School of Chemistry and Materials Science, Key Laboratory of Functional Inorganic Material Chemistry(Ministry of Education), Heilongjiang University, Harbin 150080, China
b.
Department of Mathematics, Heilongjiang Provincial Key Laboratory of Complex Systems Theory and Computation, Heilongjiang University, Harbin 150080, China
Received Date:
29 December 2019 Available Online:
15 April 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21601054, 11871198, 11801116), the University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province of China (No. UNPYSCT-2017126), and the Training Program of Innovation and Entrepreneurship for Undergraduates of Heilongjiang Province (No. 201910212073)
† These authors contributed equally to this work. Supporting information for this article is available free of charge via the Internet at http://sioc-journal.cn
Abstract:
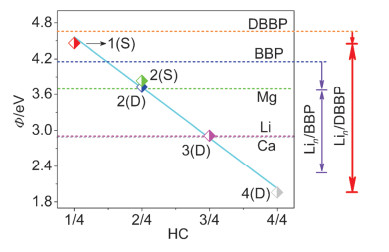
As a new member of the two-dimensional nanomaterial family, borophene is regarded as a potential material platform for nanoscale electronic devices. Especially, borophene-based electrodes have potential application values in light-emitting diodes, organic light-emitting diodes, organic solar cells and field emitters. Therefore, the work function modulation (to an optimal value) of borophene is highly important to maximize the energy conversion efficiency and performance of the device. Based on the first-principles density functional theory, the effects of Li adsorption on the structure, electronic properties and work function of double-layer α-borophene (DBBP) are studied. The calculation results show that Li adsorption can effectively adjust the work function of DBBP from 4.65 eV to 1.96~4.46 eV with different Li contents. This engineering range is superior to what are reported in the literatures for Li-adsorbed monolayer BBP (modified from 4.16 eV to 2.31~3.67 eV), and double-layer graphene with intercalated Li (3.4~3.9 eV) and K (3.3~3.8 eV). The work functions of Li2(D)/DBBP (3.73 eV) and Li3(D)/DBBP (2.91 eV) are close to the commonly used electrode materials Mg and Ca, respectively, while the work function of Li4(D)/DBBP is even lower than Ca. In addition, the factors that affect the work function reduction of Lin/DBBP relative to DBBP, such as configuration, substrate deformation, binding energy, electron transfer, charge rearrangement, electrostatic potential, vacuum and Fermi level, are systematically studied. The results demonstrate that the decrease in the Lin/DBBP work function is mainly due to the change in Fermi level, while the change in vacuum level only plays a minor role. Apart from that, the deformation of the substrate does not have a positive effect on the reduction of the Lin/DBBP work function, but the electron transfer from the adsorbed atoms to the matrix (charge redistribution caused by chemical effects) is the inherent reason for the decrease in the Lin/DBBP work function. This study shows that Li adsorption is a simple and effective method to reduce the work function of DBBP. Due to its metallic character and extremely low work function, Li-adsorbed DBBP nanomaterials can be utilized as cathode materials in electronic devices.
Figure 4.
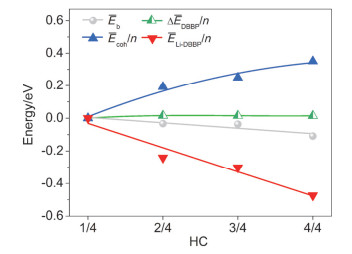
Calculated $ \Delta \bar{E}_{\text {DBBP }} / n, \bar{E}_{\text {coh }} / n, \Delta \bar{E}_{\text {Li-DBBP }} / n$ and average binding energies (Eb) of Lin/DBBP systems as a function of HC
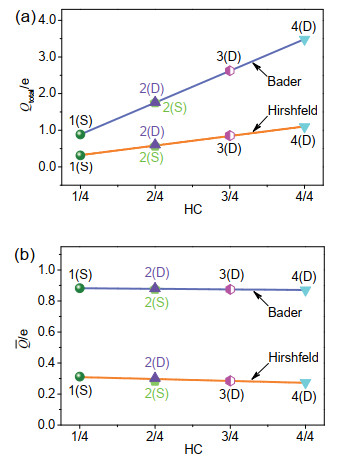
Figure 5.
(a) Total Bader and Hirshfeld charges (Qtotal) of Li adatoms in Lin/DBBP systems; (b) average Bader and Hirshfeld charges (Q) of Li adatoms in Lin/DBBP systems
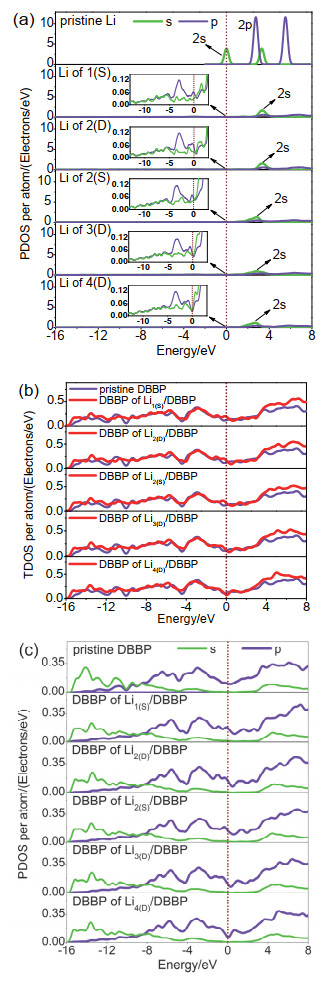
Figure 7.
(a) PDOSs of s and p orbitals of Li atoms before and after adsorption (To be brief, n(S) or n(D) is employed to simplify the system name (Lin(S)/DBBP or Lin(D)/DBBP); (b) TDOSs of B atoms of pristine DBBP and Lin/DBBP, (c) PDOSs of s and p orbitals of DBBP before and after adsorption
Figure 8.
Work functions of Lin/DBBP systems, orange, blue, green, purple and gray horizontal lines represent work functions of DBBP, BBP, Mg, Li and Ca, respectively
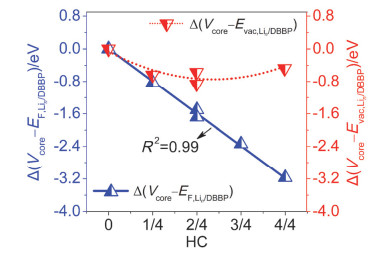
Figure 9.
Relative offsets, i.e., $\Delta\left(V_{\text {core }}-E_{\mathrm{F, Li}_{\mathrm{n}}/ \text { DBBP }}\right) $ and $\Delta\left(V_{\text {core }}-E_{\mathrm{vac, Li}_{\mathrm{n}}/ \text { DBBP }}\right) $, as a function of HC for Lin/DBBP systems. Y-axes of the left and right sides in the two figures are of uniform scale
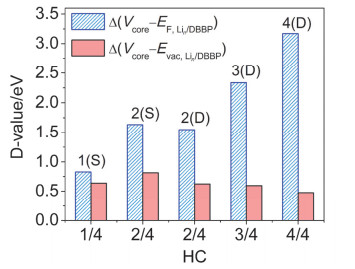
Figure 10.
The contribution of $\Delta\left(V_{\text {core }}-E_{\mathrm{F, Li}_{\mathrm{n}}/ \text { DBBP }}\right) $ and $\Delta\left(V_{\text {core }}-E_{\mathrm{vac, Li}_{\mathrm{n}}/ \text { DBBP }}\right) $ on the work function decrease of Lin/DBBP systems relative to pure DBBP, to be brief, n(S) or n(D) is employed to simplify the system name (Lin(S)/DBBP or Lin(D)/DBBP)
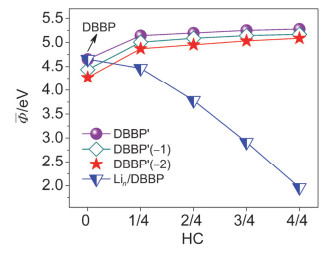
Figure 11.
Average work functions of DBBPʹ, DBBPʹ(−1), DBBPʹ(−2), and Lin/DBBP systems as a function of HC. The average work function is the average value of the work functions of different configurations under the same HC
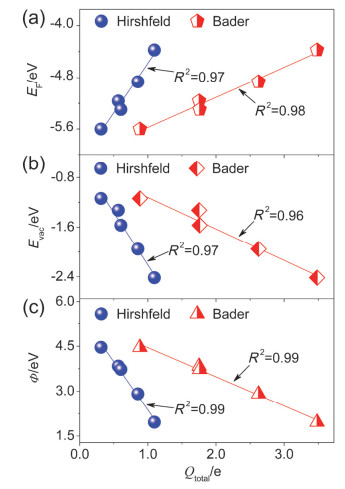
Figure 12.
(a) EF, (b) Evac and (c) work functions of Lin/DBBP systems as a function of total Bader and Hirshfeld charges (Qtotal) of Li adatoms in Lin/DBBP
Mannix, A. J.; Zhou, X. F.; Kiraly, B.; Wood, J. D.; Alducin, D.; Myers, B. D.; Liu, X. L.; Fisher, B. L.; Santiago, U.; Guest, J. R.; Yacaman, M. J.; Ponce, A.; Oganov, A. R.; Hersam, M. C.; Guisinger, N. P. Science2015, 350, 1513. doi: 10.1126/science.aad1080
Kiraly, B.; Liu, X.; Wang, L.; Zhang, Z.; Mannix, A. J.; Fisher, B. L.; Yakobson, B. I.; Hersam, M. C.; Guisinger, N. P. ACS Nano2019, 13, 3816. doi: 10.1021/acsnano.8b09339
Ranjan, P.; Sahu, T. K.; Bhushan, R.; Yamijala, S. S. R. K. C.; Late, D. J.; Kumar, P.; Vinu, A. Adv. Mater. 2019, 31, 1900353. doi: 10.1002/adma.201900353
Zhang, Z.; Yang, Y.; Gao, G.; Yakobson, B. I. Angew. Chem. Int. Ed. 2015, 54, 13022. doi: 10.1002/anie.201505425
[24]
Zhang, Z.; Mannix, A. J.; Hu, Z.; Kiraly, B.; Guisinger, N. P.; Hersam, M. C.; Yakobson, B. I. Nano Lett. 2016, 16, 6622. doi: 10.1021/acs.nanolett.6b03349
[25]
Penev, E. S.; Bhowmick, S.; Sadrzadeh, A.; Yakobson, B. I. Nano Lett. 2012, 12, 2441. doi: 10.1021/nl3004754
Figure 4
Calculated $ \Delta \bar{E}_{\text {DBBP }} / n, \bar{E}_{\text {coh }} / n, \Delta \bar{E}_{\text {Li-DBBP }} / n$ and average binding energies (Eb) of Lin/DBBP systems as a function of HC
Figure 5
(a) Total Bader and Hirshfeld charges (Qtotal) of Li adatoms in Lin/DBBP systems; (b) average Bader and Hirshfeld charges (Q) of Li adatoms in Lin/DBBP systems
Figure 7
(a) PDOSs of s and p orbitals of Li atoms before and after adsorption (To be brief, n(S) or n(D) is employed to simplify the system name (Lin(S)/DBBP or Lin(D)/DBBP); (b) TDOSs of B atoms of pristine DBBP and Lin/DBBP, (c) PDOSs of s and p orbitals of DBBP before and after adsorption
Figure 8
Work functions of Lin/DBBP systems, orange, blue, green, purple and gray horizontal lines represent work functions of DBBP, BBP, Mg, Li and Ca, respectively
Figure 9
Relative offsets, i.e., $\Delta\left(V_{\text {core }}-E_{\mathrm{F, Li}_{\mathrm{n}}/ \text { DBBP }}\right) $ and $\Delta\left(V_{\text {core }}-E_{\mathrm{vac, Li}_{\mathrm{n}}/ \text { DBBP }}\right) $, as a function of HC for Lin/DBBP systems. Y-axes of the left and right sides in the two figures are of uniform scale
Figure 10
The contribution of $\Delta\left(V_{\text {core }}-E_{\mathrm{F, Li}_{\mathrm{n}}/ \text { DBBP }}\right) $ and $\Delta\left(V_{\text {core }}-E_{\mathrm{vac, Li}_{\mathrm{n}}/ \text { DBBP }}\right) $ on the work function decrease of Lin/DBBP systems relative to pure DBBP, to be brief, n(S) or n(D) is employed to simplify the system name (Lin(S)/DBBP or Lin(D)/DBBP)
Figure 11
Average work functions of DBBPʹ, DBBPʹ(−1), DBBPʹ(−2), and Lin/DBBP systems as a function of HC. The average work function is the average value of the work functions of different configurations under the same HC
Figure 12
(a) EF, (b) Evac and (c) work functions of Lin/DBBP systems as a function of total Bader and Hirshfeld charges (Qtotal) of Li adatoms in Lin/DBBP

 下载:
下载:

 下载:
下载:













