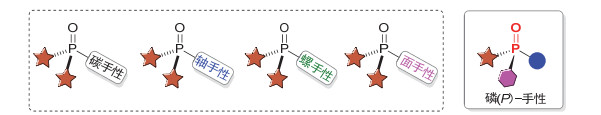
图 1.
手性膦氧化物的常见类型
Figure 1.
Classification of chiral phosphine oxides

磷(Phosphorus)是地壳中含量丰富的非金属元素之一, 也是生命机体中极为重要的、植物生长发育必须的元素之一, 因此磷在人类生命活动以及工农业生产中扮演着举足轻重的角色[1].在众多的含磷化合物中, 手性有机膦氧化物由于其在农药[2]、医药[3]、手性催化剂[4]以及材料[5]等领域具有广泛的应用, 因此受到化学家们的广泛关注.经过化学研究者的不懈努力, 大量的手性膦氧化物已被成功构建; 而根据其磷原子是否具有手性可分为两大类:一类是手性不在磷原子上的其他手性膦氧衍生物, 如碳手性[6]、轴手性[7]、螺环手性[8]或面手性[9]的膦氧化物; 另一类则是手性在磷原子上的磷(P)-手性膦氧化物(图 1).
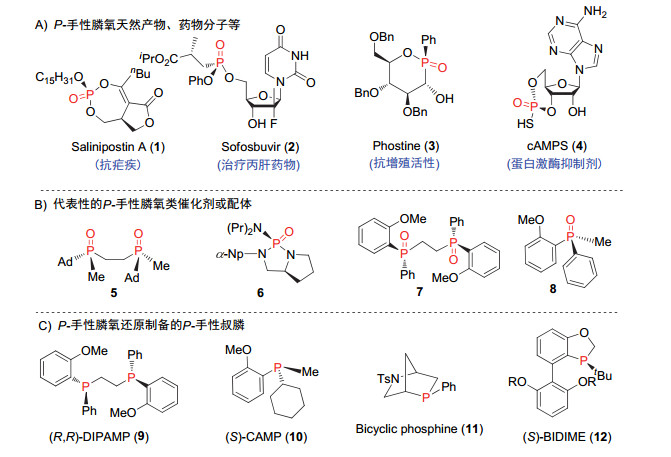
与其他手性膦氧化物相比, P-手性膦氧化物因其独特的结构特点, 在许多领域具有重要应用.一方面, P-手性膦氧结构单元广泛存在于许多天然产物、药物及生物活性分子中(图 2), 例如, 环状P-手性膦酸酯膦氧化物1是一种具有抗疟疾活性的天然产物[10]; 用于治疗慢性丙肝的特效药索非布韦2也含有这一结构单元[11].此外, 含有P-手性膦氧结构的Phostine (3)[12]具有抗增殖活性, cAMPS (4)[13]是蛋白激酶抑制剂.另一方面, 含有P-手性膦氧结构单元的化合物也是重要的手性原料, 用于发展手性催化剂或配体.例如:双齿膦氧化物5是一类有效的配体, 其与铁盐形成的配合物能有效催化不对称Diels-Alder反应[14]; (S)-脯氨酸衍生的膦酰胺6作为Lewis碱催化剂[15], 在醛的不对称烯丙基化反应中表现良好.通过选择合适的还原条件, 膦氧化合物还可进一步转化成用途更广的P-手性叔膦类催化剂和配体[16, 17], 例如Knowles等[17a]发展的(R, R)-DiPAMP (9)和(S)- CAMP (10), 以及汤文军等[16]发展的(S)-BIDIME (12).综上所述, 发展高效高选择性合成结构新颖、官能团多样性的高附加值P-手性膦氧化物的方法无疑对药物研发和合成化学等均具有重要的研究意义和应用价值.这不仅有助于设计开发结构多样化的含磷手性药物, 促进新药研发; 而且能加速新型具有P-手性的催化剂或配体的设计与应用, 进而实现一些利用其他催化剂难以实现的反应, 甚至推动更多新方法学的发展.
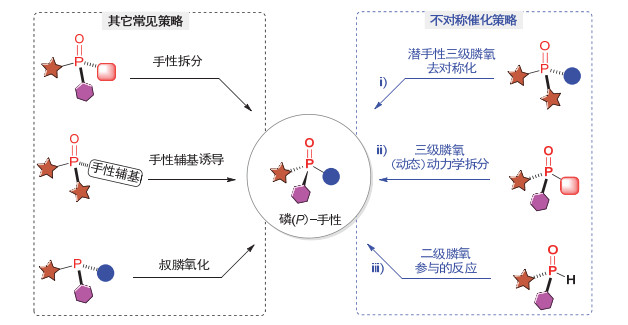
鉴于P-手性膦氧化物的重要研究价值, 化学家们对其不对称合成产生了浓厚的研究兴趣并进行了不断探索.近年来, 该领域也取得了可喜的研究进展[1a, 18], 发展了一系列构建P-手性膦氧化物的合成方法, 主要可分为以下几种策略: (1)消旋膦氧化物的手性拆分策略; (2)手性辅基诱导的策略; (3)叔膦氧化合成P-手性膦氧的策略; (4)基于膦氧化物的不对称催化策略(图 3).早期合成P-手性膦氧化物的方法主要集中在利用消旋膦氧化物的手性拆分和手性辅基诱导等策略[19].例如: Knowles等[17a]于1977年通过手性拆分的方式, 利用L-薄荷醇作为拆分试剂制备出P-手性双膦配体(R, R)-DiPAMP (9)的关键前体P-手性双膦氧化物.通过手性辅基诱导的策略, Chang等[19d]报道了Cp*-Ir(Ⅲ)催化的含手性吡咯结构的双芳基膦氧的不对称芳基邻位C—H键酰胺化反应, 实现了P-手性膦酰胺类膦氧的高效高选择性合成.此外, 利用叔膦氧化的策略, Gilheany等[19e, 19f]发现在当量的手性醇和六氯丙酮的作用下, 消旋叔膦能以中等到良好的对映选择性转化成相应的P-手性膦氧化物.
尽管利用手性拆分和辅基诱导等策略能实现一些P-手性膦氧化物的高效合成, 但这些方法普遍需要使用当量甚至过量的手性试剂, 存在经济成本较高等问题; 且手性拆分策略只能以理论50%的产率得到所需构型的产物, 而对于手性辅基诱导策略还存在额外引入和脱除辅基的反应, 导致反应步骤和原子经济性不高.相比而言, 利用不对称催化策略则是目前构建P-手性膦氧化物最为直接、高效的合成方法.近年来, 经国内外科研工作者的不懈努力, 已发展了一系列基于以下三种策略的不对称反应(图 3): ⅰ)潜手性三级膦化物参与的去对称化反应, ⅱ)三级膦氧参与的(动态)动力学拆分反应, ⅲ)二级膦氧参与的反应策略.基于此, 各种环加成、共轭加成、C—H酰胺化、C—H芳基化、关环复分解、烯丙基烷基化等反应已被成功用于结构多样性的开链和环状P-手性膦氧化物的对映选择性合成.
综上所述, 利用不对称催化构建P-手性膦氧化物近年来取得了显著进展, 但目前尚无相关方面的专题综述文章对其进行总结归纳与分析.虽然Jugé等[1a]2016年对P-手性含磷化合物的应用和立体选择性合成进行了综述; 徐利文等[18b]2017年对利用过渡金属催化的不对称C—H官能团化反应构建杂原子手性化合物进行了介绍, 但这两篇综述均只介绍了少数不对称催化合成P-手性膦氧的例子, 因为绝大部分相关的例子是近三年才被报道的.因此, 随着近几年不对称催化构建P-手性膦氧的新方法和策略越来越多, 以及该类化合物在诸多领域的应用潜能, 我们认为很有必要对构建P-手性膦氧化物的不对称催化方法进行专题报道, 总结已有反应策略及其优势, 同时分析目前仍然存在的问题以及提出一些可能的解决思路, 以期为从事有机合成及相关研究领域的化学工作者提供有益的借鉴和参考.根据图 3右侧介绍的不对称催化策略, 下面我们将从底物结构和反应类型出发, 详细介绍近十年来不对称催化合成P-手性膦氧化物的反应和方法.
催化的不对称去对称化反应由于可有效构建远离反应位点的手性中心、同时可一步构建多个手性中心等独特优势, 被认为是有机合成中合成手性化合物的重要策略之一.近几十年来, 该策略受到国内外化学家们的广泛关注并获得蓬勃发展, 且已被成功用于许多手性天然产物和药物分子的不对称合成中[20].鉴于P-手性膦氧化物在手性配体、有机催化剂以及材料等领域的应用价值, 化学家们对利用去对称化策略从潜手性三级膦氧化物出发来构建高附加值的P-手性膦氧化物进行了不断的探索.尽管这一转化存在诸多挑战, 如手性催化剂如何选择性地控制潜手性磷原子上两个完全相同的取代基中的一个参与反应, 而另一个保持不变来抑制非手性副产物的生成等; 但近年来也取得了一些重要进展, 发展了基于双炔基、双烯基、双芳基或双酚基等取代的潜手性膦氧化物的系列不对称去对称化反应, 从而高效高选择性地合成系列开链或环状的P-手性膦氧化物.
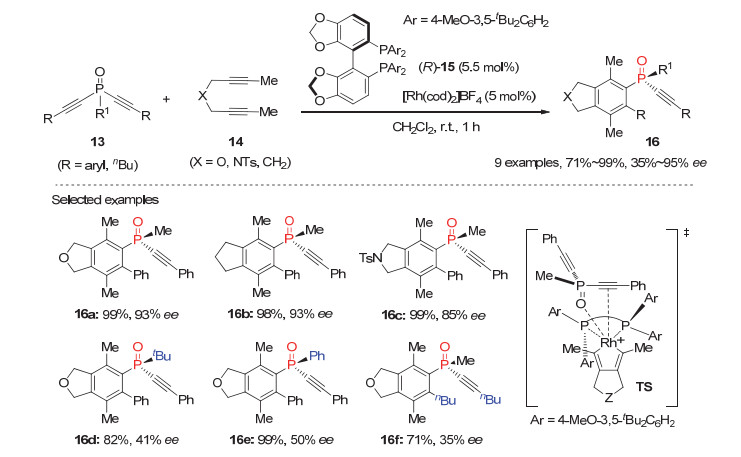
早在2008年, Tanaka小组[21]利用阳离子型手性双膦(R)-15/Rh(Ⅰ)配合物为催化剂, 实现了首例潜手性双炔基膦氧化物13与富电子1, 6-双炔14的去对称化[2+2+2]环加成反应, 以71%~99%的产率和35%~95%的对映选择性获得目标P-手性膦氧化物16(图 4).如过渡态TS所示, 该反应中1, 6-双炔首先与手性Rh配合物发生反应, 所生成的手性五元铑杂环中间体与双炔基膦氧13螯合; 随后炔基插入铑碳键, 再经还原消除得到产物16, 并再生手性Rh催化物种.然而该反应底物普适性较为有限, 当双炔基膦氧化物13的R1为叔丁基或苯基时, 产物16d~16e的对映选择性大幅下降; 且炔烃末端R取代基为丁基时, 产物16f的ee值也只有35%.
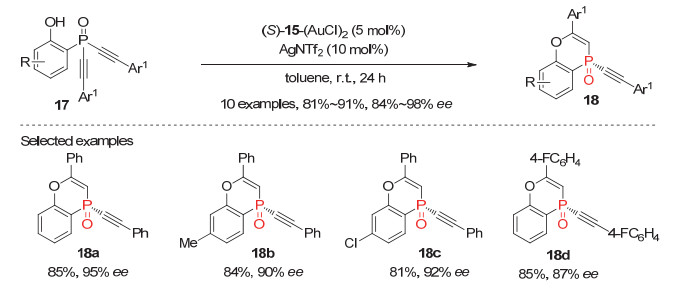
2018年, 同样利用潜手性双炔基膦氧化物的去对称化策略, 资伟伟等[22]发现在手性阳离子型金(Ⅰ)配合物催化下, 邻苯酚取代的潜手性双炔基叔膦氧化物17能高效发生炔烃的分子内的不对称氢醚化反应.使用5 mol%的(S)-Segphos[Au(NTf)2]2为催化剂, 该去对称化环化反应以81%~91%的产率和84%~98%的对映选择性获得系列环状P-手性膦氧产物18(图 5).虽然反应可以取得良好到优秀的产率和对映选择性, 但需要指出的是, 该反应的底物适用范围较窄, 仅适用于芳基取代的非末端双炔基膦氧化物.
稍后, 冯小明和刘小华等[23]利用他们发展的冯氏氮氧配体L-RaPr3与Tm(OTf)3形成的手性铥(Ⅲ)配合物, 实现了潜手性双炔基膦氧化物13与硫醇19的去对称化不对称共轭加成反应, 以高达92%的产率和97%的对映选择性获得系列开链状P-手性膦氧化物20(图 6).而且所得产物20可以进行多样性转化合成结构丰富的其它P-手性膦氧化物.需要指出的是, 该反应中硫醇的结构对产物的立体选择性有很大影响, 如巯基乙酸酯在该反应条件下与各种芳基或烷基取代的非末端双炔膦氧化物反应均能获得优秀的Z/E选择性和良好到优秀的ee值, 而相同条件下以硫酚为亲核试剂则使反应的对映选择性和Z/E选择性大幅下降.
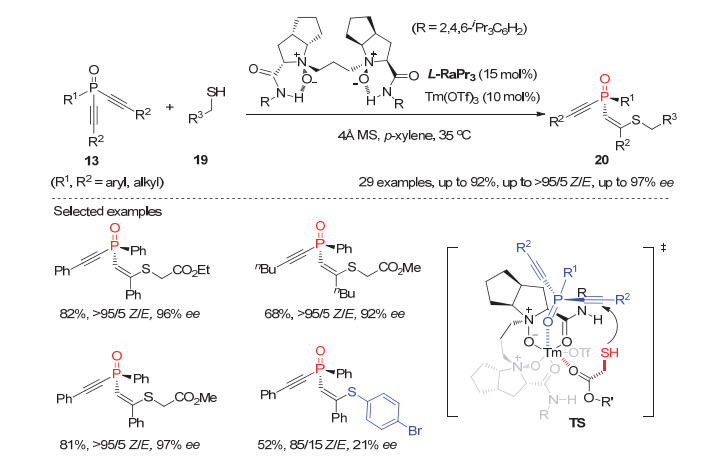
通过仔细分析手性氮氧配体与不同稀土金属配合物的单晶结构, 作者认为中心金属铥和配体的结构对区别底物中两个炔基起到至关重要的作用.如图 6中过渡态TS所示, 所形成的具有中等空腔的手性铥催化剂与双炔基膦氧化物的膦氧基团和巯基乙酸酯的羰基同时螯合, 从而对映选择性地进行加成反应.
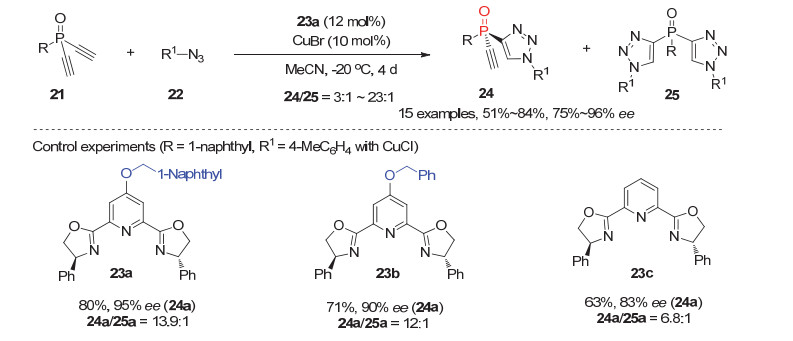
最近, 我们小组[24]利用铜催化的叠氮与炔烃的不对称环加成(CuAAC)反应策略, 实现了首例潜手性末端双炔基膦氧化物21的去对称化反应, 为P-手性膦氧化物提供了一类高效高选择性的合成方法.经过系统研究, 发现利用新设计合成的吡啶C4位带有大位阻取代基的PyBox型配体23a衍生的CuBr配合物可以高效催化潜手性双乙炔基膦氧化物21与叠氮22的对映选择性环加成反应, 以中等到优秀的单双比, 51%~84%的产率和75%~96%的ee值获得同时含有乙炔基和三氮唑的P-手性膦氧化物24(图 7).值得一提的是, 相比无取代基的PyBox配体23c, 吡啶C4位带有大位阻的PyBox配体23a不仅能有效提高反应的对映选择性, 而且能很大程度抑制非手性副产物双三氮唑25的产生.而这两点也一直是困扰潜手性双炔类底物不对称去对称化CuAAC反应中的难题[25].结合文献对CuAAC反应机理的研究[26]及我们[27]前期的工作基础, 我们认为配体23a上吡啶环C4位引入大位阻基团后, 改变了手性金属铜配合物周围的手性环境, 使得双乙炔基膦氧21进入中心金属的空间变得更狭窄, 从而有效提高反应的对映选择性, 同时抑制双三氮唑副产物的形成.根据对产物ee值随反应时间的变化曲线研究, 以及Uozumi等[28]在潜手性双炔的去对称化CuAAC反应的研究, 该反应涉及了去对称化与动力学拆分的协同组合过程.
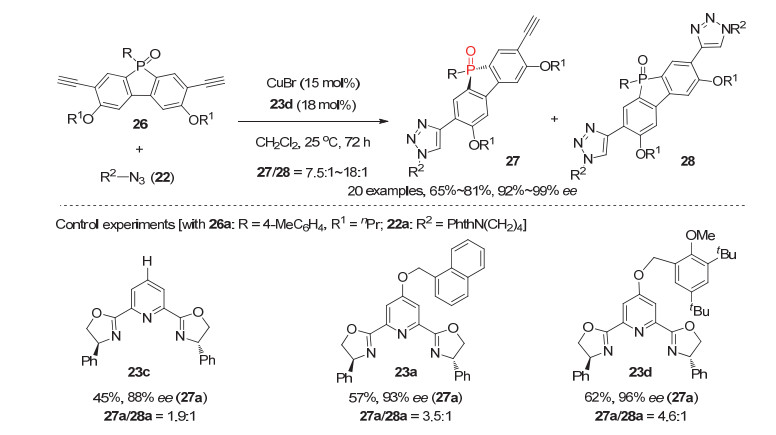
鉴于膦杂茂膦氧化物在有机光电材料等领域的重要应用价值[29], 我们进一步设想能否将这一新型大位阻PyBox配体应用于更具挑战性的膦杂茂骨架衍生的潜手性双乙炔基膦氧化物26与叠氮22的不对称去对称化CuAAC反应中, 为手性膦杂茂膦氧化物的对映选择性构建提供一类有效方法.该反应之所以更具挑战性是因为在潜手性双乙炔基膦氧化物26中, 反应位点乙炔基与潜手性磷原子中心相隔四根共价键, 距离较远; 因此如何实现该类底物的远程手性控制是反应的难点和成功的关键.令人高兴地是, 经过仔细研究, 发现使用新合成的吡啶C4位具有大位阻取代基的PyBox配体23d与CuBr形成的手性铜配合物为催化剂, 该反应能高效进行, 并以高达18:1的单双比, 81%的产率和99%的对映选择性获得含有乙炔基和三氮唑的P-手性二芳基膦杂茂膦氧化物27(图 8)[24].此外, 对照实验再次证明这一大位阻配体在控制反应对映选择性和抑制非手性副产物方面有显著优势.
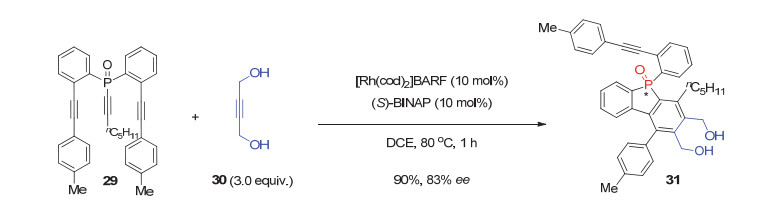
此外, 在研究铑催化的[2+2+2]环加成反应中, Shibata等[30]利用手性(S)-BINAP/Rh(Ⅰ)配合物催化剂, 报道了丁炔二醇30与潜手性非末端双炔基膦氧化物29的不对称去对称化[2+2+2]环加成反应, 以90%的产率和83% ee值获得P-手性二芳基膦杂茂31(图 9).虽然该反应只报道了一个例子, 且产物的对映选择性也不高, 但这却为P-手性膦杂茂膦氧化物的不对称合成提供了另一种策略.
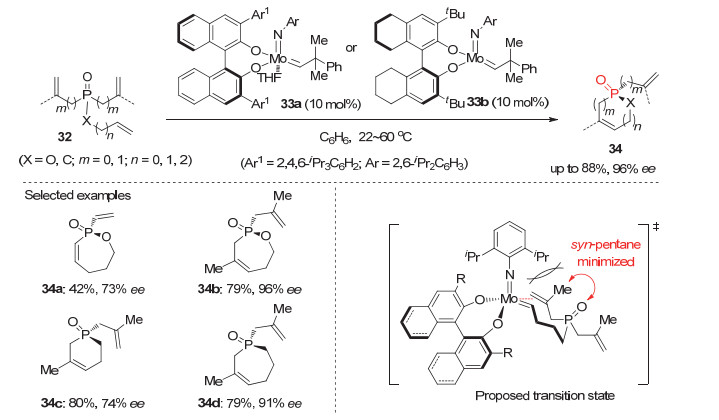
利用不对称去对称化策略, 含双烯基的潜手性膦氧化物也被成功用于开链和环状的P-手性膦氧化物的对映选择性合成. 2009年, Gouverneur和Hoveyda等[31]利用手性Mo配合物33a或33b为催化剂, 发展了首例三烯基取代的潜手性膦氧或磷酸酯32的不对称分子内关环复分解(RCM)反应, 以30%~88%的产率和27%~96%的ee值合成了系列五到七元环状P-手性膦氧化物34(图 10).有趣的是, 作者发现当潜手性膦氧化物32上的烯基为末端烯烃时, 反应均能顺利进行并以良好到优秀的产率和对映选择性获得产物; 而使用非末端烯基取代的膦氧时, 反应的活性和选择性则大幅下降.经过初步的研究, 作者提出了如图 10所示的反应过渡态.
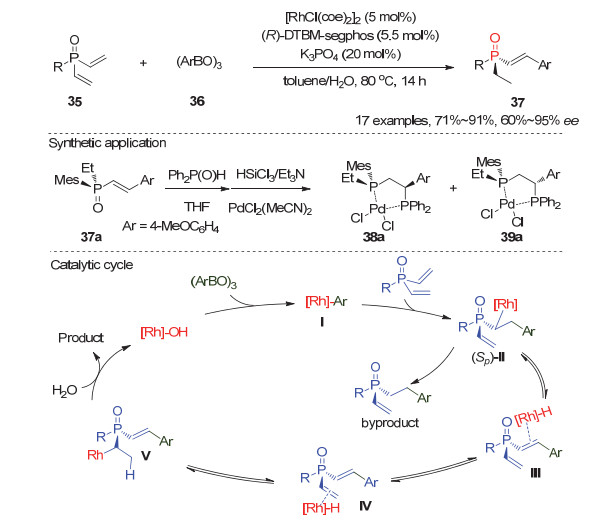
2018年, Hayashi小组[32]报道了Rh(Ⅰ)催化的双乙烯基潜手性膦氧化物35与芳基硼酸酐36的不对称氢芳基化反应, 实现了开链状烯基取代P-手性膦氧化物37的高效高对映选择性合成.他们发现在甲苯和水的混合溶剂中, 5 mol%的(R)-DTBM-Segphos/Rh(Ⅰ)配合物可以高效催化反应, 以71%~91%的产率和60%~95%的ee值得到产物37(图 11).通过初步机理研究, 作者提出如图 11所示的可能反应机理:手性Rh配合物首先与芳基硼试剂作用, 所形成的手性芳基-铑物种Ⅰ主要加成到膦氧35的pro-R乙烯基上生成烷基-铑中间体(Sp)-Ⅱ; 随后依次经历β-H消除、Rh-H迁移、铑氢化过程得到烷基-铑中间体Ⅴ, 最后质子解得到目标产物37.与此同时, 如果烷基-铑(Sp)-Ⅱ直接发生质子解则生成单乙烯基氢芳基化的副产物.此外, 该手性膦氧产物经过两步简单转化即可合成一类手性双膦配体, 且其衍生的Pd配合物38a能高效催化芳基硼酸酯对烯酮的对映选择性共轭加成反应.尤为值得一提的是, 与传统的去对称化反应不同, 在该反应中双乙烯基膦氧35中的两个乙烯基均参与了反应; 其中, 一个与芳基硼试剂发生氧化芳基化, 而另一个则通过分子内的Rh-H迁移被还原为乙基, 因此避免了生成非手性的副产物.这一新颖的去对称化过程为构建开链状烯基取代的P-手性膦氧化物的对映选择性提供了一种新的思路.
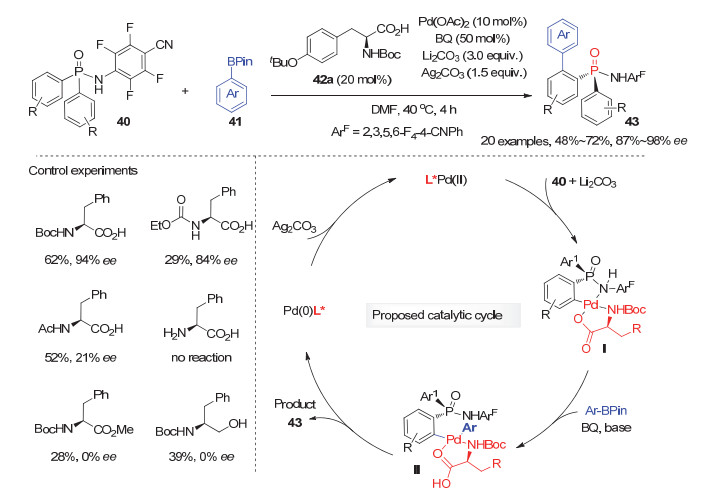
C—H键直接官能团化反应由于无须使用预先制备的官能团化底物, 具有步骤经济性、原子经济性等诸多优点, 近年来过渡金属催化的C—H键直接官能团化反应研究在国内外化学家的不懈努力下取得了迅猛发展[33].基于此, 通过C—H键官能团化策略, 利用双芳基潜手性膦氧化物的不对称去对称化反应实现P-手性膦氧化物的合成受到化学工作者的广泛关注, 并取得了一定的进展. 2014年, 韩福社等[34]发展了首例钯催化的潜手性双芳基膦酰胺40与芳基硼酸酯41的分子间不对称去对称化邻位C—H键芳基化反应, 可高效高选择性制备系列开链状的P-手性膦酰胺类化合物43.经过仔细研究, 作者发现该去对称化C—H键芳基化反应以10 mol%的醋酸钯为催化剂, 20 mol%手性N-Boc氨基酸42a为配体, 50 mol%苯醌(BQ)为氧化剂, 碳酸锂为碱, 碳酸银为添加剂的条件下可以顺利进行, 以中等到良好的产率和良好到优秀的ee值获得P-手性膦酰胺43(图 12).对照实验表明手性氨基酸配体42a上氨基保护基和羧酸基团对反应的对映选择性和产率起至关重要的作用.基于此, 根据韩福社等[35]前期工作中所获得的五元环钯物种单晶结构, 以及余金权等[36]报道的手性氨基酸-Pd(Ⅱ)配合物催化的C—H键官能团化反应的相关研究, 该反应可能经历如下历程:首先, 二芳基膦酰胺在碱性条件下与手性氨基酸衍生的Pd(Ⅱ)催化剂配位, 通过邻位C—H键活化, 形成关键的手性五元环钯物种Ⅰ; 随后与芳基硼酸酯发生偶联反应得到产物及零价钯物种; 最后, 零价钯被碳酸银氧化成二价钯实现催化循环.此外, 膦酰胺产物43可经转化合成结构多样的P-手性叔膦和硫代膦酰胺, 这进一步表明了该方法的实用性.
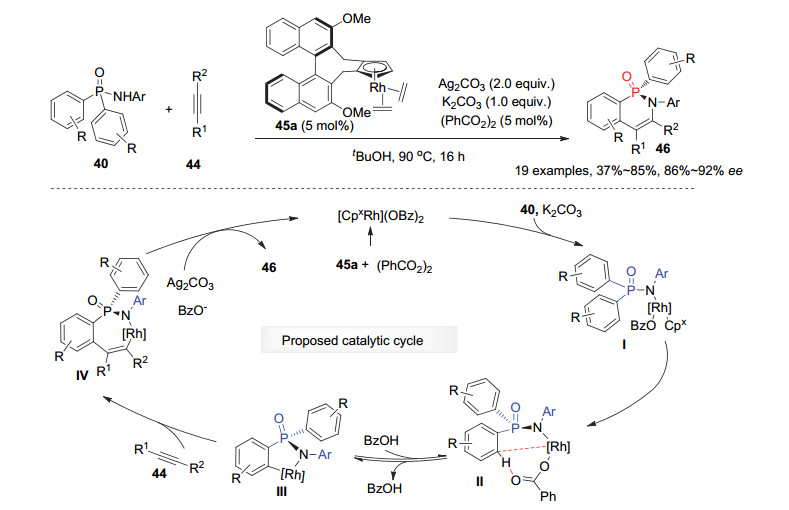
2017年, Cramer等[37]利用Rh(Ⅲ)催化的芳基C—H键活化策略实现了潜手性双芳基膦酰胺40与非末端炔烃44的不对称去对称化环化反应, 为六元环状P-手性膦酰胺类化合物提供了一种高效合成方法.使用5 mol%他们自主发展的手性Cpx-Rh(Ⅲ)配合物45a为催化剂, 该去对称化反应能以中等到良好的产率和优秀的对映选择性获得系列P-手性环状磷酰胺46.通过氘代实验等研究, 作者发现加入K2CO3降低了C—H键活化的可逆性, 从而确保了产物的高对映选择性.如图 13所示, 原位形成的手性[CpxRh](OBz)2配合物首先与底物40的导向基团酰胺N—H键作用, 顺次发生芳基邻位C—H键活化, 生成的手性五元环铑物种Ⅲ随后被炔烃44捕获发生选择性的插入反应生成七元环铑中间体Ⅳ; 最后经历还原消除得到目标产物46, 同时在Ag2CO3等的作用下再生活性[CpxRh](OBz)2催化剂.此外, 产物膦酰胺46在DBU和三氯硅氢烷(HSiCl3)的作用下能被对映选择性地还原为二芳基胺膦烷.
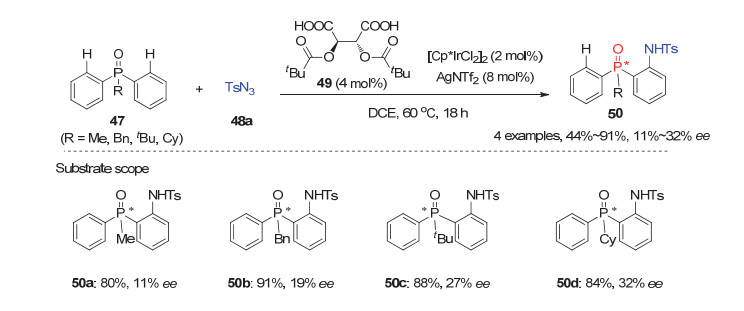
除了潜手性双芳基膦酰胺类底物以外, C—H键活化策略还被用于潜手性双芳基膦氧化物参与的不对称去对称化反应中.如2015年, Chang小组[38]实现了首例潜手性芳基膦氧化物47与对甲苯磺酰基叠氮(TsN3)的分子间去对称化C—H键酰胺化反应.他们发现该反应在非手性的Cp*Ir(Ⅲ)催化剂与手性添加剂1, 2-二羧酸49的作用下能顺利进行, 以良好的产率及11%~32%的ee值获得开链状P-手性膦氧化物50(图 14).而且作者发现手性羧酸49在该反应中具有双重作用:即作为手性配体与非手性铱催化剂形成催化活性的手性铱环物种, 也有利于最后脱金属化质解生成酰胺化产物再生手性铱催化剂的过程.虽然该反应对映选择性不够理想, 但这一研究为进一步发展高对映选择性的C—H键酰胺化反应提供了一些新的启发.
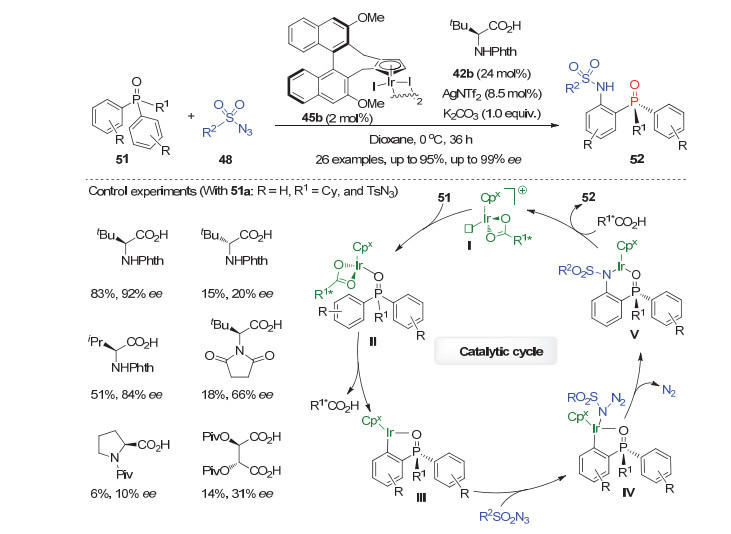
随后, Cramer等[39]利用手性Cpx-Ir(Ⅲ)催化剂与手性羧酸相结合的协同效应, 实现了高对映选择性的潜手性芳基膦氧化物51和磺酰基叠氮48的去对称化C—H键酰胺化反应.在2 mol%二聚的手性铱催化剂(IrCpxI2)2, 8.5 mol% AgNTf2, 以及24 mol%的N-邻苯二甲酰基保护的叔亮氨酸42b的共同作用下, 该反应能以高达95%的产率和99%的对映选择性获得一系列P-手性膦氧化物52(图 15).通过对一系列手性羧酸添加剂及其手性匹配与不匹配效应等进行研究, 他们发现手性羧酸的结构和绝对构型对反应活性和选择性均有很大影响.此外, 通过简单转化不仅能脱除P-手性膦氧产物上的磺酰基团, 还能对映选择性保持地将其P=O基团还原为叔膦.结合Chang等[38]对C—H键酰胺化反应机理的研究, 他们提出了如图 15所示的可能催化循环: (IrCpxI2)2首先与AgNTf2作用生成阳离子型的手性Ir配合物再与手性羧酸42b配位, 形成的手性Ir物种Ⅰ与潜手性膦氧51的P=O基团配位生成中间体Ⅱ.紧接着中间体Ⅱ通过协同的金属化-脱质子过程产生P-手性的五元环状芳基铱物种Ⅲ, 顺次与叠氮试剂48螯合, 再经历迁移插入脱氮气过程得到P-手性六元铱环Ⅴ; 最后在羧酸的协助下, 发生脱金属质解得到目标产物52并再生活性铱催化剂Ⅰ.
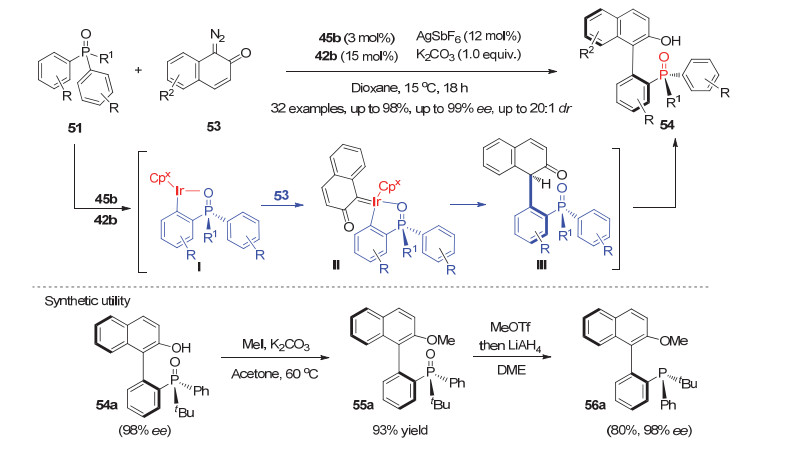
进一步利用手性Cpx-Ir(Ⅲ)配合物45b与手性羧酸42b相结合的这一催化体系, Cramer等[40]实现了潜手性双芳基膦氧化物51与重氮化合物53的不对称C—H键芳基化反应, 高效高选择性地构建了一系列高附加值含轴手性的P-手性膦氧化物54.如图 16所示, 与之前的酰胺化反应历程不同的是, 这里形成的手性五元环铱物种Ⅰ被重氮53捕获生成金属铱卡宾中间体Ⅱ, 再经脱金属化质解、芳构化生成轴手性联芳基P-手性膦氧化物54.对照实验表明手性羧酸的协同效应对该C—H键芳基化反应的选择性和活性也有明显影响.产物54a利用甲基化保护酚羟基后再经一步还原, 即可合成同时含联芳基轴手性和P-手性的叔膦化合物56a.
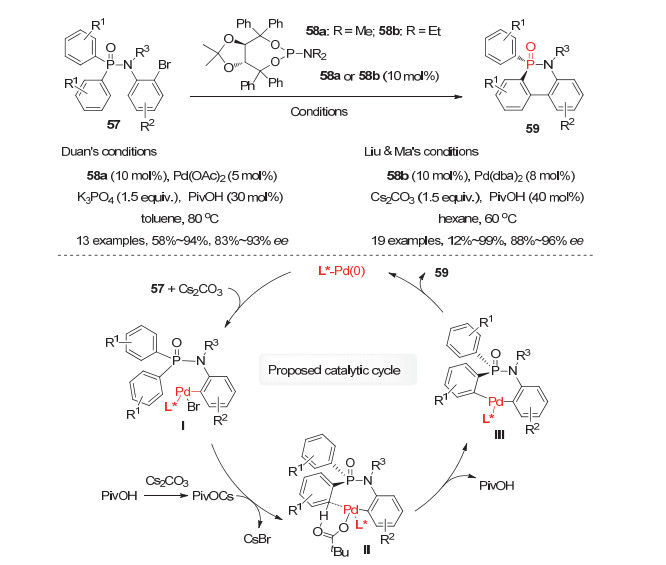
利用C—H键活化策略, 除实现上述潜手性双芳基膦氧化物参与的分子间去对称化反应以外, 邻溴芳基取代的潜手性膦氧化物参与的分子内去对称化C—H键芳基化反应也已被成功报道, 并用于六元、五元环状P-手性膦氧化物的对映选择性构建. 2015年,段伟良小组[41], 刘澜涛与马文瑾小组[42]独立地报道了手性钯催化的双芳基潜手性膦酰胺57的分子内去对称化C—H键芳基化反应(图 17).分别利用手性TADDOL亚磷酰胺衍生的钯配合物58a/Pd(OAc)2或58b/Pd(dba)2为催化剂, 磷酸钾或碳酸铯为碱, 在PivOH为添加剂的条件下, 成功实现了该反应, 均以良好到优秀的产率和对映选择性获得系列六元环状P-手性膦酰胺类化合物59.可能的催化循环如图 17所示:原位形成的手性Pd(0)配合物首先与底物57中的芳基溴发生氧化加成生成芳基-Pd(Ⅱ)-Br中间体Ⅰ, 与此同时, PivOH与Cs2CO3反应生成的PivOCs进一步与中间体Ⅰ作用生成芳基-Pd(Ⅱ)-OPiv物种Ⅱ; 随后膦氧上一个芳基的C—H键发生断裂形成二芳基钯物种Ⅲ, 最后经过还原消除得到产物59同时再生活性Pd(0)催化剂.此外, 段伟良等还发现产物59在甲基锂试剂的作用下能发生P—N键的断裂用于制备相应的联苯基单膦氧配体.
2017年, 利用相同的去对称化C—H键芳基化策略, 徐利文和崔玉明等[43]发展了手性钯催化的邻溴芳基取代的潜手性芳基膦氧化物60的不对称分子内邻位C—H键芳基化反应, 为P-手性膦杂茂类化合物的构建提供了一种新方法.以商业可得的手性配体(S, S)-Me-Duphos (61)与Pd(OAc)2形成的手性Pd配合物为催化剂, Cs2CO3为碱, 该反应能以中等的产率和对映选择性获得各种取代的P-手性芳基膦杂茂62(图 18).然而遗憾的是, 反应的对映选择性受底物具体结构影响很大, 且产物的ee值普遍不高, 仅有两例ee值大于70%.
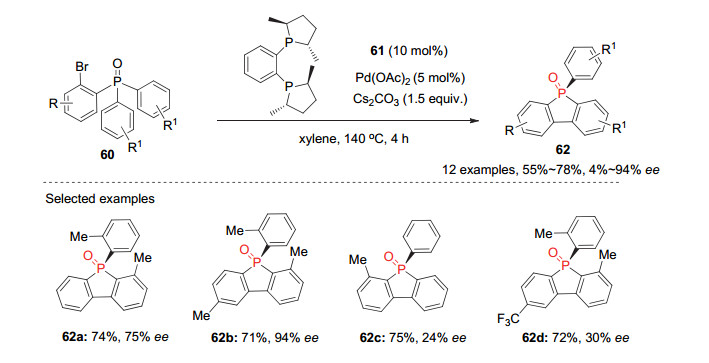
为了能高对映选择性合成在光电材料等领域具有潜在应用的这类P-手性膦杂茂, 段伟良等[44]最近对该C—H键芳基化反应进行了系统研究.发现在(R)- BINOL衍生的磷酸63和磷酰胺64与Pd(PCy3)2的催化下, 反应能以57%~90%的ee值得到(S)构型的膦杂茂(S)-62; 而以(R)-Segphos与Pd(OAc)2形成的配合物为催化剂, PivOH为酸性添加剂时, 能进一步提高该反应产物的ee值到64%~96%, 并得到构型相反的膦杂茂(R)-62(图 19).
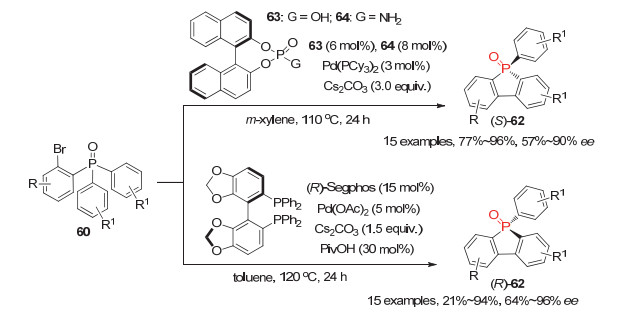
此外, 汤文军等[45]利用自主开发的P-手性联芳基单膦配体12a与Pd(OAc)2形成的手性Pd配合物为催化剂, 实现了邻溴芳基潜手性磷酸酯65的不对称分子内环化反应, 以高达92%的产率和88%的ee值获得系列六元环状P-手性磷酸酯66(图 20).经过两步与不同锂试剂的反应, 手性磷酸酯66a可手性保持地、高效地转化成用途更广的开链P-手性膦氧化物67a; 且通过改变与两种锂试剂的反应顺序可合成67a的对映体ent-67a.
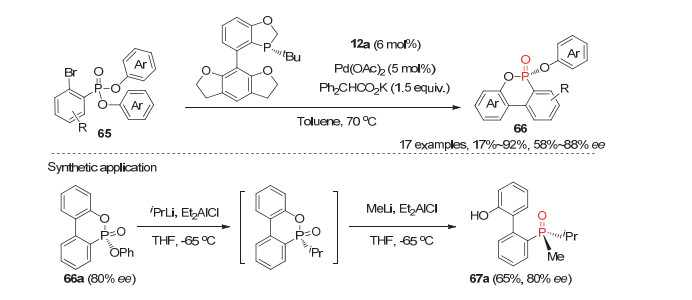
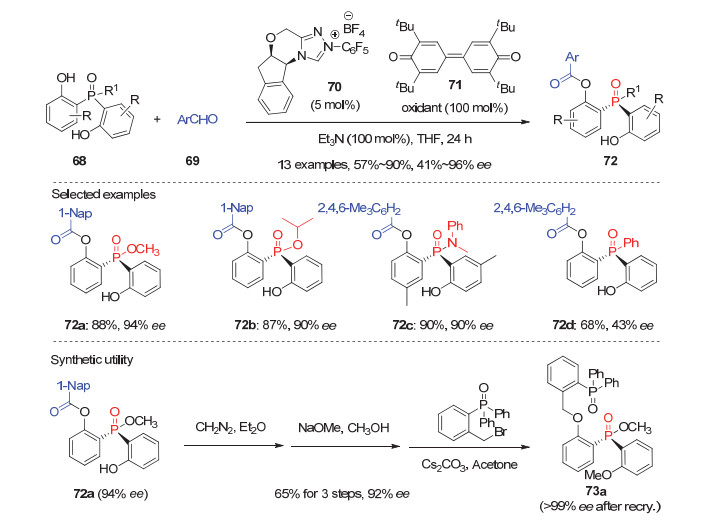
双酚羟基取代的潜手性膦氧化物, 由于反应位点在酚羟基上, 离构建的磷原子手性中心较远, 因此该类底物的去对称化反应的对映选择性较难控制.尽管如此, 经过化学家们的努力, 近年来也取得了一些可喜的结果.池永贵等[46]于2016年利用氧化氮杂环卡宾(NHC)催化实现了首例双酚取代的潜手性膦氧化物68参与的去对称化反应.他们发现在5 mol%的手性氨基茚醇衍生的NHC催化剂70, 以及当量醌71为氧化剂的作用下, 各种取代的双酚膦酸酯或膦酰胺68均能顺利与大位阻的2-萘甲醛或2, 4, 6-三甲基苯甲醛69发生去对称化酰化反应, 以57%~90%的产率和41%~96%的ee值获得相应的P-手性膦酸酯或膦酰胺类化合物72(图 21).然而三芳基取代的产物72d在该催化体系的仅只有43%的ee值; 且酚羟基的位置也至关重要, 因为当酚羟基处在间位或对位时反应产物的ee值均为0.此外, 该去对称化反应能放大到15 mmol的规模, 并仅需1 mol%的手性催化剂70即以95%的产率和88%的ee值制备5.02 g产物72a.目标产物72a经三步常规转化即可合成P-手性的双齿Lewis碱催化剂73a; 且73a能有效催化烯酮与醛的不对称共轭还原-aldol反应, 并能取得良好的ee值.
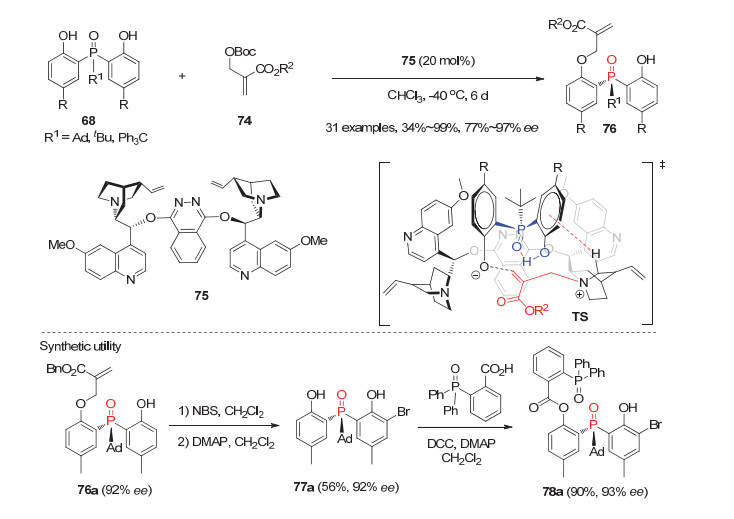
最近利用手性叔胺Lewis碱催化, 李鑫等[47]发展了双酚潜手性膦氧化物68与MBH碳酸酯74的对映选择性去对称化烯丙基烷基化反应.在20 mol%双核金鸡纳碱催化剂(DHQ)2PHAL(75)的作用下, 一系列酚羟基对位有取代基的双酚膦氧化物68均能顺利进行去对称化反应, 以高达99%的产率和97%的ee值获得各种开链状P-手性膦氧化物76(图 22).虽然该反应存在催化剂用量较高、反应时间较长、膦氧68的R1须为大位阻取代基等不足, 但这是利用不对称烯丙基烷基化反应来实现潜手性膦氧去对称化反应的一种新策略.产物经简单转化能用于合成P-手性双齿膦氧催化剂.最后作者结合线性自由能关系分析和理论计算提出了如图 22所示的可能过渡态.
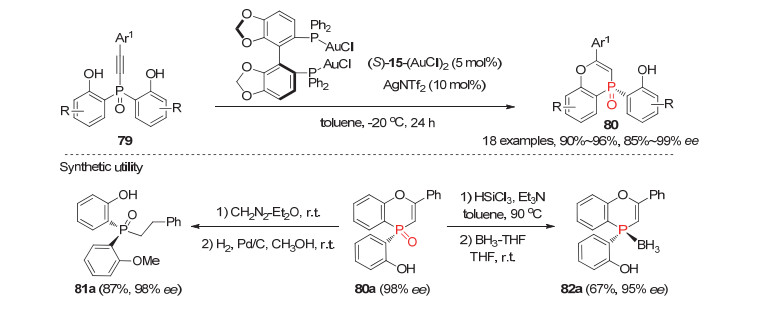
利用手性金(Ⅰ)配合物催化的炔烃的分子内氢醚化反应, 资伟伟等[22]报道了炔基取代的双酚潜手性膦氧79的分子内去对称化环化反应, 实现了环状P-手性膦氧化物80的对映选择性合成. 5 mol%的手性(S)-Segphos 15/Au(NTf2)配合物能高效催化该环化反应, 以优秀的产率和对映选择性获得环状P-手性膦氧80(图 23).该反应不仅具有良好的底物普适性, 而且所得产物能进行多样性转化.
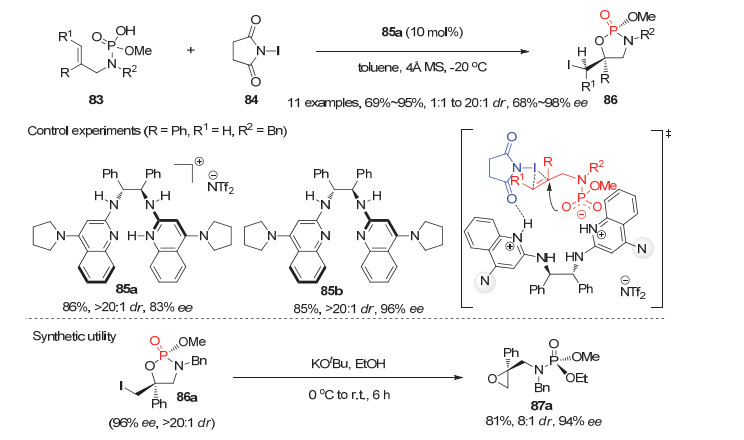
除了上述基于潜手性膦氧、膦酰胺或磷酸酯的系列不对称反应外, 最近去对称化策略还被用于从磷酸或次膦酸类底物出发构建环状或非环状P-手性膦氧化物的不对称反应.值得一提的是, 与上述利用两个相同取代基的潜手性膦氧化物的去对称化反应完全不同的是, 这里使用的是取代基完全不同的磷酸或次膦酸作为潜手性膦氧前体, 在碱的作用下形成相应的潜手性中间体进行后续的去对称化反应. 2014年, Johnston等[48]报道了手性Brønsted酸催化的烯丙基取代的氨基磷酸83与N-碘代丁二酰亚胺84的不对称碘环化反应, 提供了一种高效高选择性构建同时含C-手性和P-手性膦酰胺类膦氧化物的方法(图 24).利用10 mol%他们自主发展的双功能铵盐85a为催化剂, 该碘环化反应顺利进行并以高达95%的产率, 20:1的dr值和98%的ee值获得相应的环状P-手性膦酰胺86.同时, 产物86a在碱性的醇溶液中可以高效地发生开环反应制备含有环氧丙烷的开链P-手性膦氧化物87a.结合对照实验结果及他们前期报道的羧酸碘内酯化反应机理[49], 提出了如图 24过渡态所示的Brønsted酸和Brønsted碱双重活化模型.
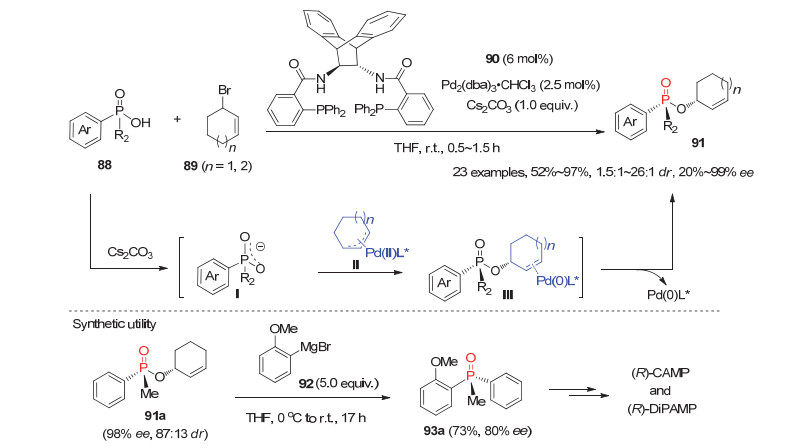
最近, Trost等[50]利用手性Pd配合物催化的不对称烯丙基烷基化反应, 实现了潜手性亲核试剂次膦酸88的去对称化反应, 为P-手性膦氧化物的高效高选择性构建提供了一种新颖的思路.运用手性1, 2-二胺衍生的双膦配体90, Pd2(dba)3•CHCl3为钯源, Cs2CO3为碱, 一系列芳基烷基次膦酸与3-溴环己(庚)烯顺利反应以良好到优秀的产率和立体选择性获得含有C-手性和P-手性次膦酸酯91.如图 25所示, 次膦酸88首先在碱Cs2CO3的作用下形成潜手性膦氧中间体Ⅰ, 接着Ⅰ上的一个氧原子选择性地进攻原位生成的手性π-烯丙基钯物种Ⅱ, 随后得到P-手性膦氧产物同时再生Pd(0)催化物种.此外, 产物91a经过一步与格氏试剂92的反应即可合成P-手性配体(R)-CAMP和(R)-DiPAMP的前体化合物93a.
基于外消旋三级膦氧化物的(动态)动力学拆分反应也是一种对映选择性构建P-手性膦氧化物的重要策略.与去对称化相比, 三级膦氧化物的(动态)动力学拆分策略具有其独特之处, 如:可使用的膦氧底物结构更为丰富, 不局限于使用含相同取代基的潜手性叔膦氧化物为原料; 动力学拆分反应通过一步反应能将消旋膦氧转化成高对映选择性的两类P-手性膦氧化物; 且动态动力学拆分反应能以100%的理论产率获得目标P-手性膦氧产物, 不存在生成非手性副产物等优点.鉴于此, 近年来利用该策略来合成P-手性膦氧化物也逐渐受到化学家们的关注, 并取得了初步的研究成果.
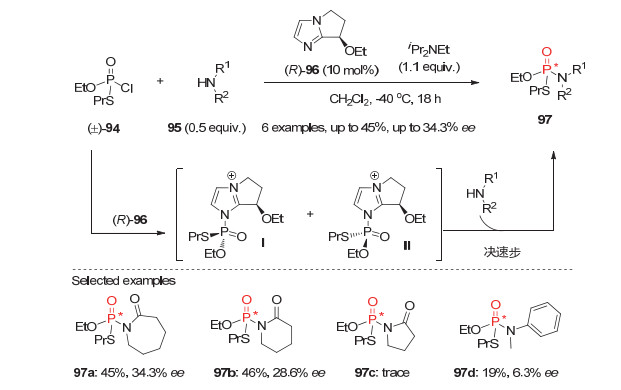
2012年, 张万斌等[51]报道了外消旋膦酰氯的不对称催化动力学拆分来构建P-手性膦酰胺类膦氧化物.他们发现在10 mol%手性双咪唑环叔胺(R)-96的作用下, 消旋膦酰氯94能与0.5 equiv.的仲胺95顺利发生反应, 以高达45%的转化率和最高34%的ee值获得相应的P-手性膦酰胺97.如图 26所示, 叔胺催化剂(R)-96首先进攻消旋膦酰氯94生成两个活性的非对映异构体中间体Ⅰ和Ⅱ, 随后与伯胺95反应选择性地得到P-手性膦酰胺97, 同时再生催化剂.虽然该反应底物普适性有限且对映选择性普遍不高, 但这却是不对称催化构建P-手性膦酰胺的首例报道.
基于前期报道的Cpx-Rh催化的潜手性膦氧化物的去对称化不对称C—H键官能团化反应基础上, Cramer小组[52]于2018年设计合成了一类新颖的三取代手性Cpx-Rh催化剂45c, 并利用其成功发展了消旋次膦酰胺105的不对称动力学拆分, 一步高效合成了开链状和六元环状P-手性次膦酰胺膦氧化物(图 27).作者研究发现, 在C—H键活化形成手性五元环铑中间体的决速步中, 次膦酰胺中的(R)-40与Cpx-Rh催化剂45c的反应速度要远远快于(S)-40, 因而很好地实现了外消旋膦酰胺40的动力学拆分, 以良好到优秀的产率和ee值同时获得P-手性膦氧产物(R)-46和回收P-手性次膦酰胺(S)-40.
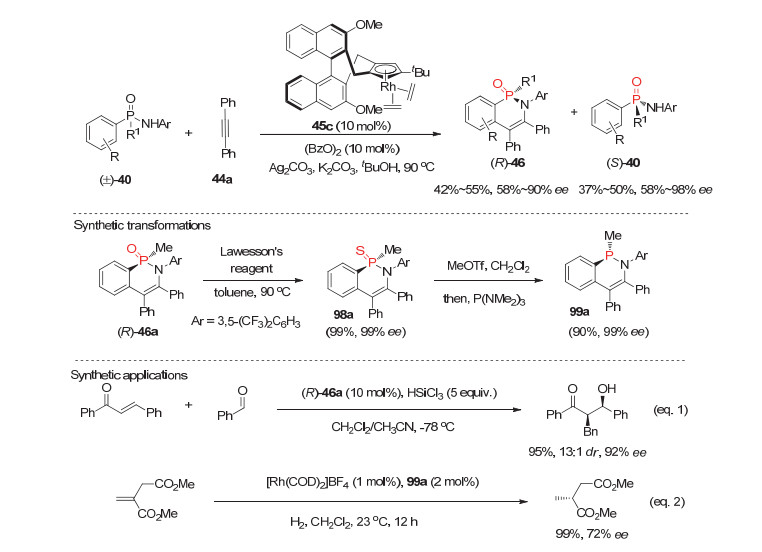
此外, 膦氧产物(R)-46a通过两步简单转化可用于合成P-手性单齿膦配体99a; 且P-手性膦氧产物(R)-46a和合成的P-手性叔膦99a分别在烯酮与醛的不对称还原aldol反应和铑催化的烯基酯的不对称氢化反应中表现出良好的手性控制能力.
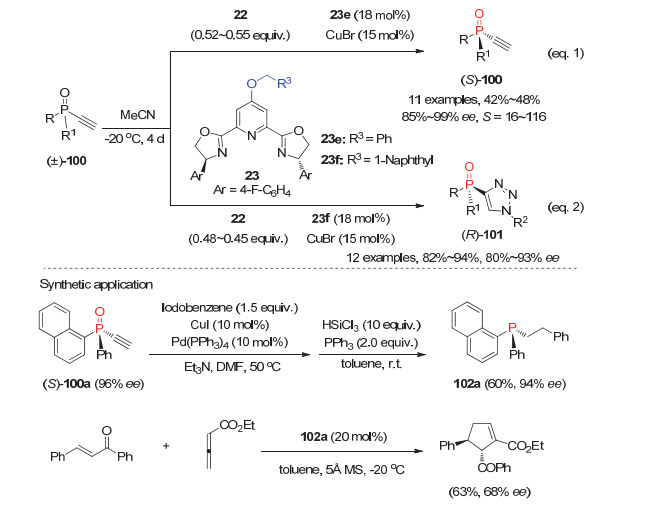
最近, 我们小组[24]在研究潜手性双乙炔基膦氧的去对称化CuAAC反应中, 发现新设计合成的吡啶C4位连有大位阻取代基的PyBox型配体衍生的手性铜配合物也能很好地催化消旋单乙炔基三级膦氧100与叠氮22的CuAAC反应, 从而实现了消旋单乙炔基膦氧的动力学拆分(图 28).通过调节叠氮22的用量, 改变PyBox配体上吡啶C4位取代基可以高效高对映选择性地获得P-手性单乙炔基膦氧(S)-100和三氮唑膦氧(R)-101.产物(S)-100a经两步反应可手性保持地合成P-手性叔膦102a, 且102a作为亲核叔膦催化剂可较好地催化烯酮和联烯酸酯的不对称环加成反应.
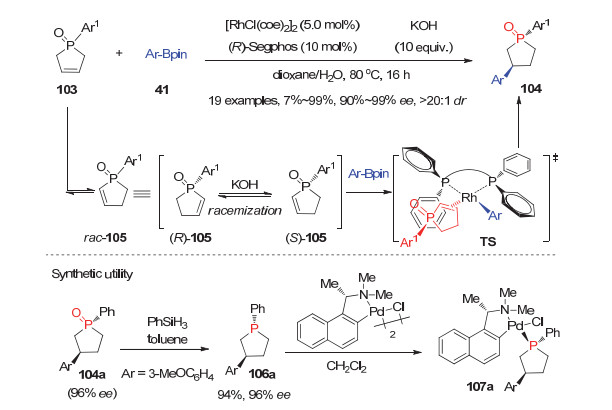
除了动力学拆分策略, 利用三级膦氧化物参与的不对称动态动力学拆分反应来合成P-手性膦氧化物这一策略也取得了初步进展. 2017年, Hayashi等[53]通过巧妙的设计, 利用手性(R)-Segphos/Rh配合物催化的不对称芳基化反应发展了环烯膦氧化物103参与的动态动力学拆分反应, 为P-手性膦杂环戊烷膦氧化物提供了一种高效高选择性的合成方法.
他们通过细致的条件考察发现, 环烯膦氧化物103中的C=C双键在KOH条件下首先会发生异构化生成更稳定的异构体rac-105, 而且它的两个对映异构体(R)-105和(S)-105在KOH条件下通过消旋化可快速转变(图 29).在10 mol%手性(R)-Segphos/Rh催化下, (S)-105表现出较高的反应活性, 优先与芳基硼酸酯发生芳基化反应, 而活性较低的(R)-105则会通过消旋化转化成(S)-105后再发生芳基化反应, 从而高选择性地获得P-手性膦氧产物104.此外, 所得产物104a可被PhSiH3高效地还原为P-手性叔膦106a, 并可进一步与Pd(Ⅱ)配位生成手性膦-钯配合物催化剂107a.
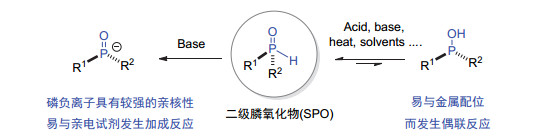
由于磷原子上含有一个氢取代基的特殊结构, 二级膦氧化物不仅在碱性条件下易被攫氢活化形成相应的亲核性的磷负离子(P-), 而且在酸性、碱性、加热或溶剂等条件下都易互变异构成羟基膦[54](图 30).因此, 二级膦氧化物一方面可与亚胺[55]、烯胺[56]、酮[57]、邻羟基苯醌中间体[58]等亲电试剂发生加成反应构建三级膦氧化物; 另一方面, 异构化生成的羟基膦容易与金属配合物配位, 随后与不同偶联试剂发生偶联反应, 从而实现三级膦氧化物的合成.基于二级膦氧化物的这些反应特性, 因此利用其参与的不对称催化反应是合成P-手性膦氧化物的另一有效策略.
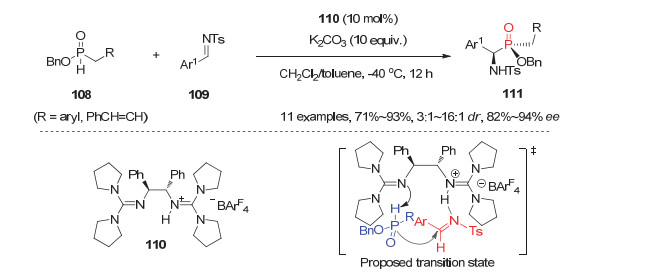
2009年, Tan等[55]利用他们发展的手性胍盐110为催化剂, 发展了二级膦氧化物108与N-Ts芳基醛亚胺109的不对称磷杂Mannich反应, 为α-氨基膦酸酯类P-手性膦氧化物提供了一种高效的合成方法(图 31).使用10 mol%的手性二胺骨架的胍盐110为Lewis碱催化剂, 该反应能以71%~93%的产率, 3:1~16:1的dr值和82%~94%的ee值获得同时含C-和P-手性的α-氨基膦酸酯111.在该反应中, 手性胍盐110是一类Brønsted酸和Brønsted碱双功能催化剂, 其中一个游离胍基作为Brønsted碱对二级膦氧108进行攫氢活化, 同时亚胺109被另一个具有Brønsted酸性质的质子化的胍基团活化, 如图 31中过渡态所示.值得一提的是, 这是首例二级膦氧化物参与的不对称催化反应构建P-手性膦氧化物的报道.
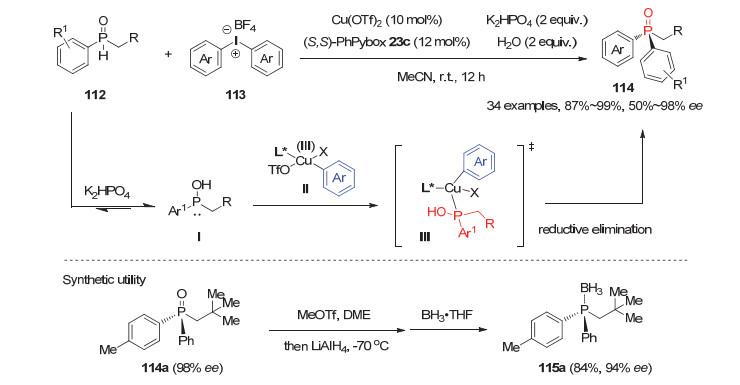
除利用手性有机催化剂对二级膦氧进行攫氢活化来对映选择性制备P-手性膦氧化物以外, 研究人员还通过其异构化形成羟基膦后与金属配位的特性发展了几例新颖的手性金属配合物催化的不对称芳基化和烯丙基化反应. 2016年, Gaunt等[59]报道了首例手性铜催化的芳基烷基二级膦氧112与芳基高价碘盐113的不对称芳基化反应.在Cu(OTf)2和(S, S)-PhPybox形成的手性铜配合物催化下, 该反应能以高达99%的产率和98%的对映选择性获得一系列二芳基取代P-手性膦氧化物114.如图 32所示, 二级膦氧112首先在碱的作用下异构化, 生成的羟基膦Ⅰ与反应体系形成的活性亲电试剂芳基- Cu(Ⅲ)物种Ⅱ进行加成反应, 经历如图中Ⅲ所示的过渡态模型, 最后还原消除得到P-手性膦氧产物114.此外, 产物114a在MeOTf与LiAlH4的还原体系下, 可一步还原为P-手性叔膦化物115a.
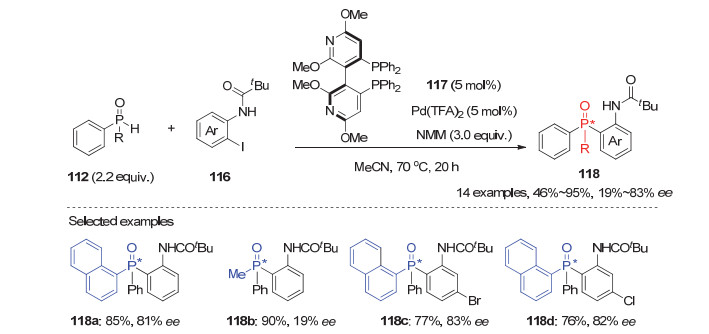
同年, 利用动力学拆分策略, 蔡倩等[60]发展了手性双膦配体/钯配合物催化的双芳基二级膦氧化物112与邻位酰胺取代的芳基碘化物116的不对称芳基化反应(图 33).经过系列优化, 该反应能以46%~95%的产率和19%~83%的ee值获得目标P-手性三芳基膦氧化物118.芳基碘化物上邻位的酰胺及其取代基对反应的活性和对映选择性均有很大影响, 最终发现当其邻位是大位阻的特戊酰胺基时, 反应能取得最优结果.虽然该反应的对映选择性不够理想, 但为合成开链P-手性三芳基膦氧化物提供了一种新的方法.
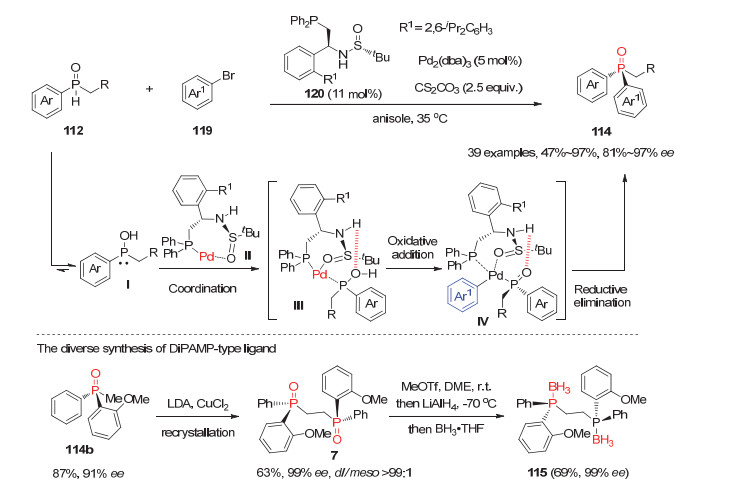
最近, 张俊良等[61]利用他们发展的叔丁基亚磺酰胺-膦配体Xiao-Phos(120)与Pd2(dba)3形成的手性钯配合物为催化剂, 成功实现了芳基烷基二级膦氧112与芳基溴化物119的不对称交叉偶联反应, 以良好到优秀的产率和ee值获得一系列P-手性二芳基烷基膦氧化物114(图 34).对照实验表明手性亚磺酰胺配体120上的N—H键和磺酰胺基团对反应活性和对映选择性至关重要.结合理论计算, 作者初步认为配体的亚磺酰胺的氧原子与叔膦基团同时与金属钯物种配位, 形成的手性Pd(0)物种Ⅱ可能优先与二级膦氧异构化生成的羟基膦Ⅰ进行螯合.产生的手性物种Ⅲ再与芳基溴化物119发生氧化加成形成关键反应中间体Ⅳ, 最后经还原消除得产物114.尤为值得一提的是, 该方法所合成的P-手性二芳基烷基膦氧化物通过两步转化即可方便地制备P-手性双膦配体DiPAMP及其系列衍生物.
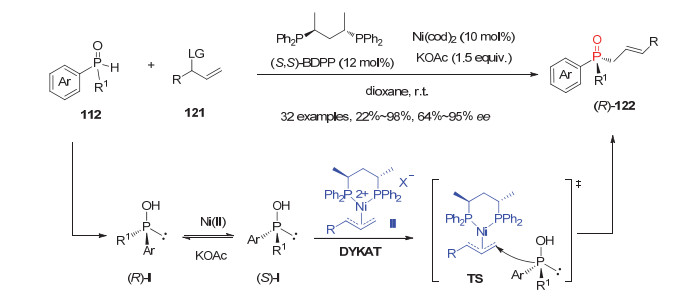
此外, 利用动态动力学不对称转化(DYKAT)策略, 张清伟等[62]发展了首例镍催化的二级膦氧化物112的不对称烯丙基化反应.使用10 mol%的(S, S)-BDPP/ Ni(cod)2配合物为催化剂, 一系列消旋二级膦氧112均能与乙酸烯丙基酯121发生反应, 以22%~98%的产率和64%~95%的ee值获得相应的烯丙基P-手性膦氧产物122(图 35).机理研究表明在KOAc作用下, 镍催化的二级膦氧的消旋化是动态动力学不对称转化的起源.其中(S)-Ⅰ在手性镍催化下会更容易与反应体系形成的关键手性膦配体-Ni-烯丙基物种Ⅱ发生亲核加成反应, 而(R)-Ⅰ与手性镍烯丙基物种Ⅱ反应较慢, 则会优先在Ni(Ⅱ)和KOAc作用下异构化成(S)构型再进行加成反应, 从而高对映选择性地生成相应的P-手性膦氧化物(R)-122.该反应不仅为烯丙基取代的P-手性膦氧化物提供了一种有效合成方法, 而且所得产物还可对映选择性保持地还原为P-手性叔膦化物.
鉴于P-手性膦氧化物在药物研究、不对称有机合成以及有机光电材料等领域中的重要应用前景, 化学家们发展了潜手性三级膦氧化物参与的去对称化反应, 三级膦氧化物参与的(动态)动力学拆分, 以及二级膦氧化物参与的不对称反应这三种不对称催化合成策略(图 3).基于上述策略, 近年来实现了一系列手性金属和有机催化的不对称新反应, 成功合成了多种开链或环状P-手性膦酸酯、膦酰胺、膦氧等化合物.这些方法不仅丰富了P-手性膦氧化物的不对称催化合成途径, 更有助于发展手性膦(氧)配体和有机膦(氧)催化剂.
尽管不对称催化构建P-手性膦氧化物的方法在近年来得到了快速发展, 但这一领域的研究仍然存在较大的局限性.首先, 绝大多数已有方法主要集中于利用潜手性三级膦氧化物的不对称去对称化反应策略, 且底物类型大部分局限于双芳基或双炔基取代的潜手性三级膦氧化物; 其次, 虽然手性有机催化和金属催化均展示了它们的催化潜能, 但仔细分析不难发现贵金属铑和钯催化在不对称构建P-手性膦氧化物中仍然起主导作用; 此外, 利用二级膦氧化物参与的催化不对称反应构筑P-手性膦氧的报道还非常有限(仅有5例).另一方面, 虽然部分实例通过选择合适的条件可将P-手性膦氧产物还原为叔膦催化剂, 但所得叔膦目前在催化一些不对称反应时并不能取得非常理想的对映选择性, 且目前还很少对P-手性膦氧类化合物在药物研究以及有机光电材料等领域的应用进行探索.因此, 发展多样性和高立体选择性构建P-手性膦氧化物的合成方法, 进一步拓展反应的类型、反应底物结构的多样性, 尝试手性有机催化或廉价金属催化剂, 以减少贵金属使用和产物金属残留等问题, 仍是今后该领域一项非常具有价值和意义的研究工作.同时, 更多地探索这些新方法合成的结构多样新颖的P-手性膦氧化物在有机合成催化剂、光电材料、药物研发等领域中的应用潜能也是需要不断努力的方向.随着不对称催化新策略和新催化剂等的不断发展, 相信经过研究者的不断努力, 不对称催化构建P-手性膦氧化物的研究及其应用在不久的将来必将迎来更具实质性的进展, 也将在有机合成、药物化学、生命科学、材料科学等相关领域创造出更广阔的应用前景.
(a) Dutartre, M.; Bayardon, J.; Jugé, S. Chem. Soc. Rev. 2016, 45, 5771. (b) Macia, E. Chem. Soc. Rev. 2005, 34, 691.
(a) Kazemi, M.; Tahmasbi, A. M.; Valizadeh, R.; Naserian, A. A.; Soni, A. Agric. Sci. Res. J. 2012, 2, 512. (b) Lamberth, C. Tetrahedron 2010, 66, 7239.
De Clercq, E. Clin. Microbiol. Rev. 2003, 16, 569. doi: 10.1128/CMR.16.4.569-596.2003
(a) Akiyama, T. Chem. Rev. 2007, 107, 5744. (b) Milo, A.; Neel, A. J.; Toste, F. D.; Sigman, M. S. Science 2015, 347, 737.
Duffy, M. P.; Delaunay, W.; Bouit, P.-A.; Hissler, M. Chem. Soc. Rev. 2016, 45, 5296. doi: 10.1039/C6CS00257A
Ohmaru, Y.; Sato, N.; Mizutani, M.; Kotani, S.; Sugiura, M.; Nakajima, M. Org. Biomol. Chem. 2012, 10, 4562. doi: 10.1039/c2ob25338k
Takaya, H.; Mashima, K.; Koyano, K.; Yagi, M.; Kumobayashi, H.; Taketomi, T.; Akutagawa, S.; Noyori, R. J. Org. Chem. 1986, 51, 629.
Xu, B.; Zhu, S.-F.; Xie, X.-L, Shen, J.-J.; Zhou, Q.-L. Angew. Chem., Int. Ed. 2011, 50, 11483. doi: 10.1002/anie.201105485
Pye, P. J.; Rossen, K.; Reamer, R. A.; Tsou, N. N.; Volante, R. P.; Reider, P. J. J. Am. Chem. Soc. 1997, 119, 6207. doi: 10.1021/ja970654g
Schulze, C. J.; Navarro, G.; Ebert, D.; DeRisi, J.; Linington, R. G. J. Org. Chem. 2015, 80, 1312. doi: 10.1021/jo5024409
Cholongitas, E.; Papatheodoridis, G. V. Ann. Gastroenterol. 2014, 27, 331.
Clarion, L.; Jacquard, C.; Sainte-Catherine, O.; Loiseau, S.; Filippini, D.; Hirlemann, M.-H.; Volle, J.-N.; Virieux, D.; Lecouvrey, M.; Pirat, J.-L.; Bakalara, N. J. Med. Chem. 2012, 55, 2196. doi: 10.1021/jm201428a
(a) Baraniak, J.; Kinas, R. W.; Lesiak, K.; Stec, W. J. J. Chem. Soc. 1979, 940. (b) Dostmann, W. R. G.; Taylor, S. S.; Genieser, H.-G.; Jastorff, B.; Døskeland, S. O.; Øgreid, D. J. Biol. Chem. 1990, 265, 10484.
Matsukawa, M.; Sugama, H.; Imamoto, T. Tetrahedron Lett. 2000, 41, 6461. doi: 10.1016/S0040-4039(00)01030-3
(a) Iseki, K.; Kuroki, Y.; Takahashi, M.; Kobayashi, Y. Tetrahedron Lett. 1996, 37, 5149. (b) Iseki, K.; Kuroki, Y.; Takahashi, M.; Kishimoto, S. Tetrahedron 1997, 53, 3513.
Xu, G.; Senanayake, C. H.; Tang, W. Acc. Chem. Res. 2019, 52, 1101. doi: 10.1021/acs.accounts.9b00029
Selected examples using P-chiral phosphines as ligands: (a) Vineyard, B. D.; Knowles, W. S.; Sabacky, M. J.; Bachman, G. L.; Weinkauff, D. J. J. Am. Chem. Soc. 1977, 99, 5946. (b) Gridnev, I. D.; Higashi, N.; Asakura, K.; Imamoto, T. J. Am. Chem. Soc. 2000, 122, 7183. (c) Tang, W.; Zhang, X. Angew. Chem., Int. Ed. 2002, 41, 1612. (d) Taylor, A. M.; Altman, R. A.; Buchwald S. L. J. Am. Chem. Soc. 2009, 131, 9900. (e) Imamoto, T.; Tamura, K.; Zhang, Z.; Horiuchi, Y.; Sugiya, M.; Yoshida, K.; Yanagisawa, A.; Gridnev, I. D. J. Am. Chem. Soc. 2012, 134, 1754. (f) Liu, G.; Liu, X.; Cai, Z.; Jiao, G.; Xu, G.; Tang, W. Angew. Chem., Int. Ed. 2013, 52, 4235. Selected examples using P-chiral phosphine as organocatalysts: (g) Sampath, M.; Loh, T.-P. Chem. Sci. 2010, 1, 739. (h) Rémond, E.; Bayardon, J.; Takizawa, S.; Rousselin, Y.; Sasai; H.; Jugé, S. Org. Lett. 2013, 15, 1870. (i) Takizawa, S.; Rémond, E.; Arteaga, F.; Yoshida, Y.; Sridharan, V.; Bayardon, J.; Jugé, S.; Sasai, H. Chem. Commun. 2013, 49, 8392. (j) Henry, C. E.; Xu, Q.-H.; Fan, Y.-C.; Martin, T. J.; Belding, L.; Dudding, T.; Kwon, O. J. Am. Chem. Soc. 2014, 136, 11890.
(a) Liu, S.; Li, Y.-M.; Wang, D.; Wei, R.; Miao, Z.-W. Chin. J. Org. Chem. 2018, 38, 341 (in Chinese). (刘双, 李玉明, 王典, 魏榕, 苗志伟, 有机化学, 2018, 38, 341.) (b) Cui, Y.-M.; Lin, Y.; Xu. L.-W. Coord. Chem. Rev. 2017, 330, 37. (c) Li, Z.; Duan, W.-L. Chin. J. Org. Chem. 2016, 36, 1805 (in Chinese). (李振, 段伟良, 有机化学, 2016, 36, 1805.) (d) Harvey, J. S.; Gouverneur, V. Chem. Commun. 2010, 46, 7477. (e) Glueck, D. S. Chem. Eur. J. 2008, 14, 7108.
Selected examples for chiral resolution: see ref. 17a, and (a) Korpiun, O.; Lewis, R. A.; Chickos, J.; Mislow, K. J. Am. Chem. Soc. 1968, 90 4842. For chiral auxiliaries: (b) Berger, O.; Montchamp, J.-L. Angew. Chem., Int. Ed. 2013, 52, 11377. (c) Han, Z. S.; Goyal, N.; Herbage, M. A.; Sieber, J. D.; Qu, B.; Xu, Y.; Li, Z.; Reeves, J. T.; Desrosiers, J.-N.; Ma, S.; Grinberg, N.; Lee, H.; Mangunuru, H. P. R.; Zhang, Y.; Krishnamurthy, D.; Lu, B. Z.; Song, J. J.; Wang, G.; Senanayake, C. H. J. Am. Chem. Soc. 2013, 135, 2474. (d) Gwon, D.; Lee, D.; Kim, J.; Park, S.; Chang, S. Chem. Eur. J. 2014, 20, 12421. For asymmetric oxidation of tertiary phosphines: (e) Bergin, E.; O'Connor, C. T.; Robinson, S. B.; McGarrigle, E. M.; O'Mahony, C. P.; Gilheany, D. G. J. Am. Chem. Soc. 2007, 129 9566. (f) Rajendran, K. V.; Kennedy, L.; Gilheany, D. G. Eur. J. Org. Chem. 2010, 5642. (g) Nikitin, K.; Rajendran, K. V.; Müller-Bunz, H.; Gilheany, D. G. Angew. Chem., Int. Ed. 2014, 53, 1906.
(a) Zeng, X.-P.; Cao, Z.-Y.; Wang, Y.-H.; Zhou, F.; Zhou, J. Chem. Rev. 2016, 116, 7330. (b) Petersen, K. S. Tetrahedron Lett. 2015, 56, 6523. (c) Willis, M. C. J. Chem. Soc., Perkin Trans. 1 1999, 1765.
Nishida, G.; Noguchi, K.; Hirano, M.; Tanaka, K. Angew. Chem., Int. Ed. 2008, 47, 3410. doi: 10.1002/anie.200800144
Zheng, Y.; Guo, L.; Zi, W. Org. Lett. 2018, 20, 7039. doi: 10.1021/acs.orglett.8b02982
Zhang, Y.; Zhang, F.; Chen, L.; Xu, J.; Liu, X.; Feng, X. ACS Catal. 2019, 9, 4834. doi: 10.1021/acscatal.9b00860
Zhu, R. Y.; Chen, L.; Hu, X. S.; Zhou, F.; Zhou, J. Chem. Sci. 2020, 11, 97. doi: 10.1039/C9SC04938J
(a) Meng, J.-C.; Fokin, V. V.; Finn, M. G. Tetrahedron Lett. 2005, 46, 4543. (b) Stephenson, G. R.; Buttress, J. P.; Deschamps, D.; Lancelot, M.; Martin, J. P.; Sheldon, A. I. G.; Alayrac, C.; Gaumont, A.-C.; Page, P. C. B. Synlett 2013, 24, 2723. (c) Song, T.; Li, L.; Zhou, W.; Zheng, Z.-J.; Deng, Y.; Xu, Z.; Xu, L.-W. Chem. Eur. J. 2015, 21, 554. (d) Chen, M.-Y.; Song, T.; Zheng, Z.-J.; Xu, Z.; Cui, Y.-M.; Xu, L.-W. RSC Adv. 2016, 6, 58698. (e) Chen, M.-Y.; Xu, Z.; Chen, L.; Song, T.; Zheng, Z.-J.; Cao, J.; Cui, Y.-M., Xu, L.-W. ChemCatChem 2018, 10, 280. For achiral version: (f) Rodionov, V. O.; Fokin, V. V.; Finn, M. G. Angew. Chem., Int. Ed. 2005, 44, 2210.
(a) Worrell, B. T.; Malik, J. A.; Fokin, V. V. Science 2013, 340, 457. (b) Díez, J.; Gamasa, M. P.; Panera, M. Inorg. Chem. 2006, 45, 10043.
Zhou, F.; Tan, C.; Tang, J.; Zhang, Y.-Y.; Gao, W.-M.; Wu, H.-H.; Yu, Y.-H.; Zhou, J. J. Am. Chem. Soc. 2013, 135 10994. doi: 10.1021/ja4066656
(a) Osako, T.; Uozumi, Y. Org. Lett. 2014, 16, 5866. (b) Osako, T.; Uozumi, Y. Synlett 2015, 26, 1475.
For reviews: (a) Ren, Y.; Baumgartner, T. Dalton Trans. 2012, 41, 7792. (b) Matano, Y.; Imahori, H. Org. Biomol. Chem. 2009, 7, 1258. For recent examples: (c) Stolar, M.; Borau-Garcia, J.; Toonen, M.; Baumgartner, T. J. Am. Chem. Soc. 2015, 137, 3366. (d) Yamaguchi, E.; Wang, C.; Fukazawa, A.; Taki, M.; Sato, Y.; Sasaki, T.; Ueda, M.; Sasaki, N.; Higashiyama, T.; Yamaguchi, S. Angew. Chem., Int. Ed. 2015, 54, 4539. (e) Reus, C.; Stolar, M.; Vanderkley, J.; Nebauer, J.; Baumgartner, T. J. Am. Chem. Soc. 2015, 137, 11710.
Tahara, Y.-K.; Sato, T.; Matsubara, R.; Kanyiva, K. S.; Shibata, T. Heterocycles 2016, 93, 685. doi: 10.3987/COM-15-S(T)57
Harvey, J. S.; Malcolmson, S. J.; Dunne, K. S.; Meek, S. J.; Thompson, A. L.; Schrock, R. R.; Hoveyda, A. H.; Gouverneur, V. Angew. Chem., Int. Ed. 2009, 48, 762. doi: 10.1002/anie.200805066
Wang, Z.; Hayashi. T. Angew. Chem., Int. Ed. 2018, 57, 1702. doi: 10.1002/anie.201712572
For selected reviews on C-H bond functionalization, see: (a) Kakiuchi, F.; Murai, S. Acc. Chem. Res. 2002, 35, 826. (b) Giri, R.; Shi, B.-F.; Engle, K. M.; Maugel, N.; Yu, J.-Q. Chem. Soc. Rev. 2009, 38, 3242. (c) Xu, L.-M.; Li, B.-J.; Yang, Z.; Shi, Z.-J. Chem. Soc. Rev. 2010, 39, 712. (d) Albrecht, M. Chem. Rev. 2010, 110, 576. (e) Song, G.; Wang, F.; Li, X. Chem. Soc. Rev. 2012, 41, 3651. (f) Newton, C. G.; Wang, S.-G.; Oliveira, C. C.; Cramer, N. Chem. Rev. 2017, 117, 8908.
Du, Z.-J.; Guan, J.; Wu, G.-J.; Xu, P.; Gao, L.-X.; Han, F.-S. J. Am. Chem. Soc. 2015, 137, 632. doi: 10.1021/ja512029x
Guan, J.; Wu, G.-J.; Han, F.-S. Chem. Eur. J. 2014, 20, 3301. doi: 10.1002/chem.201303056
(a) Shi, B.-F.; Maugel, N.; Zhang, Y.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2008, 47, 4882. (b) Shi, B.-F.; Zhang, Y.-H.; Lam, J.-K.; Wang, D.-H.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 460. (c) Yang, Y.-F.; Hong, X.; Yu, J.-Q.; Houk, K. N. Acc. Chem. Res. 2017, 50, 2853.
Sun, Y.; Cramer, N. Angew. Chem., Int. Ed. 2017, 56, 364. doi: 10.1002/anie.201606637
Gwon, D.; Park, S.; Chang, S. Tetrahedron 2015, 71, 4504. doi: 10.1016/j.tet.2015.02.065
Jang, Y.-S.; Dieckmann, M.; Cramer, N. Angew. Chem., Int. Ed. 2017, 56, 15088.
Jang, Y.-S.; Woźniak, Ł.; Pedroni, J.; Cramer, N. Angew. Chem., Int. Ed. 2018, 57, 12901. doi: 10.1002/anie.201807749
Lin, Z.-Q.; Wang, W.-Z.; Yan, S.-B.; Duan, W.-L. Angew. Chem., Int. Ed. 2015, 54, 6265. doi: 10.1002/anie.201500201
Liu, L.; Zhang, A.-A.; Wang, Y.; Zhang, F.; Zuo, Z.; Zhao, W.-X.; Feng, C.-L.; Ma, W. Org. Lett. 2015, 17, 2046. doi: 10.1021/acs.orglett.5b00122
Lin, Y.; Ma, W.-Y.; Sun, Q.-Y.; Cui, Y.-M.; Xu, L.-W. Synlett 2017, 28, 1432. doi: 10.1055/s-0036-1588983
Li, Z.; Lin, Z.-Q.; Yan, C.-G.; Duan, W.-L. Organometallics 2019, 38, 3916. doi: 10.1021/acs.organomet.9b00216
Xu, G. Q.; Li, M. H.; Wang, S. L.; Tang, W. J. Org. Chem. Front. 2015, 2, 1342. doi: 10.1039/C5QO00142K
Huang, Z.; Huang, X.; Li, B.; Mou, C.; Yang, S.; Song, B.-A.; Chi, Y. R. J. Am. Chem. Soc. 2016, 138, 7524. doi: 10.1021/jacs.6b04624
Yang, G.-H.; Li, Y.; Li, X.; Cheng, J.-P. Chem. Sci. 2019, 10, 4322.
Toda, Y.; Pink, M.; Johnston, J. N. J. Am. Chem. Soc. 2014, 136, 14734. doi: 10.1021/ja5088584
Dobish, M. C.; Johnston, J. N. J. Am. Chem. Soc. 2011, 134, 6068.
Trost, B. M.; Spohr, S. M.; Rolka, A. B.; Kalnmals, C. A. J. Am. Chem. Soc. 2019, 141, 14098. doi: 10.1021/jacs.9b07340
Liu, S.; Zhang, Z. F.; Xie, F.; Butt, N. A.; Sun, L.; Zhang, W. B. Tetrahedron Asymmetry 2012, 23, 329. doi: 10.1016/j.tetasy.2012.02.018
Sun, Y.; Cramer, N. Chem. Sci. 2018, 9, 2981. doi: 10.1039/C7SC05411D
Lim, K. M.-H.; Hayashi, T. J. Am. Chem. Soc. 2017, 139, 8122. doi: 10.1021/jacs.7b04570
Emmick, T. L.; Letsinger, R. L. J. Am. Chem. Soc. 1968, 90, 3459. doi: 10.1021/ja01015a030
Fu, X.; Loh, W.-T.; Zhang, Y.; Chen, T.; Ma, T.; Liu, H.; Wang, J.; Tan, C.-H. Angew. Chem., Int. Ed. 2009, 48, 7387. doi: 10.1002/anie.200903971
Xie, P. Z.; Guo, L.; Xu, L. L.; Loh, T.-P. Chem. Asian J. 2016, 11, 1353. doi: 10.1002/asia.201600108
(a) Zhang, H.; Sun, Y.-M.; Yao, L.; Ji, S.-Y.; Zhao, C.-Q.; Han, L.-B. Chem. Asian J. 2014, 9, 1329. (b) Wang, J.-P.; Nie, S.-Z.; Zhou, Z.-Y.; Ye, J.-J.; Wen, J.-H.; Zhao, C.-Q. J. Org. Chem. 2016, 81, 7644.
Du, J.-Y.; Ma, Y.-H.; Yuan, R.-Q.; Xin, N. N.; Nie, S.-Z.; Ma, C.-L.; Li, C.-Z.; Zhao, C.-Q. Org. Lett. 2018, 20, 477. doi: 10.1021/acs.orglett.7b03863
Beaud, R.; Phipps, R. J.; Gaunt, M. J. J. Am. Chem. Soc. 2016, 138, 13183. doi: 10.1021/jacs.6b09334
Zhang, Y.; He, H.; Wang, Q. Y.; Cai, Q. Tetrahedron Lett. 2016, 57, 5308. doi: 10.1016/j.tetlet.2016.10.048
Dai, Q.; Li, W.-B.; Li, Z.-M.; Zhang, J.-L. J. Am. Chem. Soc. 2019, 141, 20556. doi: 10.1021/jacs.9b11938
Liu, X.-T.; Zhang, Y.-Q.; Han, X.-Y.; Sun, S.-P.; Zhang, Q.-W. J. Am. Chem. Soc. 2019, 141, 16584. doi: 10.1021/jacs.9b08734

图 2 含P-手性膦氧结构的天然产物、药物、生物活性分子及相关的手性催化剂
Figure 2 Selected natural products, drugs, bioactive molecules and catalysts featuring P-chiral phosphine oxides

图 4 铑(Ⅰ)催化的潜手性双炔基膦氧13参与的[2+2+2]环加成反应
Figure 4 Rh(Ⅰ)-Catalyzed enantioselective desymmetrizative [2+2+2] cycloaddition of prochiral dialkynylphosphine oxides 13

图 5 金(Ⅰ)催化的潜手性双炔基膦氧化物17的分子内氢醚化反应
Figure 5 Au(Ⅰ)-Catalyzed intramolecular hydroetherification of prochiral dialkynylphosphine oxides 17

图 6 铥(Ⅲ)催化的膦氧双炔13与硫醇的不对称共轭加成反应
Figure 6 Tm(Ⅲ)-Catalyzed asymmetric conjugate addition of dialkynylphosphine oxides 13 with thiols

图 7 潜手性乙炔基膦氧化物21参与的不对称去对称化CuAAC反应
Figure 7 Asymmetric desymmetrization of prochiral diacetylenyl phosphine oxides 21 via CuAAC reaction

图 8 潜手性乙炔基膦氧化物26参与的不对称去对称化CuAAC反应
Figure 8 Eantioselective desymmetrization of prochiral diacetylenyl phosphine oxides 26 via CuAAC reaction

图 9 铑(Ⅰ)催化的潜手性双炔基膦氧29参与的[2+2+2]环加成反应
Figure 9 Rh(Ⅰ)-catalyzed desymmetrizative [2+2+2] cycloaddition of prochiral dialkynylphosphine oxides 29

图 10 钼(Mo)催化的潜手性双烯32参与的去对称化环化反应
Figure 10 Mo-Catalyzed desymmetrization of dialkenylphosphine oxides 32 via RCM reaction

图 11 铑(Ⅰ)催化的潜手性双乙烯基膦氧35与芳基硼试剂的氢芳基化
Figure 11 Rh(Ⅰ)-Catalyzed enantioposition-selective hydroarylation of divinylphosphine oxides 35 with aryl boroxines

图 12 钯(Ⅱ)催化的潜手性膦酰胺40参与的去对称化C—H键芳基化
Figure 12 Pd(Ⅱ)-Catalyzed desymmetric C—H arylation of prochiral diarylphosphinamides 40

图 13 铑(Ⅲ)催化的潜手性双芳基40参与的去对称化C—H键环化反应
Figure 13 Rh(Ⅲ)-catalyzed desymmetrization of prochiral dialkenyl phosphine oxides 40 via C—H cyclization

图 14 铱(Ⅲ)催化的潜手性芳基膦氧47与TsN3的C—H酰胺化反应
Figure 14 Ir(Ⅲ)-Catalyzed enantioselective C—H amidation of prochiral diarylphosphine oxides 47 with TsN3

图 15 铱催化的双芳基膦氧51和叠氮的去对称化C—H键酰胺化反应
Figure 15 Cpx-Ir(Ⅲ) catalyzed asymmetric C—H amidation of prochiral diarylphosphine oxides 51 with azides 48

图 16 铱(Ⅲ)催化的芳基膦氧51与重氮53的不对称C—H键芳基化
Figure 16 Cpx-Ir(Ⅲ) catalyzed asymmetric C—H arylation of prochiral diarylphosphine oxides 51 with diazo compound 53

图 17 钯(Ⅱ)催化的潜手性双芳基膦酰胺57的分子内去对称化C—H键芳基化
Figure 17 Pd(Ⅱ)-Catalyzed intramolecular desymmetric C—H arylation of prochiral diarylphosphinamides 57

图 18 钯(Ⅱ)催化的潜手性芳基膦氧60参与的不对称C—H键芳基化
Figure 18 Pd(Ⅱ)-Catalyzed asymmetric C—H arylation of prochiral diarylphosphine oxides 60

图 19 钯(Ⅱ)催化的潜手性双芳基60参与的去对称化C—H键芳基化
Figure 19 Pd(Ⅱ)-Catalyzed desymmetrization of diarylphosphine oxides 60 via C—H arylation

图 20 钯催化的潜手性膦氧65参与的去对称化邻位C—H键芳基化
Figure 20 Pd(Ⅱ)-Catalyzed desymmetrization of prochiral phosphine oxides 65 via C—H arylation

图 21 NHC催化的潜手性二酚68参与的去对称化酯化反应
Figure 21 NHC-catalyzed desymmetrization of prochiral bisphenols 68 via esterification reaction

图 22 (DHQ) 2PHAL催化的潜手性二酚68参与的去对称化烯丙基烷基化反应
Figure 22 (DHQ)2PHAL-catalyzed desymmetrizative allylic alkylation of prochiral bisphenols 68

图 23 金(Ⅰ)催化的双酚羟基79参与的去对称化环化反应
Figure 23 Au(Ⅰ)-Catalyzed desymmetrizative cyclization of prochiral bisphenols 79

图 24 氨基磷酸83参与的不对称碘环化反应
Figure 24 Asymmetric iodocyclization involving phosphoramidic acids 83

图 25 钯催化的次膦酸88参与的去对称化烯丙基化反应
Figure 25 Pd-Catalyzed asymmetric desymmetrizative allylic alkylation of phosphinic acids 88

图 26 外消旋膦酰氯94的不对称催化动力学拆分反应
Figure 26 Asymmetric kinetic resolutions of phosphoric chloride 94

图 27 外消旋次膦酰胺40的不对称动力学拆分反应
Figure 27 Asymmetric kinetic resolutions of phosphinic amides 40

图 28 外消旋单乙炔基三级膦氧100的不对称动力学拆分反应
Figure 28 Asymmetric kinetic resolutions of phosphine oxides 100

图 29 通过铑催化不对称芳基化实现的环烯膦氧103的动态动力学拆分反应
Figure 29 Dynamic kinetic resolution of phospholene oxides 103 via Rh-catalyzed asymmetric arylation

图 31 二级膦氧化物108与亚胺109的不对称膦杂Mannich反应
Figure 31 Phospha-Mannich reaction between SPO 108 and imines 109

图 32 铜催化的二级膦氧112与芳基高价碘盐的不对称芳基化反应
Figure 32 Cu-Catalyzed arylation of SPO 112 with diaryliodonium salts

图 34 钯催化的二级膦氧112参与的不对称芳基化反应
Figure 34 Pd-Catalyzed asymmetric arylation of SPO with aryl bromide

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:



