Shanghai Institute of Applied Physics, Division of Physical Biology, Shanghai Synchrotron Radiation Facility Bioimaging Center, CAS Key Laboratory of Interfacial Physics and Technology, Shanghai 201800
b.
Zhangjiang Laboratory, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210
Received Date:
12 December 2019 Available Online:
15 February 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 11705270, 11675251, 21390414), the Shanghai Sailing Program (No. 17YF1423600), the China Postdoctoral Science Foundation (Nos. 2018M632189, 2018M640340) and the Youth Innovation Promotion Association of CAS (Nos. 2012205, 2016236)
†These authors contributed equally to this work
Abstract:
Gold nanoparticles (Au NPs), smaller than 10 nm, have a high ratio of surface area to volume, and therefore have excellent catalytic activity. They are widely used in the field of catalysis. The concentration of small particle sized Au NPs synthesized by traditional wet chemical method is too low, and further enrichment is needed in order to meet the experimental requirements. However, small particle sized Au NPs are prone to aggregate during the concentration process and lose the catalytic activity. It is a challenge to concentrate the small Au NPs while keeping their catalytic activities. In this work, 500 nm silanized SiO2 particles which are covered by positive charges were used to adsorb 5 nm Au NPs through electrostatic interaction, and self-assemble to form Au NPs@SiO2 composite at room temperature. The loaded efficiency of Au NPs can reach 99.5% and the amount of Au NPs particles loaded on each SiO2 particle reached 800~1000, which greatly increased the effective concentration of Au NPs in the local area. Moreover, Au NPs enriched on the surface of SiO2 were bound by electrostatic action and uniformly distributed on the surface of SiO2 without agglomeration. The results showed that the catalytic activity of AuNPs@SiO2 was greatly enhanced by increasing the local concentration of AuNPs, and the catalytic activity was 3 times higher than that of AuNPs at the same concentration. After 5 times of reuse, the catalytic conversion efficiency remained at about 80%. The Au NPs@SiO2 composite could be preserved for one month with the same structure and catalytic activity. Moreover, by adjusting the molar ratio of SiO2 and Au NPs, the assembly density of Au NPs at SiO2 can be precisely regulated, and the catalytic activity of Au NPs@SiO2 can also be changed precisely. This work provides a simple method for preparing small sized Au NPs with high concentration and greatly improves the catalytic activity of Au NPs. The method has wide application in enriching other small sized nanoparticles.
本研究中, 500 nm硅烷化SiO2通过静电相互作用吸附5 nm Au NPs, 自组装形成Au NPs@SiO2复合物. Au NPs的负载效率高达100%, 每个SiO2上负载的Au NPs高达800~1000个, Au NPs局域密度高, 大大提高了Au NPs有效浓度, 并且富集到SiO2表面的Au NPs不会团聚. Au NPs@SiO2质量较大, 易从溶液中沉降, 可获得高度浓缩的Au NPs.这种自组装方法不仅高度富集了小粒径Au NPs, 且防止了Au NPs集聚并增强其催化活性. Au NPs@SiO2的结构稳定, 能保存一个月.催化活性研究结果显示, 制备得到的Au NPs@SiO2的催化活性是同浓度Au NPs的3倍. Au NPs@SiO2重复使用5次后, 催化转换效率仍能保持在80%左右.该方法简单高效, 在富集小粒径纳米颗粒、提高催化活性方面, 具有极强的应用价值.
2.
结果与讨论
2.1
Au NPs@SiO2的表征
2.1.1
紫外可见光吸收光谱、粒径和负载效率分析
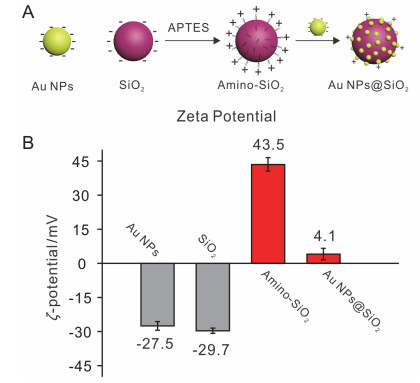
Au NPs@SiO2的制备过程如图 1A所示.首先用3-氨基丙基三乙氧基硅烷(APTES)将SiO2硅烷化, 使SiO2表面修饰上带正电的氨基(Amino-SiO2). 5 nm Au NPs表面带负电荷.通过静电相互作用, 5 nm Au NPs吸附在SiO2表面自组装形成Au NPs@SiO2复合物.
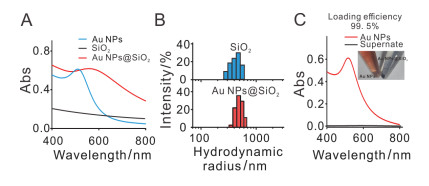
Au NPs, SiO2和Au NPs@SiO2的紫外-可见光吸收光谱如图 2A所示, SiO2在400 nm到800 nm波长范围内无吸收峰, 可以排除SiO2对Au NPs吸收的干扰.与Au NPs相比, Au NPs@SiO2的紫外-可见光吸收光谱的峰的形状发生了显著变化, 最大吸收波长红移约48 nm. Au NPs的吸收光谱与Au NPs的尺寸以及周围物质的介电常数相关[14]. Au NPs的尺寸越大, 其吸收波长越大. Au NPs组装在SiO2表面之后, Au NPs不再是分散状态, 颗粒之间的距离缩短, 相当于一个大粒径的Au NPs, 尺寸增大, 会引起吸收峰红移.另外, Au NPs组装在SiO2表面, 周围物质的介电常数发生改变, 导致了Au NPs@SiO2吸收峰的红移.
图 2
图 2.
(A) Au NPs, SiO2和Au NPs@SiO2的UV-Vis吸收光谱: Au NPs的吸收峰值在512 nm处, Au NPs@SiO2的吸收峰值在560 nm处. (B) SiO2与Au NPs @SiO2的动态光散射粒径分析结果, 其平均水合粒径分别为498 nm、513 nm. (C)组装前后上清液Au NPs的紫外吸收光谱.
Figure 2.
(A) The UV-Vis spectra of Au NPs and Au NPs@SiO2. The absorption peak of Au NPs is 512 nm and Au NPs@SiO2 is 560 nm. (B) The results of dynamic light scattering particle size analysis of SiO2 and Au NPs@SiO2 showed that the average hydration particle size is 498 nm and 513 nm, respectively. (C) The UV-vis spectra of Au NPs in the supernatant before and after assembled.
图 4.
(A) Au NPs与SiO2表面电荷在自组装过程中的变化示意图. (B) Au NPs, SiO2, Amino-SiO2以及Au NPs@SiO2的Zeta电位.
Figure 4.
(A) Scheme for the changes of surface charges of Au NPs and SiO2 in the self-assembly process. (B) The Zeta potential of Au NPs, SiO2, Amino-SiO2 and Au NPs@SiO2.
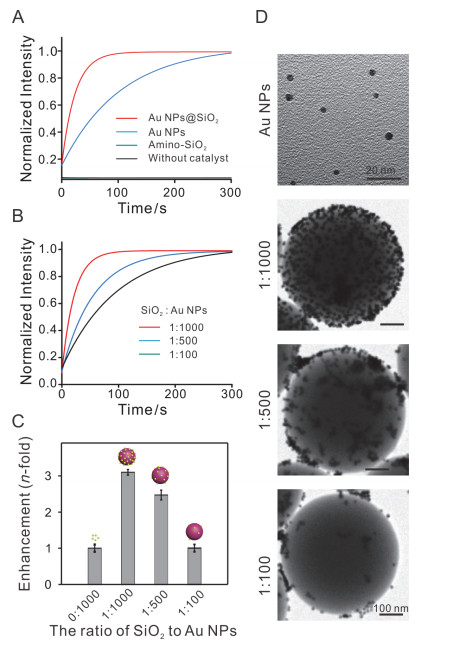
接下来, 我们进一步研究复合物Au NPs@SiO2表面的Au NPs是否具有催化活性. Au NPs可以催化经典的荧光反应: Au NPs催化非荧光底物刃天青(Rz)还原为强荧光产物试卤灵(Rf)(图 1B).我们用这个荧光反应评估Au NPs@SiO2的催化活性.如图 5A所示, 在未加催化剂或只加入Amino-SiO2时, 荧光无上升, 说明刃天青与羟胺在无催化剂的情况下不发生化学反应, Amino- SiO2对该反应无促进作用. Au NPs和Au NPs@SiO2都能加快该反应的进行.与Au NPs相比, 在Au NPs@SiO2催化下, 产物的荧光随时间增加上升更迅速, 如图 5B所示. Au NPs@SiO2催化反应速率(v1=0.0484 μmol·L-1·s-1)约为Au NPs催化反应速率(v2=0.0150 μmol·L-1·s-1)的3.1倍.这表明Au NPs@SiO2复合物不仅保持其表面Au NPs的催化活性, 还能加快化学反应速率.我们猜测, Au NPs组装到SiO2表面后, 在局部区域内, Au NPs的密度大大增高, 高密度的Au NPs使化学速率加快.实验结果表明, 与Au NPs相比, Au NPs @SiO2加速了化学反应过程.
图 5
图 5.
(A) Amino-SiO2, Au NPs和Au NPs@SiO2的催化反应的动力学过程. (B)不同SiO2与Au NPs的物质的量比时, Au NPs@SiO2的催化反应动力学过程. (C)不同SiO2与Au NPs的物质的量比时, Au NPs@SiO2的催化反应增强倍数. (D)不同SiO2与Au NPs的物质的量比时, Au NPs在SiO2表面负载量的TEM表征.
Figure 5.
(A) Kinetics reactions catalyzed by Amino-SiO2, Au NPs and Au NPs@SiO2. (B) The catalytic kinetics reactions catalyzed by Au NPs@SiO2 at different ratios of SiO2 to Au NPs. (C) Catalytic enhancement by Au NPs@SiO2 at different ratios of SiO2 to Au NPs. (D) TEM images of Au NPs@SiO2 at different ratios of SiO2 to Au NPs.
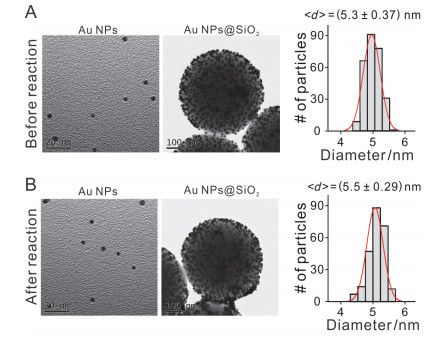
Au NPs@SiO2的稳定性对于其发挥催化功能至关重要, 我们进一步研究其催化前后稳定性变化. Au NPs@SiO2复合物中Au NPs起催化作用, 我们研究了复合物中Au NPs的形貌变化.将催化前后的Au NPs@SiO2复合物用氢氟酸刻蚀, 获得单分散的Au NPs, 然后进一步用TEM表征了Au NPs和未刻蚀的Au NPs@SiO2的形貌.如图 6所示, 催化前后, Au NPs以及Au NPs@SiO2的形貌无明显差异. Au NPs粒径统计结果显示, 催化前后, Au NPs的粒径基本保持一致.表明Au NPs@SiO2复合物中的Au NPs以及复合物本身在催化过程中能够保持自身结构的稳定.
图 6
图 6.
(A) 催化前, Au NPs@SiO2复合物中的Au NPs以及复合物本身的TEM图, 以及Au NPs的粒径统计. (B)催化后, Au NPs@SiO2复合物中的Au NPs以及复合物本身的TEM图, 以及Au NPs的粒径统计.
Figure 6.
(A) TEM images of Au NPs and Au NPs@SiO2 before catalysis, and statistics for the sizes of Au NPs. (B) TEM images of Au NPs and Au NPs@SiO2 after catalysis, and statistics for the sizes of Au NPs.
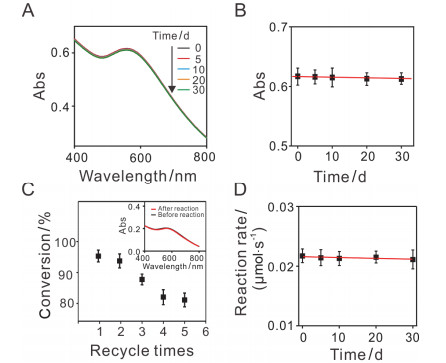
我们进一步研究Au NPs@SiO2随存放时间的变化.如图 7所示, 随着存放时间的增长(0 d、5 d、10 d、20 d、30 d), Au NPs@SiO2的紫外-可见光吸收光谱的峰形基本没有发生改变, 紫外-可见光吸收谱峰值保持不变, 紫外-可见光的吸光度不变.这表明随着存放时间的增加, Au NPs@SiO2的结构基本保持不变.实验结果表明, 自组装获得的Au NPs@SiO2结构非常稳定. Au NPs@SiO2稳定的结构使得它能够重复使用.在重复使用5次后, 其催化转化效率仍能保持在80%左右.我们对比了Au NPs@SiO2使用前后的吸光光谱, 如图 7C所示, 使用前后, Au NPs@SiO2的吸光度分别是0.208和0.20, 浓度分别为21.4 nmol·L-1和20.7 nmol·L-1.使用前后Au NPs@SiO2的吸收峰的峰形以及最大吸收峰保持一致, 因此我们认为使用后, Au NPs@SiO2以及其表面上的Au NPs基本没有流失.另外, 随着存放时间的增长, Au NPs@SiO2的反应速率基本保持不变, 说明存放一个月内, Au NPs@SiO2催化活性稳定. Au NPs@ SiO2的结构和催化活性都能稳定保持一个月.
图 7
图 7.
(A) 不同存放时间下, Au NPs@SiO2的紫外-可见光的吸收光谱. (B) Au NPs@SiO2的紫外-可见光的吸光度随存放时间的变化图. (C) Au NPs@SiO2的催化转化效率随循环使用次数的变化.插入图:催化前后Au NPs@SiO2的吸收谱. (D) Au NPs@SiO2的催化速率随存放时间的变化图.
Figure 7.
(A) The UV-vis spectra of Au NPs@SiO2 at different times. (B) The change of UV-vis absorbance of Au NPs@SiO2 at different time. (C) The catalytic conversion efficiency of Au NPs@SiO2 varies with the number of cycles. Insert: the UV-vis spectra of Au NPs@SiO2 before and after catalysis. (C) The change of catalytic activity of Au NPs@SiO2 at different time.
Zhang, P.; Qiao, Z. A.; Jiang, X.; Veith, G. M.; Dai, S. Nano Lett. 2015, 15, 823. doi: 10.1021/nl504780j
[31]
Fuerte, A.; Corma, A.; Iglesias, M.; Morales, E.; Sánchez, F. Catal. Lett. 2005, 101, 99. doi: 10.1007/s10562-004-3756-7
[32]
Amin, M. A.; Fadlallah, S. A.; Alosaimi, G. S.; Ahmed, E. M.; Mostafa, N. Y.; Roussel, P.; Szunerits, S.; Boukherroub, R. ACS Appl. Mater. Interfaces2017, 9, 30115. doi: 10.1021/acsami.7b07611
[33]
Brust, M.; Gordillo, G. J. J. Am. Chem. Soc. 2012, 134, 3318. doi: 10.1021/ja2096514
[34]
Thangavel, S.; Ramaraj, R. J. Phys. Chem. C2008, 112, 19825. doi: 10.1021/jp804310u
图 2
(A) Au NPs, SiO2和Au NPs@SiO2的UV-Vis吸收光谱: Au NPs的吸收峰值在512 nm处, Au NPs@SiO2的吸收峰值在560 nm处. (B) SiO2与Au NPs @SiO2的动态光散射粒径分析结果, 其平均水合粒径分别为498 nm、513 nm. (C)组装前后上清液Au NPs的紫外吸收光谱.
Figure 2
(A) The UV-Vis spectra of Au NPs and Au NPs@SiO2. The absorption peak of Au NPs is 512 nm and Au NPs@SiO2 is 560 nm. (B) The results of dynamic light scattering particle size analysis of SiO2 and Au NPs@SiO2 showed that the average hydration particle size is 498 nm and 513 nm, respectively. (C) The UV-vis spectra of Au NPs in the supernatant before and after assembled.
图 4
(A) Au NPs与SiO2表面电荷在自组装过程中的变化示意图. (B) Au NPs, SiO2, Amino-SiO2以及Au NPs@SiO2的Zeta电位.
Figure 4
(A) Scheme for the changes of surface charges of Au NPs and SiO2 in the self-assembly process. (B) The Zeta potential of Au NPs, SiO2, Amino-SiO2 and Au NPs@SiO2.
图 5
(A) Amino-SiO2, Au NPs和Au NPs@SiO2的催化反应的动力学过程. (B)不同SiO2与Au NPs的物质的量比时, Au NPs@SiO2的催化反应动力学过程. (C)不同SiO2与Au NPs的物质的量比时, Au NPs@SiO2的催化反应增强倍数. (D)不同SiO2与Au NPs的物质的量比时, Au NPs在SiO2表面负载量的TEM表征.
Figure 5
(A) Kinetics reactions catalyzed by Amino-SiO2, Au NPs and Au NPs@SiO2. (B) The catalytic kinetics reactions catalyzed by Au NPs@SiO2 at different ratios of SiO2 to Au NPs. (C) Catalytic enhancement by Au NPs@SiO2 at different ratios of SiO2 to Au NPs. (D) TEM images of Au NPs@SiO2 at different ratios of SiO2 to Au NPs.
图 6
(A) 催化前, Au NPs@SiO2复合物中的Au NPs以及复合物本身的TEM图, 以及Au NPs的粒径统计. (B)催化后, Au NPs@SiO2复合物中的Au NPs以及复合物本身的TEM图, 以及Au NPs的粒径统计.
Figure 6
(A) TEM images of Au NPs and Au NPs@SiO2 before catalysis, and statistics for the sizes of Au NPs. (B) TEM images of Au NPs and Au NPs@SiO2 after catalysis, and statistics for the sizes of Au NPs.
图 7
(A) 不同存放时间下, Au NPs@SiO2的紫外-可见光的吸收光谱. (B) Au NPs@SiO2的紫外-可见光的吸光度随存放时间的变化图. (C) Au NPs@SiO2的催化转化效率随循环使用次数的变化.插入图:催化前后Au NPs@SiO2的吸收谱. (D) Au NPs@SiO2的催化速率随存放时间的变化图.
Figure 7
(A) The UV-vis spectra of Au NPs@SiO2 at different times. (B) The change of UV-vis absorbance of Au NPs@SiO2 at different time. (C) The catalytic conversion efficiency of Au NPs@SiO2 varies with the number of cycles. Insert: the UV-vis spectra of Au NPs@SiO2 before and after catalysis. (C) The change of catalytic activity of Au NPs@SiO2 at different time.


 下载:
下载:


 下载:
下载:








