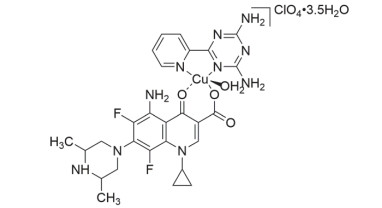
图 1.
配合物可能的分子结构图
Figure 1.
Possible molecular structure of the complex

DNA是抗肿瘤药物的重要靶点, 这类药物的作用效果与其DNA作用方式及作用力大小有关.因此, 研究药物分子与DNA之间的作用, 对开发新型、高效和低毒的靶向DNA类抗肿瘤药物有重要意义[1].
在众多靶向DNA抗肿瘤药物中, 金属配合物类药物得到人们的广泛关注.顺铂为首类被广泛应用于临床肿瘤化疗的金属配合物靶向DNA抗癌药物, 然而该药物存在着明显的不足, 如毒副作用大、易产生耐药性等[2].因此, 进一步研发新型、高效和低毒副作用的金属配合物类抗肿瘤药物有重要的理论意义和实用价值.过渡金属铜离子为生物体内源性金属, 在生命体中具有诸如稳定蛋白质结构、构成金属酶活性中心、参与线粒体呼吸及DNA合成等许多重要的功能作用.尤其是形成配合物时, 具有潜在的抗肿瘤活性等[3].研究发现, 铜配合物类抗肿瘤药物与目前广泛使用的顺铂比较, 不仅毒副作用小, 而且具有不同的作用机制, 可以潜在地避免与顺铂类药物相关的耐药性, 被认为是最具有前景的药物之一[2~5].喹诺酮类是一类重要的人工合成抗菌药物.该类化合物具有广谱、高效、无交叉耐药等特点, 其分子结构中存在多个配位点与金属离子配位, 形成配合物后不仅能够强化其原有的功能作用, 而且有可能产生新的生物活性, 如抗肿瘤作用等[6, 7].另外, 芳杂环类化合物如苯并咪唑衍生物、均三嗪类化合物等具有与生物分子中咪唑、嘌呤碱和嘧啶碱等基团类似的结构性质, 且具有杀菌、抗病毒或抗肿瘤等活性, 配合物分子中引入这类配体时将通过协同作用有助于提高其生物活性, 因而常常被用来设计、合成无机配合物药物[8].
基于以上原因, 选择第三代喹诺酮类化合物5-氨基-1-环丙基-7-(顺氏-3, 5-二甲基-1-哌嗪基)-6, 8-二氟-1, 4-二氢-4-氧-喹啉-3-羟酸(司帕沙星, Sf)及具有均三嗪结构的芳杂环化合物2, 4-二氨基-6-(2'-吡啶基)-1, 3, 5-均三嗪(PyTA)为配体, 合成和表征了新的铜(II)配合物: [Cu(Sf)(PYTA)(H2O)]•ClO4•3.5H2O, 利用电子吸收光谱、KI荧光猝灭光谱、粘度测定及分子对接技术研究了配合物与DNA的作用, 应用MTT [3-(4, 5-二甲基噻唑-2)-2, 5-二苯基四氮唑溴盐]比色法检测了配合物对多个肿瘤细胞系的抑制活性.尤为重要的是, 通过单细胞凝胶电泳、Hoechst 33342染色、Annexin V-FITC/PI双染流式细胞术、测定线粒体膜电位、检测细胞色素C和胞内Ca2+水平、以及细胞周期分析等技术和方法揭示了配合物抗肿瘤活性的作用机制.
实验表明, 配合物元素分析结果与其理论值一致; 红外光谱中配合物羰基伸缩振动νC=O较配体中低, 且∆ν=νas(COO-)-νs(COO-)=230 cm-1, 表明司帕沙星采用羰基及单齿羧基参与配位[9]; 摩尔电导率测定结果表明配合物为1:1型电解质, 表明高氯酸根未参与配位[10]; 综合以上分析及配合物电喷雾质谱和紫外可见光谱, 并参考已报道的类似配合物[11], 可推测出主题配合物的分子式为[Cu(Sf)(PyTA)(H2O)]•ClO4•3.5H2O, 可能的分子结构为变形的四方锥构型, 如图 1所示.
在研究小分子化合物(如配合物)与DNA的相互作用中, 电子吸收光谱是最常用的方法之一.一般来说, 当配合物以插入模式与DNA结合时, 其吸收光谱会出现一定的吸收峰位红移和减色效应, 并且减色效应越大, 峰位红移越多, 配合物作用越强.配合物与CT-DNA作用的电子吸收光谱如图 2所示.
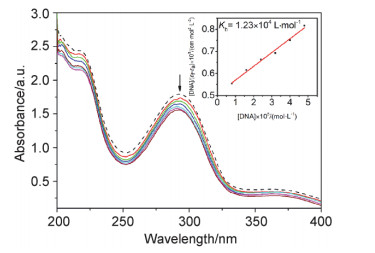
The arrow shows the absorbance changes upon increasing the DNA concentration. Inset: linear plot for the calculation of the intrinsic DNA binding constant (Kb)
配合物在200~400 nm范围内出现的吸收峰可归属于配体的π→π*跃迁.结果表明, 随着CT-DNA浓度的不断增大, 吸收峰出现明显的减色效应(13.1%)和红移现象(1 nm), 表明配合物可能以插入的模式与CT-DNA作用.
此外, 为进一步定量分析配合物与CT-DNA结合的强弱, 可应用下式(1)[12]求得结合常数Kb:
|
$ [{\rm{DNA}}]/\left( {{\varepsilon _{\rm{a}}} - {\varepsilon _{\rm{f}}}} \right) = [{\rm{DNA}}]/\left( {{\varepsilon _{\rm{b}}} - {\varepsilon _{\rm{f}}}} \right) + 1/{K_{\rm{b}}}\left( {{\varepsilon _{\rm{b}}} - {\varepsilon _{\rm{f}}}} \right) $ |
(1) |
式中[DNA]代表DNA碱基对的浓度, εa、εb和εf分别代表Aobs/CCu、与DNA完全结合后配合物的摩尔吸光系数和游离配合物的摩尔吸光系数.以[DNA]/(εf-εa)对[DNA]作图, 斜率与截距的比值为结合常数Kb.获得Kb值为: 1.23×104 L/mol, 与我们以前报道的配合物结合能力相当[13], 即具有适中的结合强度.
碘化钾(KI)荧光猝灭实验通常被用于确定荧光小分子(如配合物)与DNA作用的结合模式.当配合物以插入模式与DNA作用时, 因DNA磷酸骨架和I-之间的静电排斥作用, 配合物-DNA体系的荧光猝灭效率明显低于游离的配合物; 相反, 若为静电结合或沟槽结合, 则因配合物暴露在溶剂中, DNA对配合物的保护较少, 导致其比插入结合更易猝灭, 与游离配合物体系的猝灭率几乎相等[14]. KI在游离配合物和配合物-CT-DNA体系的荧光猝灭率如图 3所示.
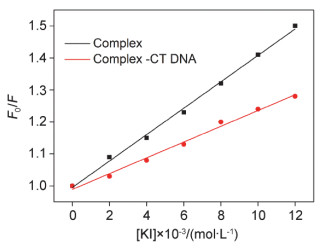
从图中可以看出, 配合物的荧光猝灭效率明显高于配合物-CT-DNA复合体系, 说明配合物以插入的模式与DNA作用, 与上述电子吸收光谱的研究结果一致.
在缺少晶体学结构数据的情况下, 粘度测定实验被认为是检测配合物与DNA结合模式最有效的方法.若DNA溶液粘度随着配合物浓度增加而增大, 则说明DNA螺旋链被拉长, 与配合物以经典的插入模式结合[14].配合物浓度对CT-DNA溶液(200 μmol/L)的粘度变化影响如图 4所示.
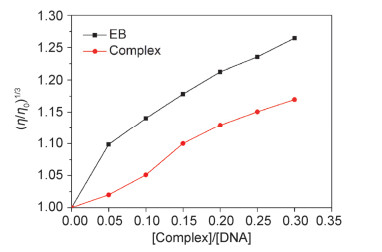
从图 4可以看出, 随着配合物的加入, CT-DNA溶液的相对粘度逐渐增加, 表明配合物以插入模式与CT-DNA作用, 进一步验证了上述电子吸收光谱和KI荧光猝灭方法所获得的结果.
分子对接技术不仅可以直观、形象地研究小分子与生物大分子之间的作用模式以及作用位点, 而且有助于小分子靶向药物以及药效机理的研究[15, 16].为进一步验证配合物与DNA之间相互作用的模式, 本工作使用双链DNA序列d(5'-G-dIU-TGCAAC-3') (PDB ID:454D)进行了分子对接计算, 结果如图 5所示.
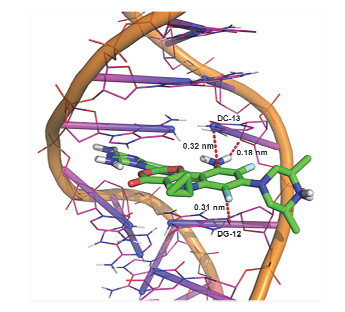
显然, 配合物分子插入到DNA碱基之间, 即配合物通过配体司帕沙星的喹啉环对DNA具有插入作用, 印证了电子吸收光谱、荧光猝灭光谱和粘度测定实验得到的结论.并且发现, 配合物与DNA之间除了疏水作用(主要为芳环堆积作用)外, 同时还具有氢键作用: Complex:H3…454D:DC-13:O4' (0.18 nm).获得配合物与DNA作用的结合能为-52.80 kJ/mol.
本工作运用MTT法测定了配合物对三种肿瘤细胞(Eca-109, Bel-7402和A549)的体外毒性, 并与对小鼠成纤维细胞(3T3)作用进行比较, 结果如表 1所示.
 下载:
导出CSV
下载:
导出CSV
| 化合物 | Cytotoxicity (IC50, μmol•L-1) | |||
| Eca-109 | Bel-7402 | A549 | 3T3 | |
| Sf | 199.5±0.7 | >200 | >200 | >200 |
| PyTA | >200 | >200 | >200 | >200 |
| Complex | 57.0±1.6 | 73.3±0.8 | 77.6±1.4 | 149.6±1.2 |
表中数据表明, 配体Sf和PyTA对所测细胞均未显示出明显的毒性作用, 但配合物细胞毒性很显著, 且具有广谱性, 这可能主要归因于配合物分子中配体间的协同作用.尤为重要的是, 该配合物对正常细胞3T3的毒性作用明显低于肿瘤细胞, 即毒副作用很小, 显示出良好的应用前景.由于配合物对Eca-109细胞最为敏感, 故选用此细胞对配合物的抗肿瘤作用机制进行探究.
药物作用于细胞后, 通过检测DNA的变化可间接反映药物是否诱导细胞发生凋亡.单细胞凝胶电泳实验是一种在单细胞水平上检测DNA损伤的研究方法, 因受损的细胞核在电场的作用下向正极移动, 形成类似彗星的尾巴, 又称彗星实验.为了确定配合物诱导细胞凋亡的存在, 本研究用不同浓度配合物处理细胞24 h后, 检测其内DNA损伤状况, 结果如图 6所示.
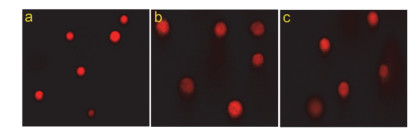
从图中可以看出, 对照组Eca-109细胞核没有发生拖尾, 说明对照组细胞核正常DNA未受损断裂; 而配合物作用过的细胞核, 均出现不同程度的拖尾现象, 表明配合物使细胞DNA受损断裂, 进而诱导细胞凋亡.同时发现, 随着配合物浓度增大细胞核拖尾长度也相应增加, 表明配合物对Eca-109细胞DNA的损伤具有浓度依赖性.
凋亡和坏死是细胞应对外来刺激的两种死亡方式, 而Hoechst 33342染色法是揭示化合物是否通过诱导凋亡的方式来杀死肿瘤细胞的一种重要方法[17].本工作采用Hoechst 33342染色法检测了配合物对Eca-109细胞的作用结果, 如图 7所示.
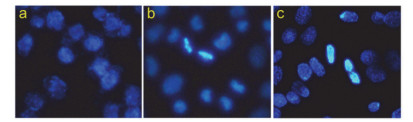
图 7表明, 对照组细胞(a)形态正常, 而加配合物组的细胞(b和c)形态固缩、染色加深变亮, 且随着浓度增大, 固缩明显增多.由此可以判定, 配合物是以诱导凋亡的方式杀死Eca-109细胞, 且具有浓度依赖性[18].
为进一步定量分析配合物诱导肿瘤细胞发生凋亡的能力, 本工作利用流式细胞仪对配合物作用后的细胞进行了Annexin V-FITC/PI染色分析, 结果如图 8所示.
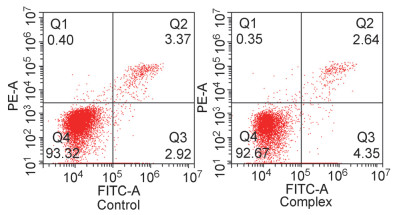
从上图可以看出, 对照组(Eca-109细胞)具有2.92%的早期凋亡细胞, 而在配合物作用24 h后, 早期凋亡细胞的比例增加为4.35%, 比对照组增加了1.43%, 表明配合物能够诱导Eca-109细胞凋亡[19], 与上述实验结果一致.
以上实验结果表明, 配合物通过诱导细胞凋亡的途径杀死肿瘤细胞.诱导细胞凋亡过程通常分为内源性(线粒体)、外源性(死亡受体)和内质网应激启动三条细胞凋亡通路, 其中线粒体通路最为常见, 并且大量研究表明, 在凋亡过程中一旦线粒体膜电位下降, 则细胞凋亡不可逆转[20, 21].为确证配合物诱导细胞凋亡的信号通路, 本文采用JC-1染色法对细胞的线粒体膜电位进行了分析, 结果如图 9所示.
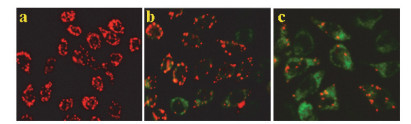
如图 9显示, 对照组Eca-109细胞发红色荧光, 但加入配合物后其红色荧光减弱, 绿色荧光增强, 最终叠合为绿色或橙色, 并且随着配合物浓度的增大, 发出的绿色荧光越强, 表明配合物能够诱导Eca-109细胞的线粒体膜电位降低, 据此推测该配合物很可能是通过线粒体途径诱导细胞凋亡[22].
细胞色素C是一类能够传递电子的含铁蛋白质, 它主要存在于线粒体内、外膜之间的空隙中.当线粒体膜电位丧失时, 细胞色素C从线粒体释放到胞浆中, 与Apaf-1结合形成复合体, 然后激活与凋亡相关的Caspase家族蛋白, 最终导致细胞凋亡[23].为了进一步证实配合物是否以线粒体这条通路诱导细胞凋亡, 本工作对细胞内的细胞色素C水平进行了检测分析(图 10).
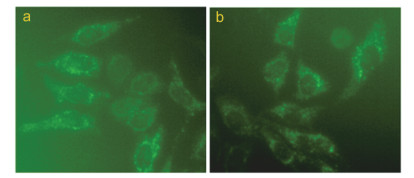
结果表明, 对照组Eca-109细胞染色后, 荧光较弱(a), 但经配合物作用后, 荧光显著增强(b).进一步定量分析结果表明, 用30 μmol/L配合物作用24 h时, Eca-109细胞的荧光强度为26.4, 比对照组增强了0.5倍, 表明配合物作用后细胞内细胞色素C水平显著增加, 有大量细胞色素C渗漏到细胞质中.这些结果进一步揭示了配合物是通过线粒体途径诱导细胞凋亡.
Ca2+为生物体内生理活动不可或缺的金属离子, 并且根据分布的位置及作用不同, 可将其分为细胞内Ca2+和细胞外Ca2+.对于正常细胞而言, 细胞内、外Ca2+的浓度始终维持在一个稳定的水平, 若打破这一平衡, 将会导致细胞死亡.已有大量研究表明, 若药物作用导致细胞内Ca2+浓度升高, 最终可导致细胞凋亡[24].本工作以Ca2+荧光探针Fluo-3Am为指示剂, 利用高内涵细胞成像系统对细胞内Ca2+浓度进行了检测并进行了定量分析, 分别如图 11和图 12所示.
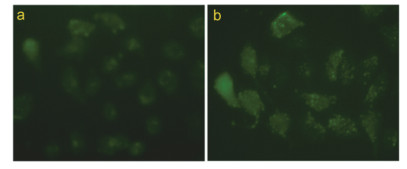
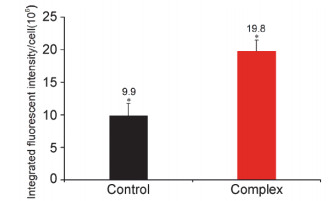
图 11表明, 与对照组Eca-109细胞内荧光(a)比较, 经过配合物作用后细胞内的荧光显著增强(b), 表明配合物作用后细胞内具有较高浓度的Ca2+. 图 12进一步定量分析结果表明, 经30 μmol/L配合物作用24 h后, Eca-109细胞的荧光强度为19.8, 比对照组增强了1.0倍, 即细胞内Ca2+浓度显著升高, 增加了线粒体对钙离子的吸收, 激活线粒体calpains, 进而触发了Eca-109细胞发生依赖于线粒体的细胞凋亡[25], 与以上实验结果一致.
细胞周期分为间期(DNA合成前期: G1期, DNA合成期: S期, DNA合成后期: G2期)和分裂期(M期)两个阶段, 是细胞生命活动的基本过程.因此, 通过对细胞周期的调控, 能阻止细胞的增殖、分化, 最终导致细胞死亡.已有研究表明, 经抗肿瘤药物作用后, 细胞周期发生紊乱, 使细胞阻滞在间期内, 阻断DNA的复制, 进而发挥抗肿瘤作用[26, 27].本文用PI染色, 应用流式细胞仪检测了配合物作用后Eca-109细胞周期的变化, 如图 13所示.
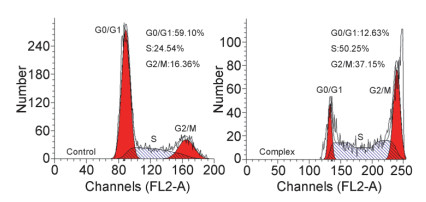
Eca-109 cells were treated with 30 μmol/L complex for 24 h
细胞周期分析结果显示, Eca-109细胞经30 μmol/L配合物作用24 h后, 与对照实验比较, G0/G1期由56.10%显著地减小到12.63%, 而S期和G2/M期分别由24.54%, 16.36%增加到50.25%和37.12%, 表明该配合物使Eca-109细胞周期阻滞在S和G2/M期, 这种作用将有可能抑制肿瘤细胞的增殖能力, 从而发挥其抗肿瘤作用.
本工作设计、合成了新的铜(II)配合物: [Cu(Sf)-(PyTA)(H2O)]•ClO4•3.5H2O.研究发现, 该配合物可通过插入模式与DNA作用, 对体外肿瘤细胞(肺癌细胞A549, 肝癌细胞Bel-7402, 食管癌细胞Eca-109)具有较强的抑制作用, 但对正常细胞毒副作用很小.更重要的是, 揭示了配合物抗肿瘤活性的作用机制, 即配合物通过DNA结合及线粒体功能障碍诱导细胞凋亡, 使细胞周期阻滞在S和G2/M期并造成DNA损伤.
2, 4-二氨基-6-(2'-吡啶基)均三嗪(PyTA)参照文献[28]方法合成.
取司帕沙星(0.25 mmol)溶于8 mL含有等物质的量NaOH的去离子水中, 待完全溶解后, 加入Cu(ClO4)2• 6H2O (0.25 mmol)室温下搅拌30 min.然后, 边搅拌边将32 mL PyTA (0.25 mmol)甲醇溶液缓慢滴入上述溶液中, 滴完后用HClO4调节pH值至7.2, 将其静置缓慢挥发数日, 得到棕黄色粉末, 然后用80%的甲醇-水混合溶剂重结晶, 收率为55%. IR (KBr) ν: 3449 (m, br, νO—H), 3357 (m, νas(N—H)), 3163 (m, br, νs(N—H)), 1637 (vs, νC=O), 1628 (sh, νas(COO-)), 1432 (s, νC=N), 1398 (sb, νs(COO-)), 1294 (s, νC—O), 1089 (s, νCl—O), 792 (w, δb(COO-)), 582 (w, νCu—O), 512 (w, νCu—N) cm-1. Anal. calcd for C27H38F2N10-O11.5ClCu: C 39.32, H 4.61, N 16.99; found C 39.12, H 4.38, N 16.69. ΛM (MeOH, S•cm2•mol-1): 104.1. UV-Vis [DMSO, λmax/nm, ε/(L•mol-1•cm-1)]: 285 (7114, π→π*), 364 (1758, π→π*), 681 (67, d→d). ESI-MS (CH3CN) m/z: 642.1 [Cu(Sf)(PyTA)]+.
称取一定量的小牛胸腺DNA (CT-DNA)溶于适量的Tris-HCl/NaCl缓冲液, 小心振荡后于4 ℃冷藏过夜.待CT-DNA完全溶解后, 稀释10倍在紫外分光光度计下测定A260∕A280=1.8~1.9, 表明CT-DNA溶液中基本上不含蛋白质[29].配制好的CT-DNA储备液置于4 ℃冷藏保存备用, 储藏时间不超过一周.
分别往参比池和样品池中加入3 mL的Tris-HCl/ NaCl缓冲溶液, 扫描基线扣除背景, 随后将样品池中缓冲液更换为等体积的配合物溶液(40 μmol/L), 测定其在200~500 nm范围内的吸收光谱.之后分别往参比池和样品池中滴加等量的CT-DNA溶液, 吹打混匀, 反应8 min后测定光谱, 重复上述操作, 直至吸收峰不再减色.
配制配合物溶液(2 μmol/L)和complex-CT-DNA溶液(complex: 2 μmol/L; DNA: 12 μmol/L).分别移取上述两种溶液3 mL于不同样品池中, 在308 nm激发波长下, 测定550~650 nm波长范围内的荧光光谱.之后, 分别依次往样品池中滴加KI溶液(2~12 mmol/L), 待其充分反应后测定荧光光谱, 以荧光猝灭效率来评价配合物与CT-DNA的作用模式.
使用乌氏粘度计测定溶液的粘度. CT-DNA溶液初始浓度为200 μmol/L, 逐渐滴加配合物溶液, 使其与CT-DNA浓度的比值为0, 0.05, 0.10, 0.15, 0.20, 0.25和0.30.置于恒温水槽恒温[(29.0±0.1) ℃]后, 测定溶液流经毛细管有效刻度的时间, 重复测定3次.然后, 按照公式η=(t-t0)/t0计算样品溶液的相对粘度(t0为缓冲液流经的时间, t为CT-DNA溶液及complex-CT-DNA溶液的流经时间), 并以(η/η0)1/3 (η0为未加配合物时CT-DNA溶液的相对粘度)对[Complex]/[CT-DNA]作图, 将得出配合物对CT-DNA溶液粘度的影响趋势.
采用Gaussian 09软件中的色散校正密度泛函理论(DFT-D3)对配合物的结构进行几何优化, 同时运用连续极化模型(CPCM)模拟水溶剂化效应, 优化后的分子几何结构和原子电荷用于对接研究[30].用于对接的DNA结构文件来源于PDB数据库(PDB ID: 454D), 其分子序列为d(5'-G-dIU-TGCAAC-3'), 对接前对其进行去水加氢.整个对接过程使用格点式的搜索方法, 在Autodock 4.2软件上进行, 以DNA片段的中心为网格的中心区域, 格点间隔为0.0375 nm, 网格大小为60×60×60, 采用经典拉马克遗传算法(Lamarckian Genetic Algorithm, LGA), 计算轮数为100, 配合物与DNA之间的能量匹配采用半经验的自由能计算方法进行评价, 其他参数遵循默认设置.计算结果由Autodock 1.5.6软件进行分析并运用PyMol软件进行可视化处理.
实验所用细胞株:人肺癌细胞(A549)、人肝癌细胞(Bel-7402)、人食管癌细胞(Eca-109)和小鼠成纤维细胞(3T3)均由中山大学实验动物中心提供.
采用MTT法测定配合物的细胞毒性.收集对数期的细胞, 调整浓度为1×105个/mL后接种至96孔板, 于37 ℃的5% CO2培养箱内培养.待细胞贴壁长至80%时, 弃去原有培养液, 加入90 μL新鲜的培养液和10 μL的配合物溶液(浓度设置为: 200, 100, 50, 25, 12.5, 6.25和3.125 μmol/L, 每个浓度设4个重复孔).配合物作用48 h后, 快速倒去板内培养液, 加入90 μL的无血清培养液和10 μL的MTT (5 mg/mL)溶液, 继续培养4 h.培养结束后, 弃上清液, 加入100 μL DMSO溶液, 在微量振荡器上振荡10 min, 随后用酶标仪测试490 nm处的吸光度值.按Eq. 2计算细胞抑制率, 并求出半抑制浓度(IC50).
|
$ 细胞抑制率\% = \left( {1 - 实验组平均{\rm{OD值/对照组平均OD值}}} \right){\rm{ \times 1}}00\% $ |
(2) |
取生长对数期的细胞以每孔约2×105个细胞接种于12孔板中培养过夜, 细胞贴壁长至80%, 加入相应浓度的配合物培养24 h后.吸弃旧培养液, PBS缓冲液清洗残余的培养基和细胞碎片, 每孔加入200 μL胰酶消化后再加入等量的无血清RPMI-1640培养基, 收集转至离心管, 离心, 弃去上清液, PBS缓冲液清洗2次, 调整细胞悬浮液浓度为2×105个/mL备用.
用正常熔点琼脂糖凝胶铺于载玻片上作为第一层胶, 将一定量细胞悬浮液和1%低熔点琼脂糖凝胶混合后立即铺于第一层胶上, 放上盖玻片置于冰箱冷却10 min.然后滴加0.5%低熔点琼脂糖凝胶, 迅速盖上盖玻片, 同样于4 ℃凝固10 min.
取出制备的玻片并移去盖玻片, 浸泡于碱性细胞裂解液中, 4 ℃裂解60 min, 取出用水清洗3次, 置于电泳槽中电泳20 min (25 V, 300 mA).电泳结束后用中和缓冲液清洗3次, 每次10 min.然后用200 μL的EB(10 μg/mL)避光染色20 min, 最后用蒸馏水清洗脱色15 min后, 立即上机检测, 全程尽量避光.实验数据用ZEN lite 2012软件进行分析.
取对数期且生长状态良好的细胞接种于12孔板, 每孔约2×105个细胞, 于37 ℃下在5% CO2培养箱内孵育过夜.细胞贴壁长至80%后, 向各个孔内加入相应浓度的配合物, 并设置对照组(未加配合物组).配合物作用24 h后, 取出培养板, 弃去旧培养液, PBS清洗2次, 加入1 mL的细胞固定液[V(甲醇):V(冰乙酸)=3:1], 于4 ℃固定10 min后, 弃去固定液, PBS清洗, 每孔再加入200 μL的Hoechst 33342染色液, 37 ℃避光染色. 10 min后弃去染色液并用PBS清洗, 于荧光显微镜下拍照, 全程尽量避光.
将细胞以每孔约6×105个细胞接种于6孔板中, 培养箱内孵育过夜, 细胞长至80%左右加入相应浓度的配合物, 继续培养24 h后, 吸除旧培养液, PBS缓冲液清洗, 每孔200 μL的胰酶消化后再加入等量的无血清RPMI-1640培养基, 收集转至相应离心管, 离心(1000 r/min, 6 min), PBS清洗, 每孔依次加入195 μL 1×Annexin V-FITC结合液5 μL Annexin V-FITC和10 μL PI工作液, 吹打混匀. 37 ℃干燥箱内避光孵育15 min, 转至指定流式管中, 上机检测, 全程尽量避光.实验数据用BD FACSEalibur软件收集并用CytExpert软件进行分析.
将细胞以每孔约2×105个细胞接种在12孔板中, 培养孵育过夜, 待长至80%左右, 加入相应配合物继续孵育.配合物作用24 h后, 吸弃旧培养液, PBS清洗, 每孔加入200 μL JC-1染色液[JC-1(200×):JC-1 buffer(5×):ddH2O=1:40:160, V/V], 于37 ℃干燥箱避光孵育20 min.染色结束并用PBS清洗后, 立即于高内涵细胞成像系统进行检测分析, 全程尽量避光.
取对数期且生长状态良好的细胞, 以每孔约2×105个细胞接种于12孔板中37 ℃孵育过夜.细胞贴壁长至80%左右, 加入相应浓度的配合物孵育24 h, 之后每孔加入300 μL固定液于4 ℃固定过夜.次日, 用TBST清洗2次, 每孔再加入300 μL的封闭液, 常温进行封闭. 60 min后, 用TBST清洗3次, 共15 min (82 r/min), 孵一抗(一抗:免疫一抗稀释液=1:200, V/V) 4 ℃过夜, 之后再次用TBST清洗3次(方法同上), 常温避光孵育二抗60 min后, TBST清洗, 每孔加入200 μL的DAPI染液, 37 ℃避光孵育20 min.染色结束后, PBS清洗, 于高内涵细胞成像系统上拍照和定量分析, 全程尽量避光.
取对数期且细胞接种于12孔板中, 于37 ℃孵育过夜.细胞贴壁长至80%左右, 加入相应浓度的配合物, 继续孵育24 h后取出, PBS清洗2次, 每孔加入200 μL的Fluo-3Am染液(Fluo-3Am:PBS=1:2000, V/V), 37 ℃避光孵育. 30 min后, 每孔加入500 μL的PBS于37 ℃静置30 min, 弃去PBS后每孔加入200 μL的Hoechst 33342染液(10 μg/mL), 37 ℃避光染色30 min, PBS清洗后上高内涵细胞成像系统检测并进行定量分析, 全程尽量避光.
采用6孔板进行细胞周期测试, 每孔约4×105个细胞, 培养至80%左右, 加入相应配合物孵育24 h.之后取出培养板, PBS清洗, 每孔加入200 μL的胰酶消化后再加入等量的无血清RPMI-1640培养基, 收集离心(1000 r/min, 6 min), PBS清洗后加入800 μL 75%的冷酒精, 于4 ℃固定, 5 h后离心弃去酒精, 用冷PBS清洗, 再加入200 μL的PI染液(1 mg/mL PI, 200 μL; 10 mg/mL RNaseA, 200 μL; Triton X-100, 10 μL; 用PBS溶解并定容至10 mL)染色30 min后, 即可上机检测, 全程尽量避光.实验数据用BD FACSCalibur软件收集并用FlowJo X软件进行分析.
Rehman, S. U.; Sarwar, T.; Mohammed, A. H.; Hassan, M. I.; Mohammad, T. Arch. Biochem. Biophys. 2015, 576, 49. doi: 10.1016/j.abb.2015.03.024
Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Chem. Rev. 2014, 114, 815. doi: 10.1021/cr400135x
Tardito, S.; Bassanetti, I.; Bignardi, C.; Elviri, L.; Tegoni, M.; Mucchino, C.; Bussolati, O.; Franchi-Gazzola, R.; Marchiò, L. J. Am. Chem. Soc. 2011, 133, 6235. doi: 10.1021/ja109413c
Qi, Y. Y.; Gan, Q.; Liu, Y. X.; Xiong, Y. H.; Mao, Z. W.; Le, X. Y. Eur. J. Med. Chem. 2018, 154, 220. doi: 10.1016/j.ejmech.2018.05.023
Shi, X. C.; Fang, H. B.; Guo, Y.; Yuan, H.; Guo, Z. J.; Wang, X. Y. J. Inorg. Biochem. 2019, 190, 38. doi: 10.1016/j.jinorgbio.2018.10.003
Turel, I. Coord. Chem. Rev. 2002, 232, 27. doi: 10.1016/S0010-8545(02)00027-9
Dorotíková, S.; Kožíšková, J.; Malček, M.; Jomová, K.; Herich, P.; Plevová, K.; Briestenská, K.; Chalupková, A.; Mistríková, J.; Milata, V.; Dvoranová, D.; Bučinský, L. J. Inorg. Biochem. 2015, 150, 160. doi: 10.1016/j.jinorgbio.2015.06.017
Akhtar, J.; Khan, A. A.; Ali, Z.; Haider, R.; Yar, M. S. Eur. J. Med. Chem. 2017, 125, 143. doi: 10.1016/j.ejmech.2016.09.023
Xiao, Y.; Wang, Q.; Huang, Y. M.; Ma, X. L.; Xiong, X. N.; Li, H. Dalton Trans. 2016, 45, 10928. doi: 10.1039/C6DT00915H
Geary, W. J. Coord. Chem. Rev. 1971, 7, 81. doi: 10.1016/S0010-8545(00)80009-0
Efthimiadou, E. K.; Katsarou, M. E.; Karaliota, A.; Psomas, G. J. Inorg. Biochem. 2008, 102, 910. doi: 10.1016/j.jinorgbio.2007.12.011
郭琼, 李连之, 董建方, 刘鸿雁, 薛泽春, 许涛, 化学学报, 2012, 70, 1617. doi: 10.6023/A12040114Guo, Q.; Li, L. Z.; Dong, J. F.; Liu, H. Y.; Xue, Z. C.; Xu, T. Acta Chim. Sinica 2012, 70, 1617. doi: 10.6023/A12040114
Fu, X. B.; Liu, D. D.; Lin, Y.; Hu, W.; Mao, Z. W.; Le, X. Y. Dalton Trans. 2014, 43, 8721. doi: 10.1039/c3dt53577k
Fu, X. B.; Zhang, J. J.; Liu, D. D.; Gan, Q.; Gao, H. W.; Mao, Z. W.; Le, X. Y. J. Inorg. Biochem. 2015, 143, 77. doi: 10.1016/j.jinorgbio.2014.12.006
Trott, O.; Olson, A. J. J. Comput. Chem. 2010, 31, 455.
Chen, J. J.; Ye, H. N.; Zhang, M. J.; Li, J. Y.; Liu, J. Y.; Xue, J. P. Chin. J. Chem. 2016, 34, 983. doi: 10.1002/cjoc.201600481
Feng, C. G.; Gan, Q.; Liu, X.; He, H. Y. Chin. J. Chem. 2012, 30, 1589. doi: 10.1002/cjoc.201100744
Hong, X. L.; Zhou, Y. H.; Zeng, C. C.; Wu, X. C.; Liu, Y. J. J. Organomet. Chem. 2017, 846, 312. doi: 10.1016/j.jorganchem.2017.07.004
Weng, H.; Tan, Z. J.; Hu, Y. P.; Shu, Y. J.; Bao, R. F.; Jiang, L.; Wu, X. S.; Li, M. L.; Ding, Q.; Wang, X. A.; Xiang, S. S.; Li, H. F.; Cao, Y.; Tao, F.; Liu, Y. B. Cancer Cell Int. 2014, 14, 96. doi: 10.1186/s12935-014-0096-6
Huang, H. Y.; Zhang, P. Y.; Yu, B. L.; Chen, Y.; Wang, J. Q.; Ji, L. N.; Chao, H. J. Med. Chem. 2014, 57, 8971. doi: 10.1021/jm501095r
Tang, B.; Wan, D.; Lai, S. H.; Yang, H. H.; Zhang, C.; Wang, X. Z.; Zeng, C. C.; Liu, Y. J. J. Inorg. Biochem. 2017, 173, 93. doi: 10.1016/j.jinorgbio.2017.04.028
Wan. D.; Tang, B.; Wang, Y. J.; Guo, B. H.; Yin, H.; Yi, Q. Y.; Liu, Y. J. Eur. J. Med. Chem. 2017, 139, 180. doi: 10.1016/j.ejmech.2017.07.066
Min, X.; Heng, H.; Yu, H. L.; Dan, M.; Jie, C.; Zeng, Y.; Ning, H.; Liu, Z. G.; Wang, Z. Y.; Lin, W. Oncology Lett. 2018, 15, 2459.
Clapham, D. E. Cell 2007, 131, 1047. doi: 10.1016/j.cell.2007.11.028
Kar, P.; Samanta, K.; Shaikh, S.; Chowdhury, A.; Chakraborti, T.; Chakraborti, S. Arch. Biochem. Biophys. 2010, 495, 1. doi: 10.1016/j.abb.2009.12.020
Zou, H. H.; Wang, L.; Long, Z. X.; Qin, Q. P.; Song, Z. K.; Xie, T.; Zhang, S. H.; Liu, Y. C.; Lin, B.; Chen, Z. F. Eur. J. Med. Chem. 2016, 108, 1. doi: 10.1016/j.ejmech.2015.11.005
刘莹, 陈小曼, 张朗棋, 孙冬冬, 周艳晖, 陈兰美, 刘杰, 化学学报, 2014, 72, 473. doi: 10.6023/A13101092Liu, Y.; Chen, X. M.; Zhang, L. Q.; Sun, D. D.; Zhou, Y. H.; Chen, L. M.; Liu, J. Acta Chim. Sinica 2014, 72, 473. doi: 10.6023/A13101092
Zhao, Q. H.; Fan, A. L.; Li, L. N.; Xie, M. J. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, M622. doi: 10.1107/S1600536809016055
Waring, M. J. J. Mol. Biol. 1965, 13, 269. doi: 10.1016/S0022-2836(65)80096-1
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi: 10.1063/1.3382344

图 2 Tris-HCl/NaCl缓冲溶液中, 配合物在不同CT-DNA浓度下的电子吸收光谱图
Figure 2 Electronic absorption spectra of the complex (40 μmol/L) upon addition of CT-DNA in 5 mmol/L Tris-HCl buffer at room temperature ([DNA]×105=0.8, 1.6, 2.4, 3.2, 4.0, 4.8 mol/L)
The arrow shows the absorbance changes upon increasing the DNA concentration. Inset: linear plot for the calculation of the intrinsic DNA binding constant (Kb)

图 3 KI在游离配合物和配合物-CT-DNA体系的荧光猝灭率
Figure 3 Quenching effects of KI to the complex in the presence and absence of CT DNA at room temperature ([Complex]=2 μmol/L; [DNA]=12 μmol/L; [KI]=2, 4, 6, 8, 10, 12 μmol/L)

图 4 (29.0±0.1) ℃, 配合物及EB浓度的增加对CT-DNA相对粘度的影响
Figure 4 Effect of increasing amounts of the complex and EB on the relative viscosity of CT-DNA at (29±0.1) ℃ ([DNA]=200 μmol/L; [Complex]/[DNA]=0, 0.05, 0.10, 0.15, 0.20, 0.25, 0.30)

图 5 配合物与DNA (PDB ID:454D)的分子对接模型图
Figure 5 The molecular docked model of the complex with DNA (PDB ID: 454D)

图 6 用彗星实验检测配合物对Eca-109细胞的作用
Figure 6 Comet assay of EB-stained Eca-109 cells in the control (a) and treated by 15 and 30 μmol/L of the complex (b, c) after 24 h incubation

图 7 Hoechst 33342染色法检测配合物对细胞形态的影响
Figure 7 Change of the morphology of Eca-109 cells after incubation with 15 and 30 μmol/L of the complex (b and c) for 24 h was determined by Hoechst 33342 staining method

图 8 Annexin V-FITC/PI双染检测配合物诱导Eca-109细胞凋亡图
Figure 8 Apoptosis rates of Eca-109 in the control (a) and treated with 30 μmol/L complex (b) for 24 h at 37 ℃ were detected by Annexin V-FITC/PI double staining

图 9 JC-1探针染色法测定配合物对Eca-109细胞内线粒体膜电位的影响
Figure 9 The mitochondrial membrane potentials of Eca-109 cells exposed to cccp (positive control, a) and 6.25 and 12.5 μmol/L of the complex (b and c) for 24 h were determined with JC-1 as fluorescence probe staining method

图 10 配合物对Eca-109细胞内细胞色素C影响的水平分析
Figure 10 Release of cyt-C was examined after Eca-109 cells were exposed to 30 μmol/L complex for 24 h

图 11 配合物对Eca-109胞内Ca2+水平影响的检测分析
Figure 11 Intracellular Ca2+level was assayed after Eca-109 cells were exposed to 30 μmol/L complex for 24 h

图 12 配合物作用24 h后, 胞内Ca2+水平的定量分析
Figure 12 Intracellular Ca2+level analysis of quantitatively assayed after Eca-109 cells were exposed to 30 μmol/L complex for 24 h

图 13 流式细胞仪定量分析配合物诱导Eca-109细胞凋亡
Figure 13 Quantitative analysis of complex-induced apoptotic cell death by flow cytometry
Eca-109 cells were treated with 30 μmol/L complex for 24 h
表 1 配体及配合物对细胞的IC50值(平均值±标准误差)
Table 1. IC50 values of the ligands and complex toward the tested cell lines (mean value±standard error)
| 化合物 | Cytotoxicity (IC50, μmol•L-1) | |||
| Eca-109 | Bel-7402 | A549 | 3T3 | |
| Sf | 199.5±0.7 | >200 | >200 | >200 |
| PyTA | >200 | >200 | >200 | >200 |
| Complex | 57.0±1.6 | 73.3±0.8 | 77.6±1.4 | 149.6±1.2 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
