Key Laboratory of Chemical Biology & Traditional Chinese Medicine Research(Ministry of Education of China), College of Chemistry and Chemical Engineering, Hunan Normal University, Changsha 410081
Received Date:
12 November 2019 Available Online:
15 April 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21675050, 21475041 and 21775137), Hunan Lotus Scholars Program (2011) and Foundation of the Science & Technology Department of Hunan Province (No. 2016SK2020)
Abstract:
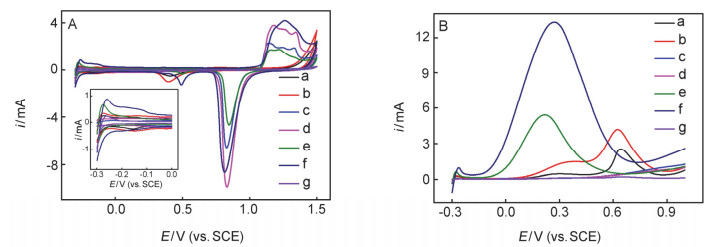
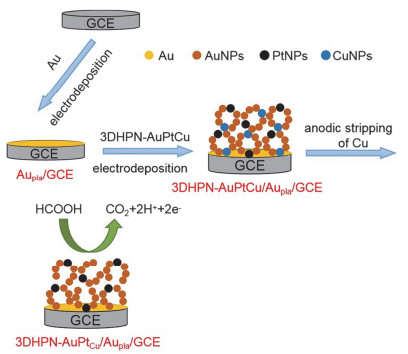
Improving the performance of electrocatalytic formic acid oxidation is the key issue to develop high-performance direct formic acid fuel cells (DFAFC). Pt-based and Pd-based materials are the important electrocatalysts for formic acid oxidation. Micro/nano-porous metal materials are widely concerned in the electrochemistry field due to the high specific electrode-surface area. The dynamic hydrogen bubble template (DHBT) method has been widely used for preparing the three-dimensional honeycomb-like porous nano-metals (3DHPNMs). However, as far as we know, the use of a sacrificial metal template to prepare the 3DHPNMs with improved performance for the electrocatalytic oxidation of small organic molecules has not been reported. Herein, a three-dimensional honeycomb-like porous nano-AuPtCu (3DHPN-AuPtCu) composite was electrodeposited on a gold-plated glassy carbon electrode (Aupla/GCE) by the DHBT method, followed by anodic stripping of Cu to yield a 3DHPN-AuPtCu/Aupla/GCE. The relevant modified electrodes were characterized by cyclic voltammetry (CV), metallographic microscopy, scanning electron microscopy (SEM), energy dispersive spectroscopy and inductively coupled plasma-atomic emission spectrometry. The SEM results clearly revealed that the use of the sacrificial Cu template can modulate the metal-honeycomb structure, and the 3DHPN-AuPtCu/Aupla/GCE can thus possess the better micro/nano-porous structure and the improved electrocatalytic performance than a Cu-template-free 3DHPN-AuPt/Aupla/GCE. In our opinion, the simultaneous electrodeposition of Cu can intervene in the electrodeposition of Au and Pt, and thus a new structure with more active sites exposed and the electrocatalysis performance improved can be obtained after the anodic stripping of electrodeposited Cu. As a result, the 3DHPN-AuPtCu/Aupla/GCE exhibited high anti-poisoning nature and high stability, because many discontinuous Pt atoms on this electrode can suppress the formation of adsorption-state COads during the electrocatalytic oxidation of formic acid. The electrocatalytic oxidation peak current density on 3DHPN-AuPtCu/Aupla/GCE in 0.5 mol/L aqueous H2SO4 containing 0.2 mol/L HCOOH was 12.5 mA·cmPt-2 (CV, -0.3~1.0 V, 50 mV/s), which is superior to the control electrodes and many reported Pt-based electrocatalysis electrodes. The suggested double- template method for preparing honeycomb-structured micro/nano-porous metal materials with improved performance has the potential for wider electrocatalysis and electroanalysis applications.
Figure 1.
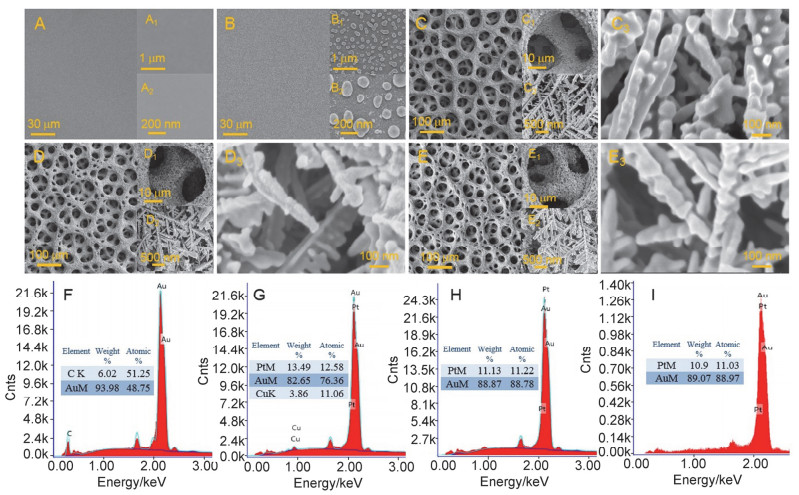
SEM images and EDS results of bare GCE (A, A1 and A2), Aupla/GCE (B, B1, B2 and F), 3DHPN-AuPtCu/Aupla/GCE (C, C1, C2, C3 and G), 3DHPN-AuPtCu/Aupla/GCE (D, D1, D2, D3 and H) and 3DHPN-AuPtCu/Aupla/GCE (E, E1, E2, E3 and I, after 1200 s catalytic test at 0.2 V in stirred 0.2 mol/L HCOOH+0.5 mol/L H2SO4)
Figure 2.
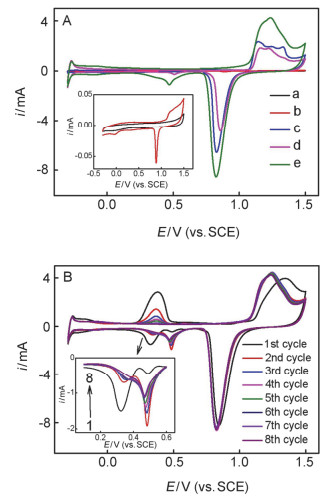
MM pictures of Aupla/GCE after electrodeposition at -4.0 V for 200 s in 0.5, 1, 2, 3, 4, 5, 7 or 9 mol/L H2SO4 containing 20 mmol/L HAuCl4+0.3 mmol/L H2PtCl6+1.5 mmol/L CuSO4, and then anodic stripping of Cu
Kim, J.; Roh, C.-W.; Sahoo, S. K.; Yang, S.; Bae, J.; Han, J. W.; Lee, H. Adv. Energy Mater. 2018, 8, 1701476. doi: 10.1002/aenm.201701476
[2]
Marcinkowski, M. D.; Murphy, C. J.; Liriano, M. L.; Wasio, N. A.; Lucci, F. R.; Sykes, E. C. H. ACS Catal. 2015, 5, 7371. doi: 10.1021/acscatal.5b01994
[3]
Fan, H. s.; Cheng, M.; Wang, L.; Song, Y. j.; Cui, Y.; Wang, R. m. Nano Energy2018, 48, 1. doi: 10.1016/j.nanoen.2018.03.018
Kottakkat, T.; Klingan, K.; Jiang, S.; Jovanov, Z. P.; Davies, V. H.; El-Nagar, G. A. M.; Dau, H.; Roth, C. ACS Appl. Mater. Inter. 2019, 11, 14734. doi: 10.1021/acsami.8b22071
[26]
Yuan, W.; Zhang, J.; Shen, P. K.; Li, C. M.; Jiang, S. P. Electrochim. Acta2016, 190, 817. doi: 10.1016/j.electacta.2015.12.152
[27]
Pilapil, B. K.; van Drunen, J.; Makonnen, Y.; Beauchemin, D. Jerkiewicz, G.; Gates, B. D. Adv. Funct. Mater. 2017, 27, 1703171. doi: 10.1002/adfm.201703171
[28]
Xi, Z.; Lv, H.; Erdosy, D. P.; Su, D.; Li, Q.; Yu, C.; Li, J.; Sun, S. Nanoscale2017, 9, 7745. doi: 10.1039/C7NR02711G
[29]
Wang, C.; van der Vliet, D.; More, K. L.; Zaluzec, N. J.; Peng, S.; Sun, S.; Daimon, H.; Wang, G.; Greeley, J.; Pearson, J.; Paulikas, A. P.; Karapetrov, G.; Strmcnik, D.; Markovic, N. M.; Stamenkovic, V. R. Nano Lett. 2011, 11, 919. doi: 10.1021/nl102369k
Figure 1
SEM images and EDS results of bare GCE (A, A1 and A2), Aupla/GCE (B, B1, B2 and F), 3DHPN-AuPtCu/Aupla/GCE (C, C1, C2, C3 and G), 3DHPN-AuPtCu/Aupla/GCE (D, D1, D2, D3 and H) and 3DHPN-AuPtCu/Aupla/GCE (E, E1, E2, E3 and I, after 1200 s catalytic test at 0.2 V in stirred 0.2 mol/L HCOOH+0.5 mol/L H2SO4)
Figure 2
MM pictures of Aupla/GCE after electrodeposition at -4.0 V for 200 s in 0.5, 1, 2, 3, 4, 5, 7 or 9 mol/L H2SO4 containing 20 mmol/L HAuCl4+0.3 mmol/L H2PtCl6+1.5 mmol/L CuSO4, and then anodic stripping of Cu

 下载:
下载:


 下载:
下载:






