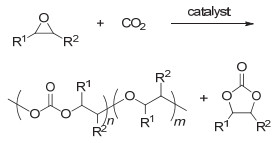
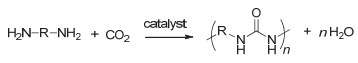
图 1.
CO2和环氧化物的聚合反应
Figure 1.
The polymerization of CO2 and epoxides

二氧化碳(CO2)是导致“温室效应”的主要气体, 而在化学合成领域却是一种丰富、廉价、无毒以及可再生的一碳资源.经过几十年的发展, CO2已经可以转化为多种高附加值的化学品, 例如:尿素、水杨醛、环状碳酸酯、甲醇、甲酸和异氰酸酯等[1~8].
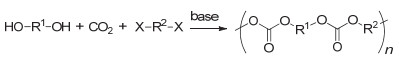
将CO2转变为性能优良的高分子材料是其利用与转化非常重要的途径.目前被广泛研究的是CO2和环氧单体的聚合反应用于制备聚碳酸酯, 其反应通式如图 1所示.该聚合反应最早由Inoue等[9]于1969年报道, 所使用的催化体系是二乙基锌和水的混合物.该聚合反应的发现开辟了利用CO2制备高分子材料的先河.之后的半个世纪中, 各国科学家纷纷投身于CO2和环氧单体聚合反应的研究中.研究内容主要包括: (1)深入研究聚合反应机理; (2)发展新的催化体系, 不断提高反应活性; (3)设计制备结构可控聚合物等.经过科学家们几十年的不断努力, 这一聚合反应已在国内外成功实现工业化.在本综述中, CO2和环氧单体的聚合反应不详细展开, 请参考一些系统全面的代表性综述[10~24].
近些年, 除了利用CO2和环氧单体的聚合反应制备聚碳酸酯之外, 科学家们相继报道了多种CO2参与的新型聚合反应并制备了新型CO2基聚合物, 为CO2的利用与转化开辟了新途径.利用CO2制备高分子材料大致有两条途径: (1)首先将CO2转化为可聚合的单体, 例如:内酯、环状碳酸酯和2, 5-二呋喃甲酸等, 然后通过开环或者逐步聚合将CO2基单体聚合得到高分子材料; (2)直接以CO2为单体和其他种类单体共聚合得到高分子材料.这两种途径对于丰富和拓展CO2基高分子材料均具有重要意义, 接下来将详细进行讨论.
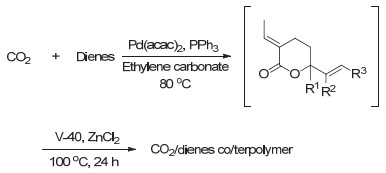
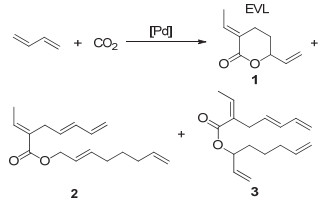
Inoue等[25]在1976年发现CO2和丁二烯在钯基催化剂催化下可以生成少量的五元环内酯, 然而由于产率极低, 且有多种复杂的副产物生成, 并未引起大家的重视. 1978年, Musco等[26]发现在无溶剂或者苯中, CO2可与丁二烯反应生成六元环内酯3-Ethylidene-6-vinyltetra- hydro-2H-pyran-2-one (EVL), 同时还会生成两种线形酯的副产物2和3(如图 2).由于EVL环外具有两种烯键, 主体是内酯结构, 因此具有丰富的化学反应活性, 引起了科研工作者们的广泛关注.此后, Behr, Braunstein, Dinjus, Pitter和Beller等[27~31]也先后开展了CO2和丁二烯聚合反应的研究, 通过不断的优化, 目前这一反应得到的EVL选择性可高达90%, 产率高达70%.
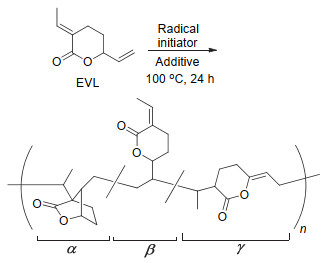
CO2和丁二烯制备得到的EVL由于丰富的化学性质, 可发生催化加氢、氢化甲酰化以及氢化胺化等反应得到多种高附加值化学品.然而EVL作为单体通过自由基聚合得到聚合物还是一个没有得到解决的难题.直到2014年, Nozaki等[32]首次实现了EVL的自由基聚合, 并得到了高分子量的聚合物(如图 3).当使用传统自由基引发剂偶氮二异丁腈(AIBN)引发聚合时, 产物的产率和分子量都很低, 而当使用半衰期更长的1, 1'-偶氮(氰基环己烷)(V40)作为引发剂时, 产物的聚合产率和分子量均得到了明显提升.作者还探索了溶剂和路易斯酸对聚合反应的影响, 发现当采用氯化锌为路易斯酸和碳酸乙烯酯为溶剂时, 聚合产率可提升至59%, 数均分子量可提升至62000 g/mol.值得注意的是, 在添加路易斯酸的聚合反应中, 聚合物结构中出现了如图 3所示的β与γ结构.通过进一步研究, 他们发现CO2和丁二烯通过反应得到的中间体EVL不必进行分离提纯即可进一步聚合得到目标聚合物, 即可以通过“一锅、两步”法实现CO2和丁二烯的间接聚合.此外, 通过引入第三单体异戊二烯和1, 3-戊二烯, 作者还实现了三元共聚(图 4), 得到CO2含量很高的新型聚合物.通过差示扫描量热(DSC)表征, 作者发现制备的新型CO2基聚合物具有很高的玻璃化转变温度, 最高可达192 ℃, 将在工程塑料方面具有很大的潜力.
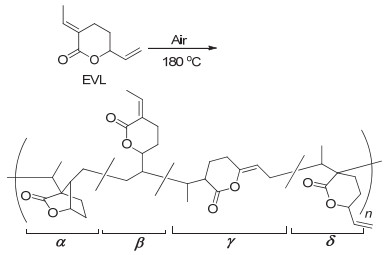
2017年, Lin等[33]报道了更加简便高效的EVL自由基聚合反应. EVL在氧气存在的条件下, 不需要添加任何引发剂和溶剂即可高效聚合得到高分子量的、具有4种不同重复单元的聚合物(图 5).得到的聚合物由于含有大量未反应的烯键, 还可以通过巯基-烯点击反应进行高效后修饰, 从而获得含硫聚合物.
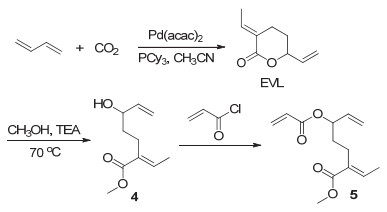
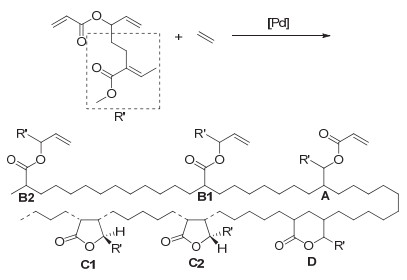
2019年, Jian等[34]通过三步反应将CO2和丁二烯转化成了一种全新的多烯单体5(图 6), 然后其可与乙烯单体在钯催化下进行自由基聚合得到具有区域和立构规整性的类聚乙烯产物(图 7).这种聚合物中含有大量酯键、烯键以及环状内酯, 从而具有特殊的性能.
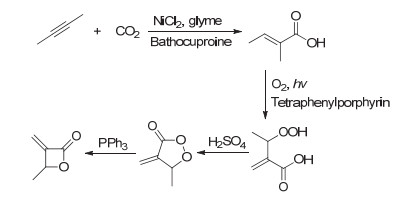
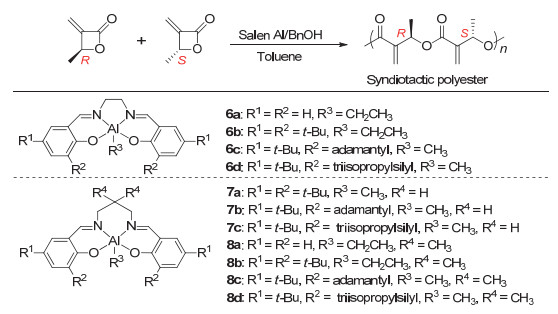
CO2不仅可以和烯类化合物反应生成内酯单体, 也可以与炔类化合物反应得到可用于进一步聚合的内酯单体. 2016年, Lu等[35]利用CO2和2-丁炔通过4步反应制备得到了α-亚甲基-β丁内酯(图 8), 然后通过Salen-Al催化开环聚合得到了聚酯(图 9).结果表明, Al催化剂中不同取代基对于聚合反应的活性和选择性都有重要影响, 采用6c作为催化剂表现出活性聚合的特性.
作者随后对制得的立构规整聚合物进行了热重分析(TGA)和DSC表征, 间规聚合物玻璃化转变温度约为20 ℃, 熔点最高可达109 ℃; 全同等规聚合物熔点可达103 ℃, 热分解温度为276 ℃, 具有良好的加工窗口.广角X-射线衍射表明这是一种结晶性高分子.
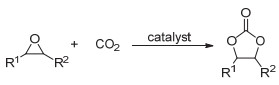
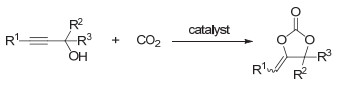
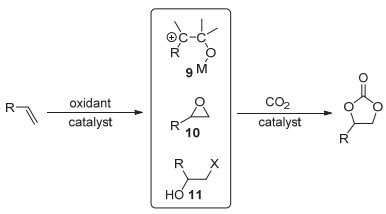
五元环状碳酸酯是一种可利用CO2制得的重要化工产品, 在高分子合成方面也具有广泛用途.目前将CO2转化为五元环状碳酸酯的方法主要有: (1) CO2和环氧化合物反应(图 10, 所用催化体系包括季铵盐、过渡金属和离子液体等[36~39]); (2) CO2和炔丙醇反应(图 11, 可用离子液体或者氮杂卡宾等催化[40~42]); (3) CO2和烯烃反应(图 12, 这实际上是一个“一锅、两步”的反应过程).第一步是烯烃的氧化反应, 第二步是氧化中间体和CO2的环加成反应, 根据所采用的氧化剂的不同可得到不同的氧化中间体(9~11, 图 12)[43, 44].
五元环状碳酸酯具有丰富的化学性质, 可以和胺类化合物反应, 开环得到氨酯类化合物.因此, 二官能度的五元环状碳酸酯常被用作非异氰酸酯路线合成聚氨酯的重要单体. 2017年, Detrembleur等[45]利用CO2和炔丙醇制备了含有环外双键的二官能度环状碳酸酯单体.这些单体可以在室温条件下进一步和一级胺、二级胺以及醇类单体反应, 区域选择性开环聚合得到聚氨酯和聚碳酸酯.值得注意的是, 当使用一级胺单体反应时, 制备得到的聚氨酯会发生分子链内的环化作用, 最终得到聚噁唑啉酮(图 13).
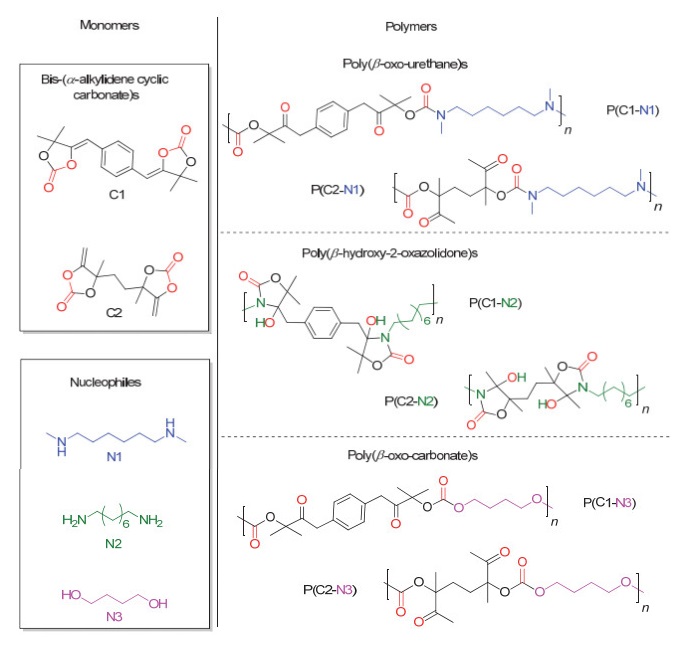
此外, Detrembleur等[46]还利用从CO2和炔丙醇制得的五元环状碳酸酯先和双官能度的二级胺反应, 区域选择性开环得到氨酯类化合物, 然后再与双官能度的一级胺反应, 最终得到具有酸降解性能的新型聚合物(图 14).通过在不同pH下聚合物的降解实验可以得出, 新型CO2基聚合物可在pH为1的条件下放置24 h即可完全降解.
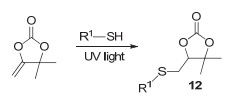
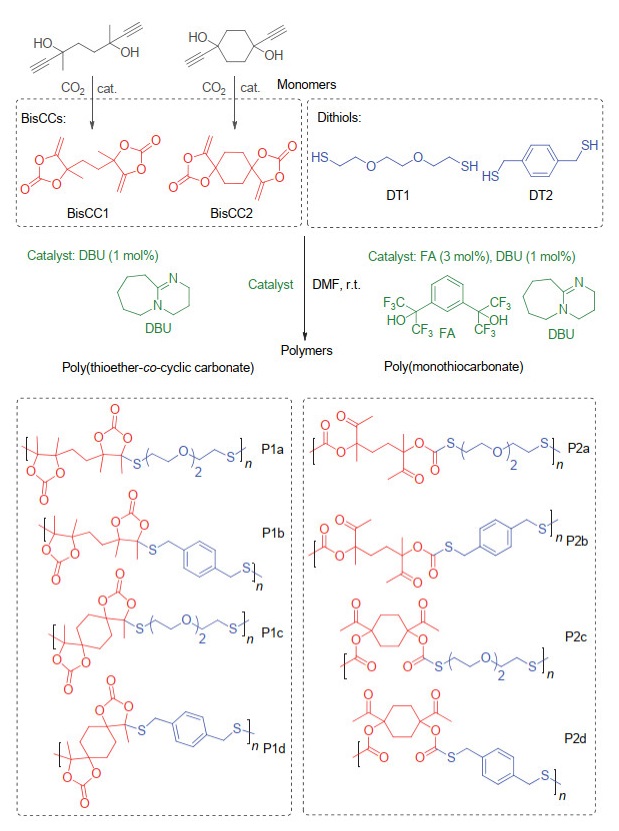
五元环状碳酸酯除了能与胺类化合物反应外, 还可以与硫醇类化合物反应. 2018年, Zhang等[47]发现硫醇可以和五元环状碳酸酯的环外双键加成, 得到含硫的环状碳酸酯12(图 15). 2019年, Detrembleur等[48]再次研究了硫醇和五元环状碳酸酯的反应, 他们意外发现该反应在1, 8-二氮杂二环十一碳-7-烯(1, 8-diazabicyclo[5.4.0]- undec-7-ene, DBU)催化下存在先开环后成环的串联反应过程(图 16), 通过对反应的时间监控发现, 反应为1 min时, 五元环状碳酸酯的转化率已经大于99%, 此时主要存在的是开环产物13, 随着时间的延长, 该产物逐渐成环转化为新的环状产物14; 在反应24 h之后, 最终得到的几乎全部为环状产物14.随后他们通过对反应机理的研究指出, 可以通过添加1, 3-双(六氟-羟异丙基)苯(1, 3-bis(2-hydroxyhexa fluoroisopropyl)benzene, FA)来控制最终聚合产物的结构, 如图 17所示.当仅仅通过DBU来催化聚合时, 最终得到的是聚(硫醚-环状碳酸酯); 而当使用FA和DBU共同催化时, 最终得到的产物是聚(硫代碳酸酯).因此, 这是一种高效、温和以及可控的制备CO2基含硫高分子的新途径.

2, 5-呋喃二甲酸(furan-2, 5-dicarboxylic acid, FDCA)作为一种新型生物基原材料最大的用途是作为单体和乙二醇进行聚合反应得到聚呋喃二甲酸乙二醇酯(polyethylene furandicarboxylate, PEF).而PEF是石油基工程塑料聚对苯二甲酸乙二醇酯(polyethylene terephthalate, PET)潜在的大规模替换产品.现有的工业化制备FDCA的途径是将果糖(fructose)转化为5-羟甲基糠醛(5-hydroxymethyl furfural, HMF), 再通过氧化作用得到FDCA(图 18).
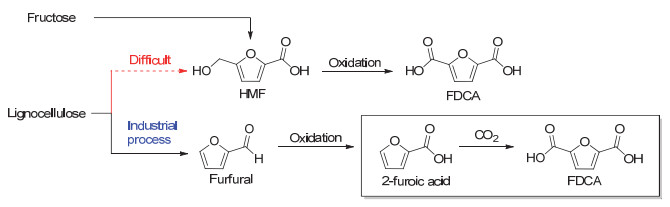
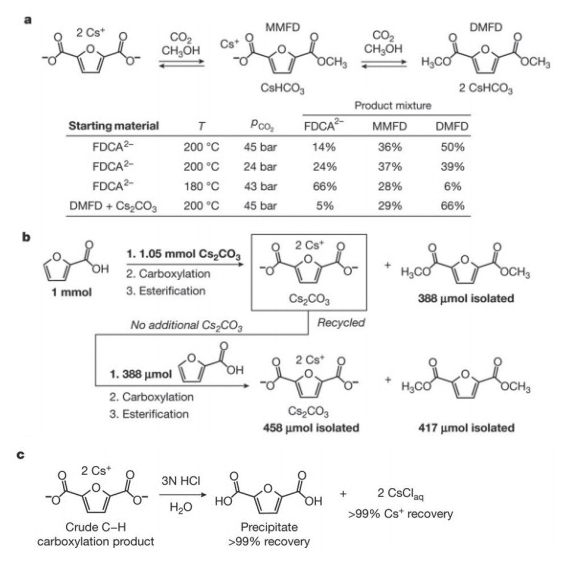
2016年, Kanan等[49]发展了一种从木质纤维素(lignocellulose)和CO2到FDCA的更加经济高效的路线(图 18).木质纤维素难以转化为HMF, 却可以通过成熟的工业化路线转化为2-呋喃甲醛, 再经过氧化得到2-呋喃甲酸(2-furoic acid). 2-呋喃甲酸可以在180~200 ℃下, 通过碳酸铯催化, 促使C—H键发生羧化反应最终得到FDCA(图 19).为了测试碳酸铯是否可以回收再利用, 作者进行了连续的羧化和酯化实验, 通过两次连续的实验, 作者计算出羧化产物总产率为91%, 而铯离子的回收率大于99%.另外, 铯离子可通过简单的双极膜电渗析转化为氢氧化铯, 进而和2-呋喃甲酸以及CO2生成起始原料碳酸铯进入下一次循环中.在这项工作中, Kanan等发展了一种全新的从CO2制备PEF的路线, 并详细评估了工业化生产的成本, 在学术和工业领域均有重大意义.
CO2可直接和二醇(酚)类单体聚合生成聚碳酸酯(图 20).早在1998年, Kadokawa等[50]就利用苯二甲基乙二醇和CO2聚合制备得到了聚碳酸酯, 但产率和分子量都不高. 2016年, Tamura等[51]报道了稀土催化剂二氧化铈催化CO2和二醇类单体的聚合.该工作不仅极大地拓展了二醇类单体的种类, 而且显著提高了反应活性.
CO2和二醇单体如果再引入二卤代物即可发生三组分聚合反应, 生成聚碳酸酯. Inoue等[52]在1994年便发现这一三组分聚合反应可以在常压CO2下, 通过碳酸钾催化进行(图 21).实验表明, 只有极性非质子溶剂才适用于这个体系, 例如, N-甲基吡咯烷酮, N, N-二甲基甲酰胺(DMF), N, N-二甲基乙酰胺(DMAc)等.而在四氢呋喃(THF)这样较小极性的溶剂中, 聚合反应则不能发生. 2016年, Gnanou等[53]发现碳酸铯也能催化这类单体的聚合反应, 并且要比碳酸钾表现出更高的反应活性, 得到聚合物的分子量和产率都有了一定程度的提高.
CO2也可以和二胺类单体聚合生成聚脲(图 22). Yamazaki等[54, 55]采用亚磷酸酯和吡啶的催化体系, 实现了温和条件下CO2和二胺类单体的缩聚反应.单体种类对聚合反应影响很大, 芳香二胺与CO2的聚合反应活性高, 可得到高分子量的聚合物.而当采用脂肪二胺作为单体时, 由于其碱性过强会引起中间产物结构的变化和溶解度的减小, 产率和分子量都很低.
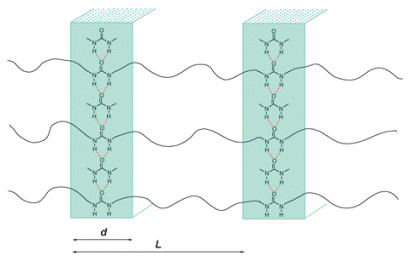
2012年, Zhao等[56]发现在不添加任何催化剂和溶剂的情况下, CO2和二胺类单体就可以聚合生成聚脲.他们还发现, 聚脲分子链之间存在双配位的氢键作用, 使得聚合物表现出半结晶的特殊性质(图 23).
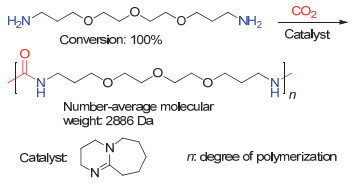
2019年, Wu等[57]发现DBU可以催化CO2和4, 7, 10-三氧-1, 13-十三烷二胺(TOTDDA)的聚合并产生聚脲(图 24).通过对聚合反应的在线红外监测, 作者认为DBU对于二胺单体和CO2均具有活化作用(图 25).
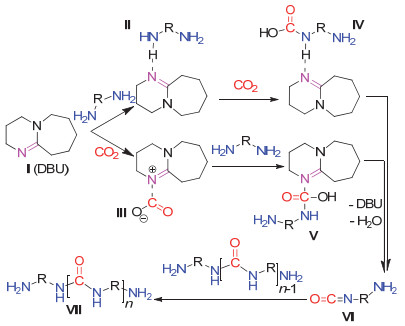
CO2和二胺单体也可与二卤代物发生聚合反应. 2017年, Gnanou等[58]就实现了CO2、二胺单体与二卤代物在碳酸铯和四丁基溴化铵催化下的三组分聚合反应(图 26).通过对得到的聚合物的结构表征发现, 聚合物链中除了三组分反应产物氨酯基之外, 还有一定含量的碳酸酯基.为此作者设计了控制实验, 使CO2、二胺和二卤代物单体分别两两在标准反应条件下反应, 结果表明, 聚合物主链中的碳酸酯基是由CO2和二卤代物反应而生成的(图 27).
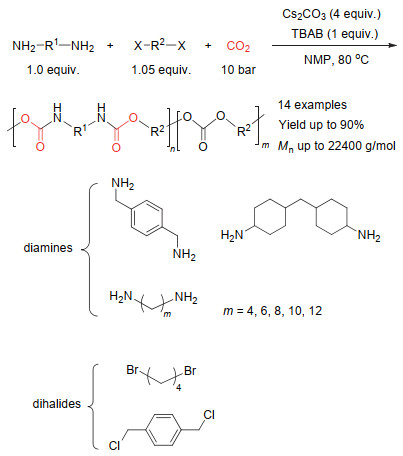
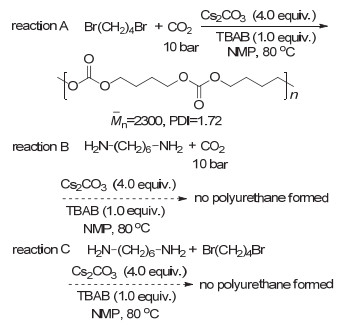
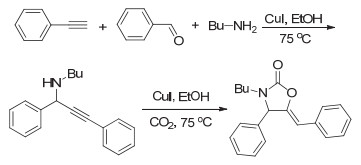
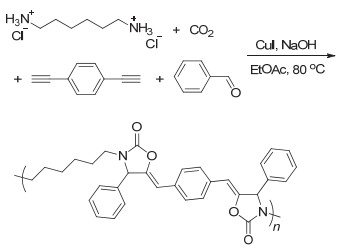
CO2和胺还可以引入炔和醛类单体进行四组分反应.早在2008年, Li等[59]便报道了常压下CO2和胺、炔、醛的四组分反应.该反应在碘化亚铜的催化下以很高的收率得到噁唑啉酮.进一步研究发现, 反应过程首先是炔、醛和胺类化合物反应生成炔丙胺中间体, 然后该中间体再和CO2反应得到最终的产物(图 28).随后, Jiang等[60]报道了CO2和炔、酮、胺的四组分反应, 通过引入二水合氯化亚锡和碘化亚铜共同催化, 从而使反应收率更高. Yu等[61]采用新的反应策略, 先将CO2和伯胺反应生成固态的盐, 再使之与炔和醛类化合物反应得到最终产物.新的反应策略显著减少了碘化亚铜的使用, 仅仅5 mol%的催化剂即可得到很高的产物收率.通过借鉴有机小分子反应的催化体系, Li等[62]将CO2和胺、炔、醛的四组分反应成功发展为聚合反应(图 29).他们采用对苯二炔、二铵盐、苯甲醛和CO2进行四组分聚合反应制备得到了聚噁唑啉酮.不过由于聚合物不溶于常见有机溶剂中, 聚合物的结构并未得到清晰的表征.
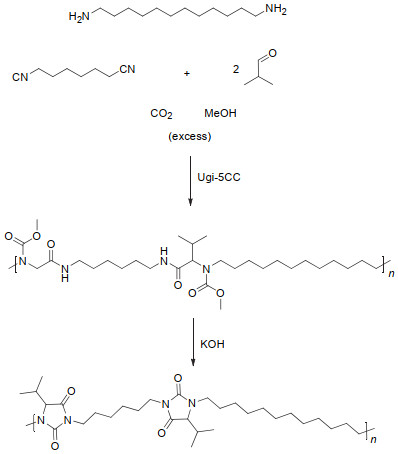
Ugi等[63]最早报道了酸、醛、胺和异腈的四组分反应, 并用于α-酰胺基酰胺类化合物的制备, 因此该反应被称作Ugi反应.随后他们将四组分Ugi反应中的酸替换成醇和CO2, 发展了CO2、醇、醛、胺和异腈的五组分Ugi反应[64]. 2014年, Meier等[65]基于该反应发展了CO2参与的新型聚合反应.如图 30所示, 作者将CO2、1, 12-二氨基十二烷、1, 6-二异腈基己烷、异丁醛和甲醇作为单体进行聚合得到了新型聚氨酯, 当加入氢氧化钾时, 聚氨酯主链会发生成环反应, 最终得到聚乙内酰脲.
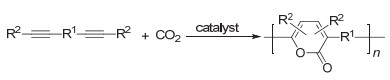
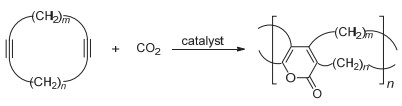
CO2和炔烃的有机反应已经有众多报道, 但是CO2作为单体和炔类单体的聚合反应却鲜有研究. 1992年, Tsuda等[66]报道了CO2和二炔类单体交替共聚用来制备聚(2-吡喃酮)的工作(图 31).该聚合反应的催化剂是零价镍, 溶剂是THF和乙腈的混合物, 制得的聚合物数均分子量为2100~17900 g/mol, 为防止发生分子内环化反应, 二炔单体中的R1基团设为芳基或者碳原子数为5个以上的脂肪烃基.在相同的催化条件下, CO2还能和环二炔单体进行共聚合制备梯形聚(2-吡喃酮)(图 32).这是首次报道的CO2和不饱和烷烃的共聚反应.所得聚合物具有优异的耐热性能, 氮气氛围下热分解温度为420 ℃.环二炔单体的反应活性和环的大小有关, 当m=n=6时, 其反应活性最高.
通过引入第三单体, CO2和炔类单体也可以进行三组分聚合反应. 1996年, Oi等[67]报道了碘化亚铜和碳酸钾催化的CO2、二炔和二卤代物的三组分聚合反应, 但得到聚炔酯的分子量和产率都不高.
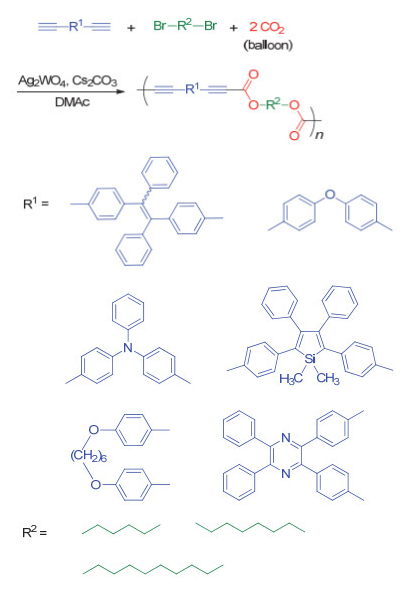
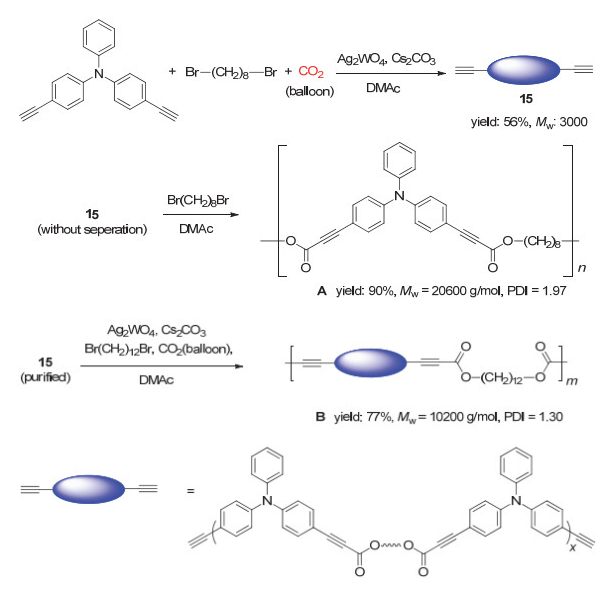
2018年, 我们课题组[68]借鉴He等的工作, 成功发展了钨酸银和碳酸铯催化的CO2、二炔和二卤代烷单体的三组分聚合反应(图 33), 并以最高可达95%的产率得到重均分子量最高可达31400 g/mol的聚炔酯.我们所选用的钨酸银是一种双功能催化剂, 其中, 银离子会与端炔基团反应生成炔银中间体, 而钨酸根离子对CO2具有一定的络合作用, 因此聚合反应可在常压下进行.由于制备的聚炔酯主链中炔基被邻近的酯基所活化, 从而具有较高的反应活性, 因此我们可以通过炔-胺点击反应进行后功能化, 最终以100%转化率得到区域和立构规整的聚烯胺酯(图 34).
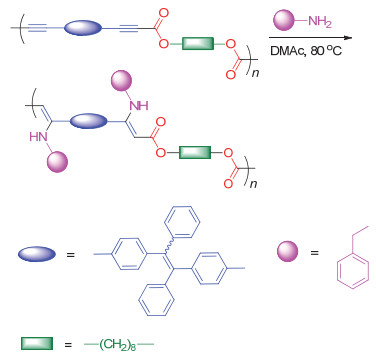
此外, 我们还利用该聚合反应的特点, 制备了末端为端炔基团的低分子量遥爪聚合物15.如图 35所示, 该遥爪聚合物仍然具有反应活性, 可以作为大分子单体进一步和CO2以及二卤代物单体聚合得到更高分子量的聚炔酯.由于该聚合反应具有很好的官能团耐受性, 因此可以将具有聚集诱导发光(AIE)特性的四苯基乙烯(TPE)、四苯基吡嗪(TPP)以及噻咯等基元引入聚合物主链中得到同样具有AIE特性的聚炔酯.所制备的聚合物薄膜绝对荧光量子效率最高可达61%.另外, 聚合物主链中由于含有大量酯基, 在碱性条件下具有很好的降解性能, 有望成为一类环境友好的新材料.
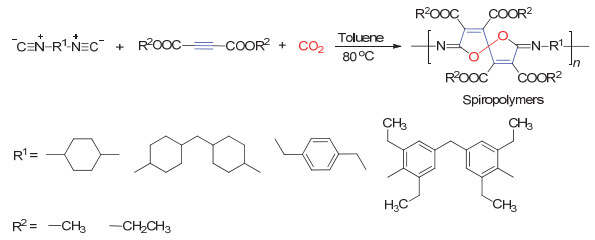
2018年, Dong等[69]报道了活化炔、CO2和二异腈单体的新型聚合反应.该反应在甲苯溶剂中, 80 ℃下, 不添加任何催化剂即可高效聚合得到螺旋聚合物(图 36).值得指出的是不同类型的单体聚合均有较好的结果, 产率最高可达88%, 重均分子量最高可达59000 g/mol.
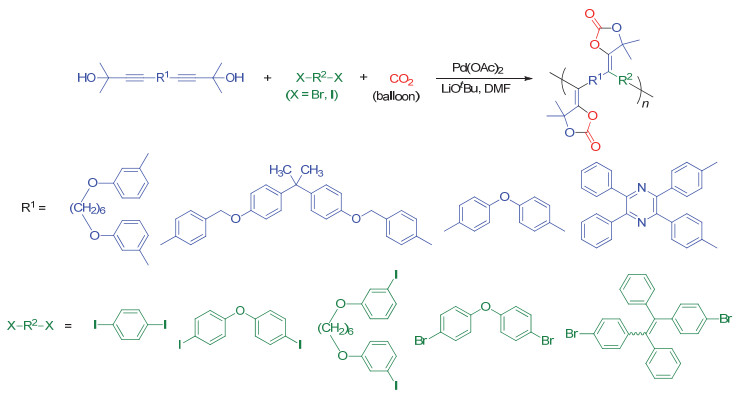
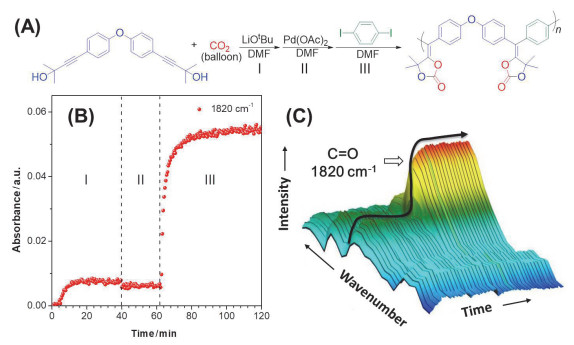
上文提到二官能度的五元环状碳酸酯已经被广泛应用于制备聚氨酯, 而聚五元环状碳酸酯还鲜有报道.已报道的文献中, 聚五元环状碳酸酯合成路线繁琐, 产率较低.因此, 发展一种新型简便、高效制备聚五元环状碳酸酯的聚合反应至关重要.近期, 我们[70]发展了CO2, 炔丙醇和卤代芳烃单体的三组分聚合反应, 可“一锅法”制备聚五元环状碳酸酯.该聚合反应具有很高的反应活性, 可在30 min内以95%的产率得到重均分子量高达42500 g/mol的聚合物(图 37).我们通过密度泛函理论和在线红外光谱技术(图 38)对聚合反应机理进行了探究.结果表明三种单体在聚合过程中存在协同效应, 卤代芳烃单体在环状碳酸酯成环过程中发挥了至关重要的作用.制备得到的聚合物可以与胺类化合物以接近100%的效率, 区域选择性开环得到聚氨酯(图 39).由于单体普适性广, 我们还制备了具有AIE和手性特性的聚合物.此外, 我们还利用多官能单体策略构筑了超支化聚合物以及多孔聚合物.
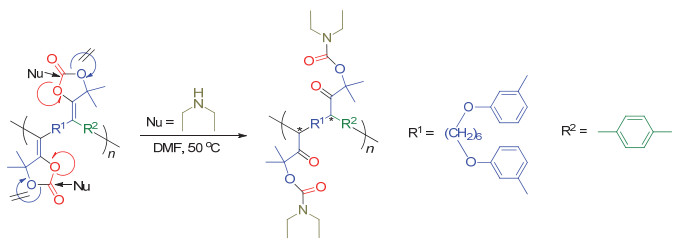
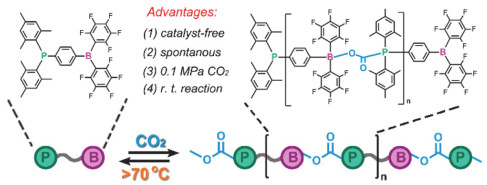
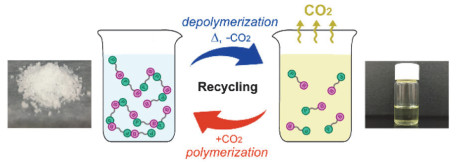
受阻路易斯酸碱对(frustrated Lewis pair, FLP)是指分子内或者混合体系中同时存在路易斯酸和路易斯碱两个位点, 由于较大的空间位阻使得这两个位点不能结合成路易斯酸碱加和物, 从而具有特殊的反应活性. Stephan等[71~73]已经证明了受阻路易斯酸碱对可以紧密结合CO2. 2019年, Yan等[74]利用这一特性, 将受阻路易斯酸碱对集中在一个分子上, 并将其作为单体和CO2在常压、室温和无催化剂的条件下进行聚合反应(图 40).作者通过基质辅助激光解吸电离飞行时间质谱法(MALDI-TOF)和尺寸排除色谱法(SEC)对聚合物分子量进行表征, 最高重均分子量可达54200 g/mol, 多分散性为2.51.该聚合物还可以在加热条件下释放CO2并解聚为初始单体.有意思的是, 解聚单体还可以与CO2再次聚合, 循环聚合-解聚多次之后仍具有很高的转化效率(图 41).
综上所述, 除了被广泛研究的CO2和环氧单体聚合反应之外, 近些年涌现出多种CO2参与的新型聚合反应.这些工作或者将CO2先转化为可聚合的单体, 再通过经典的聚合反应制备聚合物; 或者直接将CO2作为单体和不同种类单体直接聚合得到多种新型CO2基聚合物.这些工作都为CO2向高分子材料的转变开辟了新的途径, 为CO2的利用和转化提供了新的思路.
根据近年来的这些工作, 以下几个方面可能会是之后的研究重点: (1)发展更多CO2参与的新型聚合反应, 使反应过程更加绿色环保, 反应条件更加温和高效, 并进一步提升聚合物中CO2的含量; (2)关注CO2基高分子材料的性能, 除了热学和力学性能优异的材料可被用作工程塑料之外, 还可以开发其他方面的功能材料, 拓宽CO2基高分子材料的应用; (3)发展更多CO2聚合反应的催化体系, 降低催化剂成本, 提高反应效率, 实现催化剂的回收利用, 为大规模工业化生产提供可能性.
Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Nat. Commun. 2015, 6, 5933. doi: 10.1038/ncomms6933
Sakakura, T.; Choi, J.-C.; Yasuda, H. Chem. Rev. 2007, 107, 2365. doi: 10.1021/cr068357u
Du, P.; Su, T.; Luo, X.; Zhou, X.; Qin, Z.; Ji, H.; Chen, J. Chin. J. Chem. 2018, 36, 538. doi: 10.1002/cjoc.201700761
Zhang, F. H.; Liu, C.; Li, W.; Tian, G. L.; Xie, J. H.; Zhou, Q. L. Chin. J. Chem. 2018, 36, 1000. doi: 10.1002/cjoc.201800278
张文珍, 张宁, 郭春晓, 吕小兵, 有机化学, 2017, 37, 1309. doi: 10.6023/cjoc201701031Zhang, W.; Zhang, N.; Guo, C.; Lü, X. B. Chin. J. Org. Chem. 2017, 37, 1309. doi: 10.6023/cjoc201701031
Cao, Y.; He, X.; Wang, N.; Li, H. R.; He, L. N. Chin. J. Chem. 2018, 36, 644. doi: 10.1002/cjoc.201700742
Qi, C.; Yan, D.; Xiong, W.; Jiang, H. Chin. J. Chem. 2018, 36, 399. doi: 10.1002/cjoc.201700808
徐佩, 汪顺义, 方毅, 纪顺俊, 有机化学, 2018, 38, 1626. http://www.cnki.com.cn/Article/CJFDTotal-YJHU201807006.htmXu, P.; Wang, S. Y.; Fang, Y.; Ji, S. J. Chin. J. Org. Chem. 2018, 38, 1626. http://www.cnki.com.cn/Article/CJFDTotal-YJHU201807006.htm
Inoue, S.; Koinuma, H.; Tsuruta, T. J. Polym. Sci., Part C: Polym. Lett. 1969, 7, 287. doi: 10.1002/pol.1969.110070408
Lu, X. B.; Darensbourg, D. J. Chem. Soc. Rev. 2012, 41, 1462. doi: 10.1039/C1CS15142H
Lu, X. B.; Ren, W. M.; Wu, G. P. Acc. Chem. Res. 2012, 45, 1721. doi: 10.1021/ar300035z
Li, Y.; Zhang, Y. Y.; Liu, B.; Zhang, X. H. Chin. J. Polym. Sci. 2018, 36, 139. doi: 10.1007/s10118-018-2047-5
Li, Y.; Zhang, Y. Y.; Hu, L. F.; Zhang, X. H.; Du, B. Y.; Xu, J. T. Prog. Polym. Sci. 2018, 82, 120. doi: 10.1016/j.progpolymsci.2018.02.001
Darensbourg, D. J.; Mackiewicz, R. M.; Phelps, A. L.; Billodeaux, D. R. Acc. Chem. Res. 2004, 37, 836. doi: 10.1021/ar030240u
Darensbourg, D. J. Chem. Rev. 2007, 107, 2388. doi: 10.1021/cr068363q
Trott, G.; Saini, P. K.; Williams, C. K. Philos. Trans. R. Soc., A 2016, 374, 20150085. doi: 10.1098/rsta.2015.0085
Cheng, M.; Lobkovsky, E. B.; Coates, G. W. J. Am. Chem. Soc. 1998, 120, 11018. doi: 10.1021/ja982601k
Coates, G. W.; Moore, D. R. Angew. Chem., Int. Ed. 2004, 43, 6618. doi: 10.1002/anie.200460442
Sugimoto, H.; Inoue, S. J. Polym. Sci., Part A: Polym. Chem. 2004, 42, 5561. doi: 10.1002/pola.20319
Noh, E. K.; Na, S. J.; Sujith, S.; Kim, S. W.; Lee, B. Y. J. Am. Chem. Soc. 2007, 129, 8082. doi: 10.1021/ja071290n
Ohkawara, T.; Suzuki, K.; Nakano, K.; Mori, S.; Nozaki, K. J. Am. Chem. Soc. 2014, 136, 10728. doi: 10.1021/ja5046814
Wang, Y.; Qin, Y.; Wang, X.; Wang, F. ACS Catal. 2015, 5, 393. doi: 10.1021/cs501719v
Zhuo, C.; Qin, Y.; Wang, X.; Wang, F. Chin. J. Chem. 2018, 36, 299. doi: 10.1002/cjoc.201800019
Luo, M.; Li, Y.; Zhang, Y. Y.; Zhang, X. H. Polymer 2016, 82, 406. doi: 10.1016/j.polymer.2015.11.011
Sasaki, Y.; Inoue, Y.; Hashimoto, H. J. Chem. Soc., Chem. Commun. 1976, 15, 605.
Musco, A.; Perego, C.; Tartiari, V. Inorg. Chim. Acta 1978, 28, 147. doi: 10.1016/S0020-1693(00)87385-5
Behr, A.; Juszak, K.-D. J. Org. Chem. 1983, 255, 263. doi: 10.1016/0022-328X(83)87028-4
Braunstein, P.; Matt, D.; Nobel, D. J. Am. Chem. Soc. 1988, 110, 3207. doi: 10.1021/ja00218a033
Dinjus, E.; Leitner, W. Appl. Org. Chem. 1995, 9, 43. doi: 10.1002/aoc.590090108
Pitter, S.; Dinjus, E. J. Mol. Catal. Chem. 1997, 125, 39. doi: 10.1016/S1381-1169(97)00081-2
Beller, M.; Bolm, C. ChemInform 1998, 36, 211.
Nakano, R.; Ito, S.; Nozaki, K. Nat. Chem. 2014, 6, 325. doi: 10.1038/nchem.1882
Liu, M.; Sun, Y.; Liang, Y.; Lin, B. L. ACS Macro Lett. 2017, 1373.
Zhang, Y.; Xia, J.; Song, J.; Zhang, J.; Ni, X.; Jian, Z. Macromolecules 2019, 52, 2504. doi: 10.1021/acs.macromol.9b00195
Xu, Y.-C.; Zhou, H.; Sun, X.-Y.; Ren, W.-M.; Lu, X. B. Macromolecules 2016, 49, 5782. doi: 10.1021/acs.macromol.6b01372
Calo, V.; Nacci, A.; Monopoli, A.; Fanizzi, A. Org. Lett. 2002, 4, 2561. doi: 10.1021/ol026189w
Kihara, N.; Hara, N.; Endo, T. J. Org. Chem. 1993, 58, 6198. doi: 10.1021/jo00075a011
Song, J. L.; Zhang, Z. F.; Han, B. X.; Hu, S. Q.; Li, W. J.; Xie, Y. Green Chem. 2008, 10, 1337. doi: 10.1039/b815105a
Xie, Y.; Zhang, Z. F.; Jiang, T.; He, J. L.; Han, B. X.; Wu, T. B.; Ding, K. L. Angew. Chem. In. Ed. 2007, 46, 7255. doi: 10.1002/anie.200701467
Gu, Y.; Shi, F.; Deng, Y. J. Org. Chem. 2004, 69, 391. doi: 10.1021/jo0351365
Song, Q. W.; Yu, B.; Li, X. D.; Ma, R.; Diao, Z. F.; Li, R. G.; Li, W.; He, L. N. Green Chem. 2014, 16, 1633. doi: 10.1039/c3gc42406e
Song, Q.-W.; He, L.-N. Adv. Synth. Catal. 2016, 358, 1251. doi: 10.1002/adsc.201500639
Eghbali, N.; Li, C. J. Green Chem. 2007, 9, 213. doi: 10.1039/B615612F
Wang, J. L.; Wang, J. Q.; He, L. N.; Dou, X. Y.; Wu, F. Green Chem. 2008, 10, 1218. doi: 10.1039/b807108j
Gennen, S.; Grignard, B.; Tassaing, T.; Jerome, C.; Detrembleur, C. Angew. Chem., Int. Ed. 2017, 56, 10394. doi: 10.1002/anie.201704467
Gennen, S.; Grignard, B.; Jérôme, C.; Detrembleur, C. Adv. Synth. Catal. 2019, 361, 355. doi: 10.1002/adsc.201801230
Liu, B.; Zhang, X. H. Gen. Chem. 2018, 4, 180007. doi: 10.21127/yaoyigc20180007
Ouhib, F.; Grignard, B.; Van Den Broeck, E.; Luxen, A.; Robeyns, K.; Van Speybroeck, V.; Jerome, C.; Detrembleur, C. Angew. Chem., Int. Ed. 2019, 58, 11768. doi: 10.1002/anie.201905969
Banerjee, A.; Dick, G. R.; Yoshino, T.; Kanan, M. W. Nature 2016, 531, 215. doi: 10.1038/nature17185
Kadokawa, J.; Habu, H.; Fukamachi, S.; Karasu, M.; Tagaya, H.; Chiba, K. Macromol. Rapid Commun. 1998, 19, 657. doi: 10.1002/(SICI)1521-3927(19981201)19:12<657::AID-MARC657>3.0.CO;2-R
Tamura, M.; Ito, K.; Honda, M.; Nakagawa, Y.; Sugimoto, H.; Tomishige, K. Sci. Rep. 2016, 6, 24038. doi: 10.1038/srep24038
Oi, S.; Nemoto, K.; Matsuno, S.; Inoue, Y. Macromol. Rapid Commun. 1994, 15, 133. doi: 10.1002/marc.1994.030150207
Chen, Z. L.; Hadjichristidis, N.; Feng, X. S.; Gnanou, Y. Polym. Chem. 2016, 7, 4944. doi: 10.1039/C6PY00783J
Yamazaki, N.; Higashi, F.; Iguchi, T. J. Polym. Sci. Polym. Lett. Ed. 1974, 12, 517. doi: 10.1002/pol.1974.130120904
Yamazaki, N.; Nakahama, S.; Higashi, F. Ind. Eng. Chem. Prod. Res. Dev. 1979, 18, 249.
Wu, C. Y.; Wang, J. Y.; Chang, P. J.; Cheng, H. Y.; Yu, Y. C.; Wu, Z. J.; Dong, D. W.; Zhao, F. Y. Phys. Chem. Chem. Phys. 2012, 14, 464. doi: 10.1039/C1CP23332G
Wu, P.-X.; Cheng, H.-Y.; Shi, R.-H.; Jiang, S.; Wu, Q.-F.; Zhang, C.; Arai, M.; Zhao, F.-Y. Adv. Synth. Catal. 2019, 361, 317. doi: 10.1002/adsc.201801134
Chen, Z.; Hadjichristidis, N.; Feng, X.; Gnanou, Y. Macromolecules 2017, 50, 2320. doi: 10.1021/acs.macromol.7b00142
Yoo, W. J.; Li, C. J. Adv. Synth. Catal. 2008, 350, 1503. doi: 10.1002/adsc.200800232
Zhao, J. W.; Huang, H. W.; Qi, C. R.; Jiang, H. F. Eur. J. Org. Chem. 2012, 29, 5665.
Yu, B.; Cheng, B. B.; Liu, W. Q.; Li, W.; Wang, S. S.; Cao, J.; Hu, C. W. Adv. Synth. Catal. 2016, 358, 90. doi: 10.1002/adsc.201500921
Teffahi, D.; Hocine, S.; Li, C. J. Lett. Org. Chem. 2012, 9, 585. doi: 10.2174/157017812802850221
Ugi, I.; Meyr, R.; Fetzer, U. Angew. Chem. 1959, 71, 386.
Ugi, I.; Steinbruckner, C. Chem. Ber. 1961, 94, 2802. doi: 10.1002/cber.19610941032
Sehlinger, A.; Schneider, R.; Meier, M. A. Macromol. Rapid Commun. 2014, 35, 1866.
Tsuda, T.; Maruta, K.; Kitaike, Y. J. Am. Chem. Soc. 1992, 114, 1498. doi: 10.1021/ja00030a065
Oi, S.; Fukue, Y.; Nemoto, K.; Inoue, Y. Macromolecules 1996, 29, 2694. doi: 10.1021/ma9514784
Song, B.; He, B.; Qin, A.; Tang, B. Z. Macromolecules 2018, 51, 42. doi: 10.1021/acs.macromol.7b02109
Fu, W.; Dong, L.; Shi, J.; Tong, B.; Cai, Z.; Zhi, J.; Dong, Y. Polym. Chem. 2018, 9, 5543. doi: 10.1039/C8PY01336E
Song, B.; Bai, T.; Xu, X.; Chen, X.; Liu, D.; Guo, J.; Qin, A.; Ling, J.; Tang, B. Z. Macromolecules 2019, 52, 5546. doi: 10.1021/acs.macromol.9b00898
Mömming, C. M.; Otten, E.; Kehr, G.; Fröhlich, R.; Grimme, S.; Stephan, D. W.; Erker, G. Angew. Chem., Int. Ed. 2009, 48, 6643. doi: 10.1002/anie.200901636
Stephan, D. W.; Erker, G. Angew. Chem., Int. Ed. 2010, 49, 46. doi: 10.1002/anie.200903708
Stephan, D. W. Science 2016, 354, aaf7229. doi: 10.1126/science.aaf7229
Liu, R.; Liu, X.; Ouyang, K.; Yan, Q. ACS Macro Lett. 2019, 8, 200. doi: 10.1021/acsmacrolett.9b00066

图 4 CO2和1, 3-二烯单体“一锅、两步”法二/三元共聚反应
Figure 4 "One-pot, two-step" co/terpolymerization of CO2 and 1, 3-dienes

图 6 从丁二烯和CO2原料制备得到多烯单体5
Figure 6 Synthesis of the monomer 5 derived from 1, 3-butadiene and CO2 feedstocks

图 8 CO2和2-丁炔制备α-亚甲基-β丁内酯路线图
Figure 8 Synthetic route of α-methylene-β-butyrolactone from CO2 and 2-butyne

图 9 α-亚甲基-β丁内酯通过Salen-Al催化开环聚合制备间同立构聚酯
Figure 9 Synthesis of syndiotactic polyester by ring-opening polymerization of α-methylene-β-butyrolactone using Salen-Al complexes (Reproduced with permission from ref. 35. Copyright 2016, American Chemical Society)

图 10 CO2和环氧化物反应制备五元环状碳酸酯
Figure 10 Synthesis of five-membered cyclic carbonates by the reaction of CO2 and epoxides

图 11 CO2和炔丙醇反应制备五元环状碳酸酯
Figure 11 Synthesis of five-membered cyclic carbonates by the reaction of CO2 and propargyl alcohol

图 12 CO2和烯烃“一锅、两步”法制备五元环状碳酸酯
Figure 12 Synthesis of five-membered cyclic carbonates by "one-pot, two-step" reaction of CO2 and olefins

图 13 环状碳酸酯、醇、胺类单体以及相应的聚合物
Figure 13 Cyclic carbonate, alcohol and amine monomers and their corresponding polymers (Reproduced with permission from ref. 45. Copyright 2017, Wiley-VCH)

图 14 可水解CO2基聚合物的合成
Figure 14 The synthesis of hydrolysable CO2-based polymer (Reproduced with permission from ref. 46. Copyright 2019, Wiley-VCH)

图 15 紫外光诱导的硫醇和五元环状碳酸酯的反应
Figure 15 Ultraviolet (UV) light-induced reaction of thiols and five-membered cyclic carbonates

图 16 DBU诱导的硫醇和五元环状碳酸酯的串联反应
Figure 16 DBU-induced tandem reaction of thiols and five-membered cyclic carbonates

图 17 硫醇和环状碳酸酯单体的聚合反应
Figure 17 The polymerization of thiol and cyclic carbonate monomers (Reproduced with permission from ref. 48. Copyright 2019, Wiley-VCH)

图 18 FDCA合成路线图
Figure 18 The synthetic routes of FDCA (Reproduced with permission from ref. 49. Copyright 2016, Nature)

图 19 FDCA产物分离和铯离子回收
Figure 19 Product of FDCA isolation and Cs+ recovery (Reproduced with permission from ref. 49. Copyright 2016, Nature)

图 23 聚脲的形态示意图
Figure 23 The schematic picture of the morphology of polyurea (Reproduced with permission from ref. 56. Copyright 2012, Royal Society of Chemistry)

图 24 DBU催化的CO2和TOTDDA的聚合反应
Figure 24 DBU catalyzed polymerization of CO2 and TOTDDA (Reproduced with permission from ref. 57. Copyright 2019, Wiley-VCH)

图 25 聚合反应机理
Figure 25 Polymerization mechanisms (Reproduced with permission from ref. 57. Copyright 2019, Wiley-VCH)

图 26 CO2、二胺和二卤代物的三组分聚合反应
Figure 26 Three-component polymerization of CO2, diamines and dihalides (Reproduced with permission from ref. 58. Copyright 2017, American Chemical Society)

图 27 在标准反应条件下的控制实验
Figure 27 Control experiment under standard reaction conditions (Reproduced with permission from ref. 58. Copyright 2017, American Chemical Society)

图 28 CO2、炔、醛和胺的四组分反应
Figure 28 Four component reaction of aldehydes, amines, alkynes and CO2 (Reproduced with permission from ref. 59. Copyright 2008, Wiley-VCH)

图 29 CO2、二铵盐、二炔和醛的四组分聚合反应
Figure 29 Four component polymerization of CO2, di(ammonium salt), diyne and aldehyde

图 30 CO2、1, 12-二氨基十二烷、1, 6-二异腈基己烷、异丁醛和甲醇的五组分聚合反应
Figure 30 Five component polymerization of CO2, 1, 12-diamino- dodecane, 1, 6-diisocyanohexane, isobutyraldehyde and methanol (Reproduced with permission from ref. 65. Copyright 2014, Wiley-VCH)

图 33 CO2、二炔和二卤代物的聚合反应
Figure 33 The polymerization of CO2, diynes and dihalides (Reproduced with permission from ref. 68. Copyright 2018, American Chemical Society)

图 34 聚炔酯的后修饰
Figure 34 Post-polymerization modification (PPM) of poly(alkynoate)s (Reproduced with permission from ref. 68. Copyright 2018, American Chemical Society)

图 35 遥爪聚合物的合成以及聚合
Figure 35 Synthesis and polymerization of telechelic polymers (Reproduced with permission from ref. 68. Copyright 2018, American Chemical Society)

图 36 CO2、活化炔和二异腈单体的三组分聚合反应
Figure 36 Three component polymerization of CO2, activated alkynes and diisocyanides (Reproduced with permission from ref. 69. Copyright 2018, Royal Society of Chemistry)

图 37 CO2、炔丙醇和卤代芳烃的三组分聚合反应
Figure 37 Three component polymerization of CO2, bis(propargylic alcohol)s and aryl dihalides (Reproduced with permission from ref. 70. Copyright 2019, American Chemical Society)

图 38 在线红外监测三步反应
Figure 38 Monitoring three-step reaction via in situ FT-IR (Reproduced with permission from ref. 70. Copyright 2019, American Chemical Society)

图 39 聚合物的区域选择性开环
Figure 39 Regioselective ring opening of polymers (Reproduced with permission from ref. 70. Copyright 2019, American Chemical Society)

图 40 CO2和FLP单体的聚合反应
Figure 40 The polymerization of CO2 and FLP monomer (Reproduced with permission from ref. 74. Copyright 2019, American Chemical Society)

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:









