Chem., Int. Ed. 2019, 58, 6708. (k) Zhan, B.-B.; Wang, L.; Luo, J.; Shi, B.-F. Angew. Chem., Int. Ed. 2020, 59, 3568. (l) Zhou, T.; Jiang, M.-X.; Yang, X.; Yue, Q.; Han, Y.-Q.; Ding, Y.; Shi, B.-F. Chin. J. Chem. 2020, 38, 242.

图式 1 位点选择性碳氢键官能团化策略
Scheme 1 Diverse strategy for site-selective C—H functionalization

图式 2 叔丁氧羰基(Boc)保护苯胺及苯胺的邻位选择性碳氢键硼化
Scheme 2 otho-Selective C—H borylation of N-Boc anilines and free anilines

图式 3 氢键控制的苯甲酰胺间位碳氢键硼化及其转化
Scheme 3 Hydrogen-bonding-enabled meta-selective C—H borylation and transformations of benzamides

图式 4 氢键控制的间位选择性碳氢键硼化的配体
Scheme 4 Relationship between ligand structure and reactivity for hydrogen-bonding-enabled meta-selective C—H boryaltion

图式 5 氢键控制的苄胺、苯乙胺、苯丙胺衍生的三氟乙酰胺间位碳氢键硼化
Scheme 5 Hydrogen-bonding-enabled meta-C—H borylation of benzylamine-, phenethylamine- and phenylpropylamine-derived trifluoroacetamides

图式 6 氢键控制的芳香酰胺的邻位碳氢键硼化
Scheme 6 Hydrogen-bonding-enabled ortho-selective C—H borylation of secondary aromatic amides

图式 7 离子对控制的季胺盐间位碳氢键硼化反应
Scheme 7 Ion pair-enabled meta-C—H borylation of quaternary ammonium salts

图式 8 铱催化苯酚、苄醇和苯胺的对位碳氢键硼化
Scheme 8 Ir-catalyzed para-C—H borylation of phenols, benzyl alcohols and anilines

图式 9 铱催化芳香醛间位选择性碳氢键硼化
Scheme 9 Iridium-catalyzed meta-selective C—H borylation of aromatic aldehydes

图式 10 路易斯酸碱对控制的芳烃硫醚的邻位选择性碳氢键硼化
Scheme 10 Lewis acid-base interaction-enabled ortho-selective C—H borylation of aryl sulfides

图式 11 铱/铝催化的芳环对位选择性碳氢键硼化
Scheme 11 para-Selective C—H borylation of (hetero)arenes by cooperative iridium/aluminum catalysis

图式 12 铱/铝催化的苯甲酰胺及吡啶的间位选择性碳氢键硼化
Scheme 12 meta-Selective C−H borylation of benzamides and pyridines by an iridium−aluminum bifunctional catalyst

图式 13 铱催化芳香酯对位碳氢键硼化
Scheme 13 Iridium-catalyzed para-C—H borylation of aromatic esters

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:

