图 1.
自交联PEI-PSF的合成示意图
Figure 1.
Scheme for the synthesis of self-crosslinked PEI-PSF membrane

质子交换膜燃料电池(Proton Exchange Membrane Fuel Cell, PEMFC)作为一种清洁能源的技术, 具有高效、环保和功率密度高等优点[1~9], 在环境问题日益严峻的今天已经引起了人们的广泛关注.高温质子交换膜燃料电池(High Temperature Proton Exchange Membrane Fuel Cells, HT-PEMFCs)运行温度区间为100~200℃[10], 具有电极反应活性高[11, 12]、催化剂抗CO中毒能力强[13]以及水、热管理简单[14]等优点.作为HT-PEMFCs的关键材料, 高温质子交换膜(High Temperature Proton Exchange Membrane, HT-PEM)直接决定燃料电池的输出性能和使用寿命.
目前, 磷酸掺杂聚苯并咪唑膜[15](PA/PBI)因具有良好的热稳定性和优异的质子电导率而成为研究最为广泛的HT-PEM.然而, PBI合成步骤耗时繁琐且成本较为高昂, 成为限制高温质子交换膜燃料电池发展的重要因素之一.近年来, 含吡啶的主链或将含N官能团接枝到聚芳醚酮/砜等高分子膜材料在高温质子膜领域崭露头角, 迅速成为关注热点. Xu等[16]采用聚乙烯基吡咯烷酮(polyvinylpyrrolidone, PVP)作为磷酸吸附基体, 以聚偏氟乙烯(polyvinylidene fluoride, PVDF)为骨架材料合成了性能优异的高温质子交换膜.研究表明此类膜磷酸掺杂水平随着PVP含量的增加而提高.当PVP的质量百分含量为80%时, 磷酸掺杂水平(ADL)为一个重复单元包含9.1个磷酸分子, 质子电导率在200 ℃时达到9.3×10-2 S•cm-1, 用此膜组装的电池在180℃的输出性能为530 mW•cm-2.然而, 磷酸同时也作为塑化剂, 削弱了高分子链之间的范德华力, 大幅降低了膜的力学性能.
近年来, 研究者通过优化聚合物分子结构以及其他改性方法以平衡高温质子交换膜的质子电导率和机械性能.化学交联改性是提高质子交换膜化学稳定性、机械性能和长期耐久性的有效方法[17]. Pan等[18]将磺化苯并咪唑与3, 3', 5, 5'-四甲基-二苯基二缩水甘油醚进行化学交联, 与未交联膜相比, 制备的交联膜的机械性能以及氧化和水解稳定性得到显著改善.其中交联膜的拉伸强度在53.6~108.4 MPa范围内, 最高达到未交联膜的1.6倍; 其在80 ℃下通过Fenton试剂(3% H2O2, 3 mg/L Fe2+)测试抗自由基氧化稳定性为未交联膜的4倍. Zhang等[19]合成了一系列含三甲氧基硅烷基柔性侧链的可交联磺化聚酰亚胺, 交联膜拉伸强度达到77.6 MPa, 远高于未交联膜的48.3 MPa; 并且与原始膜相比, 交联膜的氧化和水解稳定性显著提高.然而, 传统的化学交联方法因为官能团与交联剂化学反应而减少了磷酸吸附位点的数量, 导致交联膜离子电导率下降明显. Yang等[20]将六氟亚丙基聚苯并咪唑和氯甲基聚砜进行共价交联反应制备了交联的高温质子交换膜, 当交联度由4.6%增加到18.5%时, 其磷酸掺杂水平由13.5降低到8.7.因此, 交联剂的选择对平衡高温质子交换膜力学性能和质子电导率尤为重要.
平均分子量为200的枝状聚乙烯亚胺(Polyethylene- imine, PEI)是一种结构单元含有4个伯仲叔胺含N官能团组成的弱碱性水溶性高分子, 可以同时提供多个发生交联反应的活性位点和吸收磷酸的位点[21, 22], 其结构式如图 1所示.因此, 本文将PEI作为自交联剂添加至氯甲基化聚砜(Polysulfone, PSF)的聚合物溶液中.成膜过程中PEI上的胺基基团与聚砜侧链上的苄氯基团发生反应, 从而形成自交联结构的聚合物膜.采用核磁共振(NMR)、傅里叶变换红外光谱(FT-IR)和X-射线光电子能谱(XPS)对聚合物的交联结构进行表征, 研究聚砜的氯甲基化程度对所制备的高温质子交换膜的力学性能和离子传导率的影响, 并在电池器件中考察所制备的自交联结构膜材料的电池输出性能和电池稳定性.
PEI-PSF膜的制备路线如图 1所示.本研究采用PSF为聚合物主体材料, 通过氯甲基化反应将氯甲基接枝到苯环上, 生成氯甲基化聚砜(chloromethylated polysulfone, CMPSF). CMPSF中苄氯基团与支链结构PEI中的伯胺和仲胺基团反应生成自交联型PEI-PSF膜.
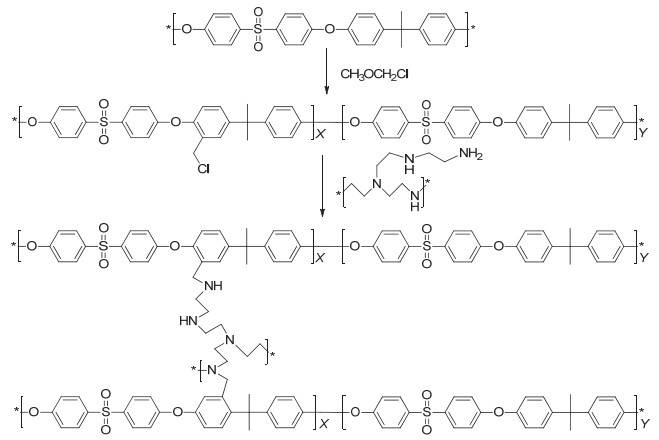
图 2a为PSF的核磁谱图, 其中Ar-H存在三种不同的化学环境, 对应图中数字1~4, 其化学位移分布在δ 8~6.8. C-(CH3)2化学位移δ为1.6, 对应数字5.经过氯甲基化反应后, Ar-H的化学环境稍微改变, 如图 2b所示, 其具体结果如下: δ 7.8 (O2S-Ar-H meta position), 7.2 (Me2C-Ar-H ortho position), 7.0 (O2S-Ar-H ortho position), 6.9 (Me2C-Ar-H meta position)[23].同时, δ 4.6处出现了氯甲基上CH2-Cl峰, 证明聚砜成功进行氯甲基化反应.聚砜的氯甲基化程度(氯甲基官能团与聚砜高分子链重复单元的摩尔百分比, GD)通过核磁共振氢谱中峰5和峰9的峰面积的比值可以计算得到, 具体计算方法参见支持信息.将所制备的具有不同氯甲基化程度的聚砜膜标记为CMPSF-x, 其中x代表PSF的氯甲基化程度.
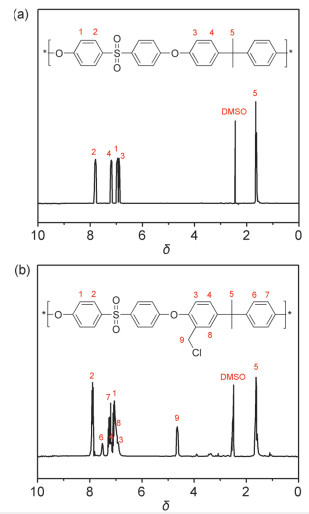
由于交联后的PEI-PSF膜难溶于有机溶剂, 因此无法采用1H NMR进一步对所制备的PEI-PSF聚合物进行表征.为此, 本研究采用傅里叶变换红外光谱和X-射线光电子能谱对PEI-PSF膜进行了表征. 图 3为PSF、CMPSF和PEI-PSF膜傅里叶变换红外光谱图.可以看出, PSF的FT-IR谱图上均出现了芳环氢峰(1011 cm-1), O=S=O的对称(1151和1175 cm-1)和不对称伸缩振动峰(1322和1293 cm-1)及C—O—C的伸缩振动峰(1235 cm-1)[24].经过氯甲基化反应后, CMPSF红外谱线上出现了C—Cl的伸缩振动峰(761 cm-1)[25], 这进一步证明聚砜已成功进行了氯甲基化反应.交联剂PEI加入后, 该处的峰消失, 说明苄氯基团全部参与了自交联反应.
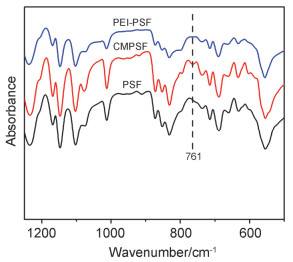
CMPSF上的苄氯基团与PEI发生交联反应之后造成Cl元素和N元素化学环境发生变化, 如图 4所示. 图 4a为CMPSF-58和PEI-PSF-58膜的Cl元素XPS精细谱.对于未加入PEI的CMPSF-58膜, 出现了Cl 2p的峰, 分别由Cl 2p3/2的199.8 eV和Cl 2p1/2的201.5 eV峰组成, 这是共价键合的氯物种的特征峰[26].而与PEI发生交联反应后形成的PEI-PSF-58中Cl元素的能谱峰基本消失, 说明CMPSF中以共价键存在的Cl元素基本上全部转为离子态的氯离子, 并通过离子交换后被交换出膜材料. 图 4b为CMPSF和PEI-PSF膜的N元素XPS精细谱.由图 4b可知, PEI-PSF中出现了Ⅰ、Ⅱ两个N 1s峰, 位于399.07 eV处的Ⅰ代表着仲胺N的特征峰, 而位于400.22 eV处的Ⅱ代表着叔胺N的特征峰[27].峰Ⅰ、Ⅱ面积基本相同, 即交联后叔胺和仲胺的数量基本相等, 表明苄氯基团主要是和PEI中的伯胺和仲胺发生交联反应.另外, 未见伯胺N的特征峰, 说明了PEI中伯胺N全部与CMPSF-58中的苄氯基团发生了交联反应.
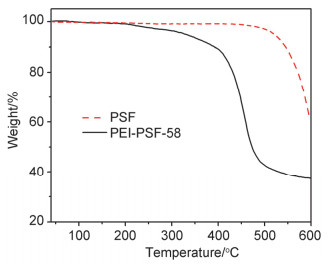
图 5为PSF和PEI-PSF-58在氮气气氛下的热重分析曲线. PSF作为一种热稳定性优异的工程塑料[28~30], 其起始热分解温度为480 ℃左右.所制备的PEI-PSF-58膜材料在210 ℃后开始失重, 主要是由于侧链PEI的热分解所致[31].因此, 所制备的PEI-PSF可以满足高温质子交换膜150 ℃左右的温度范围使用要求.
在膜制备过程中发现, 当氯甲基化程度大于60%以后制备的交联膜变脆, 难以得到大片完整的膜材料用于后续测试, 因此本研究中分别采用氯甲基化程度为40%, 50%及58%的CMPSF来制备自交联膜, 并研究其相关性能.
图 6a为PEI-PSF膜的吸酸量和体积溶胀率随氯甲基化程度的变化关系.与传统交联高温质子交换膜吸酸量随GD的提高而降低不同[32], PEI-PSF膜的吸酸量随着GD增加而快速提升.这是由于GD的提高, 与CMPSF发生交联反应的PEI含量增加, 进而使得PEI-PSF中含N功能基团的数量急剧增加, 致使复合膜的吸酸量也随之快速提高.当CMPSF的GD从40%提升至58%时, PEI-PSF膜的磷酸掺杂水平从1 mol重复单元包含8.7 mol磷酸分子提高至17.1 mol.同时, PEI-PSF-x浸泡浓磷酸后体积溶胀率也随GD的升高而增大.这是由于GD增加使得磷酸吸附位点增多, 导致磷酸掺杂水平提高, 进而体积溶胀率增大.此外, 磷酸掺杂水平变化趋势与聚合物膜体积溶胀变化趋势相同, 这是因为聚合物膜溶胀的体积即为吸附磷酸占据的体积.
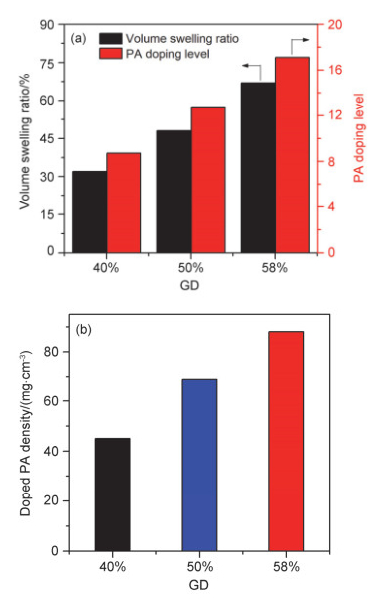
图 6b为PEI-PSF-x浸泡磷酸后的单位体积磷酸含量.当GD从40%提升至58%时, 交联PEI-PSF膜单位体积磷酸含量由45 mg•cm-3增加至88 mg•cm-3.尽管PEI-PSF-x的体积溶胀随着吸酸量的增加同时增大, 但单位体积的磷酸含量依然明显提升.
高温质子交换膜应有良好的力学性能以保证燃料电池长期运行的稳定性.对于传统磷酸掺杂型高温质子交换膜材料而言, 未浸泡浓磷酸的聚合物膜具有良好的力学性能; 但是浸泡磷酸之后, 由于磷酸分子对聚合物高分子链的塑化作用降低了高分子链间的范德华力, 导致聚合物膜力学性能的降低[33]. 图 7a为PEI-PSF-x浸泡磷酸后的应力应变曲线.由图 7a可知, 随着聚合物氯甲基化程度增加, PEI-PSF膜的杨氏模量和拉伸强度逐渐降低, 断裂伸长率逐渐增加.其中, PEI-PSF-40膜的杨氏模量最高, 为1.1 GPa, 拉伸强度为58 MPa, 断裂伸长率为17.8%;当GD增加至58时, PEI-PSF-58的杨氏模量和拉伸强度均有所降低, 但其杨氏模量仍可达到0.6 GPa, 拉伸强度仍可维持在25 MPa以上, 完全可以满足电池组装测试时所需的力学性能要求.
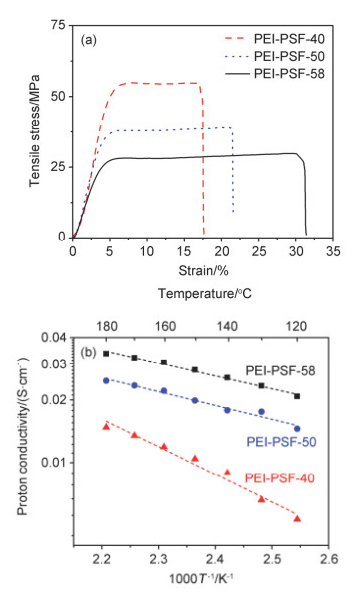
质子电导率是高温质子交换膜性能最重要的参数之一. 图 7b为磷酸掺杂后PEI-PSF-x膜质子电导率随温度的变化曲线. PEI-PSF-x的离子电导率均随着温度的上升而提高, 而且随着氯甲基化程度的增加而提高. PEI-PSF-40, PEI-PSF-50和PEI-PSF-58在120 ℃无水条件下的质子电导率分别为5.3×10-3 S•cm-1, 1.5×10-2 S•cm-1和2.1×10-2 S•cm-1, 而升温至180 ℃时分别提升至1.5×10-2 S•cm-1, 2.5×10-2 S•cm-1和3.4×10-2 S•cm-1.这是因为温度的上升加快了质子传输速率[34].此外, 随着GD提高PEI-PSF膜的磷酸吸附量增加, 单位体积中的磷酸含量增加(如图 6b), 从而缩短磷酸分子间质子的传递距离, 同时形成更多的质子传递通路[35].同时根据阿伦尼乌斯公式计算得到磷酸掺杂的PEI-PSF-40, PEI-PSF-50和PEI-PSF-58膜的质子传导活化能分别为25.1 kJ•mol-1, 13.1 kJ•mol-1和11.81 kJ• mol-1, 表明交联膜的质子传导活化能随GD的增加而降低.此外, PEI-PSF-50和PEI-PSF-58膜的质子传导活化能与85%浓磷酸的活化能14.3 kJ•mol-1接近[36], 表明在高GD的PEI-PSF膜中, 质子的传输行为类似于其在浓磷酸中的传输行为, 主要是沿着磷酸阴离子路线(
表 1比较了本研究所制备的PEI-PSF-58与文献中报道的PSF基的高温质子交换膜磷酸吸收量、离子电导率和拉伸强度等参数.与其他PSF基膜相比, PEIPSF-58兼具优异的力学性能和较高的质子传导率.
 下载:
导出CSV
下载:
导出CSV
| PA doping sample |
PA uptake/ wt% |
σ/(mS• cm-1) @160℃ |
Tensile strength/ MPa |
Power Densitya/ (mW•cm−2)@150 ℃ |
Ref. |
| EtPSU | 168.5 | 20b | 5.1c | 204 | [38] |
| ImPSF | 132 | 34.5d | 2.4 | — | [39] |
| PSU-DMIm | ADL=13 | 38 | 7.2 | 269e | [40] |
| PSf-Im | 235.8 | 48 | 4.2 | — | [41] |
| TDPA-PSF-51 | 79.7 | 17 | 8.4 | 160 | [25] |
| TDPA-PSF-75 | 159.8 | 35 | 3.7 | 310 | [25] |
| PEI-PSF-58 | 122 | 27.9b | 26.1 | 200 | This work |
| a H2和O2下测试, b 150 ℃下测试, c 130 ℃下测试, d 140 ℃下测试, e 160℃下测试. | |||||
将磷酸掺杂PEI-PSF-x膜作为电解膜组装高温质子交换膜燃料电池, 考察其在实际电池中应用的可行性. 图 8a为采用磷酸掺杂PEI-PSF-x膜的燃料电池在150℃下的极化曲线和功率密度曲线.从图中可以看出, 基于PEI-PSF-x组装的燃料电池的开路电压均在0.9 V以上, 说明膜的气体渗透率较低[42].此外, 随着GD从40%提高到58%, 燃料电池在150 ℃时的最大输出功率密度从31 mW•cm-2迅速提升到200 mW•cm-2, 这主要归因于PEI-PSF-58膜具有更高的质子电导率, 即具有更低电池内阻. 图 8b为基于PEI-PSF-58的燃料电池在不同温度下的极化曲线.电池工作温度由120 ℃升至150 ℃, 燃料电池的最大输出功率密度从174 mW•cm-2提高到200 mW•cm-2, 这归因于温度的升高提升了电极电化学反应动力学速度, 以及提升了膜的质子传输效率和降低了膜电阻.
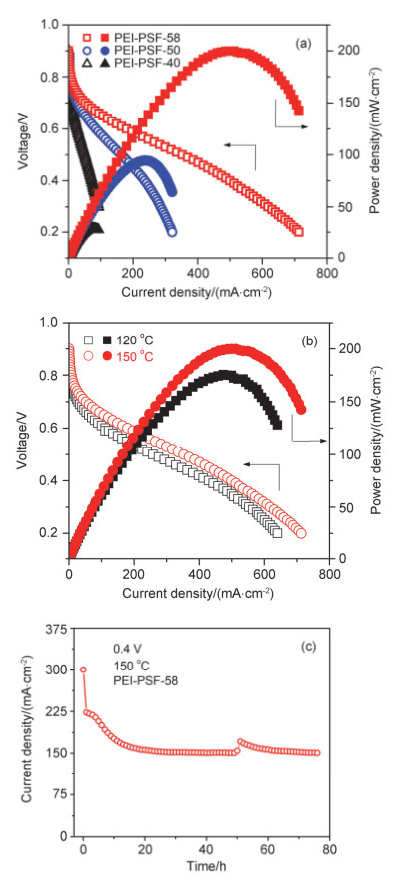
图 8c为基于PEI-PSF-58的燃料电池在150 ℃和0.4 V恒压条件下放电稳定性曲线.由图可知, 燃料电池在放电初期有一个较大的衰减, 电流密度从350 mA•cm-2迅速降低到223 mA•cm-2, 这是由于在放电初始改变电压产生充电电流导致.随后, 电流密度缓慢衰减, 主要原因为燃料电池催化剂在高温强酸条件下引起的腐蚀和团聚造成的[43, 44].在随后的18~80 h的测试时间内, 放电电流密度趋于一个相对稳定的数值, 约为150 mA•cm-2.以上结果初步表明磷酸掺杂的PEI-PSF膜在高温质子交换膜燃料电池工作环境下展现出了良好的工作稳定性.
为了获得力学性能与质子电导率兼顾的高温质子交换膜, 本研究通过氯甲基化和自交联反应制备了一种自交联结构的PEI-PSF聚合物膜. PEI既作为交联剂提高聚合物膜力学性能, 同时作为功能基团提供磷酸吸附位点. FT-IR及XPS证明功能分子PEI成功地交联在PSF高分子链之间.浸泡磷酸之后, PEI-PSF膜的磷酸吸附量及高温质子电导率随GD的提高而增加, 其中PEI-PSF-58的磷酸吸附量为122 wt%, 质子电导率在180℃时达到最高, 为3.4×10-2 S•cm-1.力学性能测试表明, 磷酸掺杂PEI-PSF膜拉伸强度随GD的升高而降低, 而PEI-PSF-58的杨氏模量仍然高达0.6 GPa, 远高于同类型磷酸掺杂高温质子交换膜.单电池测试表明, PEI-PSF能够良好地应用于高温燃料电池中, 并且以PEI-PSF-58膜组装的燃料电池在150 ℃不加湿条件下最大输出功率密度达到200 mW•cm-2, 而且具有良好的放电稳定性.
本研究中的自交联PEI-PSF膜的制备方法如下:将10 g聚砜溶解于100 mL二氯乙烷中, 置于35 ℃油浴锅中, 待聚合物完全溶解后依次加入1 g锌粉和3.5 mL三氟乙酸作为催化剂, 之后采用恒压漏斗缓慢加入氯甲基氯甲醚, 反应时间为1 h.控制加入氯甲基氯甲醚的量获得不同GD的氯甲基化聚砜.反应完成后, 将溶液边搅拌边倒入甲醇中, CMPSF析出.将产物依次过滤、水洗和干燥后, 再溶解于二氯乙烷中, 再次析出洗涤, 反复三次.将最终洗净的产物置于真空干燥箱中40 ℃干燥24 h去除水分、甲醇和二氯乙烷, 得到干燥样品.称取一定质量的CMPSF聚合物溶于DMF中, 按照1:2的结构单元当量比(PEI结构单元为2)加入平均分子量为200的PEI, 室温下持续搅拌, 待聚合物完全溶解混合均匀后将溶液浇铸于平整的玻璃槽中, 置于真空干燥箱中70℃干燥24 h, 溶剂挥发干净后得到PEI-PSF聚合物膜.
正文中的试剂、仪器以及膜的性能表征等信息详见“支持信息”(Supporting Information)中.
Wang, M.; Chen, M.; Yang, Z. Y.; Liu, G. C.; Lee, J. K.; Yang, W.; Wang, X. D. Energy Convers. Manage. 2019, 191, 132. doi: 10.1016/j.enconman.2019.04.014
彭思侃, 徐鑫, 张劲, 刘祎阳, 卢善富, 相艳, 化学学报, 2015, 73, 137. doi: 10.3866/PKU.WHXB201411171Peng, S.-K.; Xu, X.; Zhang, J.; Liu, Y.-Y.; Lu, S.-F.; Xiang, Y. Acta Chim. Sinica 2015, 73, 137. doi: 10.3866/PKU.WHXB201411171
黄文姣, 张浩宇, 胡硕真, 钮东方, 张新胜, 化学学报, 2018, 76, 723. doi: 10.6023/A18060231Huang, W.-J.; Zhang, H.-Y.; Hu, S.-Z.; Niu, D.-F.; Zhang, X.-S. Acta Chim. Sinica 2018, 76, 723. doi: 10.6023/A18060231
钟国玉, 王红娟, 余皓, 彭峰, 化学学报, 2017, 75, 943. doi: 10.6023/A17040183Zhong, G.-Y.; Wang, H.-J.; Yu, H.; Peng, F. Acta Chim. Sinica 2017, 75, 943. doi: 10.6023/A17040183
朱红, 洛明川, 蔡业政, 孙照楠, 物理化学学报, 2016, 32, 2462. doi: 10.3866/PKU.WHXB201606293Zhu, H.; Luo, M.-C.; Cai, Y.-Z.; Sun, Z.-N. Acta Phys-Chim. Sinica 2016, 32, 2462. doi: 10.3866/PKU.WHXB201606293
崔丽瑞, 张劲, 孙一焱, 卢善富, 相艳, 化学学报, 2019, 77, 47. doi: 10.7503/cjcu20180421Cui, L.-R.; Zhang, J.; Sun, Y.-Y.; Lu, S.-F.; Xiang, Y. Acta Chim. Sinica 2019, 77, 47. doi: 10.7503/cjcu20180421
Li, H.; Li, L.; Chen, S.; Zhang, Y.; Li, G. Chinese J. Chem. 2017, 35, 903. doi: 10.1002/cjoc.201600740
陈鑫, 鄢慧君, 夏定国, 化学学报, 2017, 75, 189. doi: 10.3969/j.issn.0253-2409.2017.02.008Chen, X.; Yan, H.-J.; Xia, D.-G. Acta Chim. Sinica 2017, 75, 189. doi: 10.3969/j.issn.0253-2409.2017.02.008
徐鑫, 彭思侃, 张劲, 卢善富, 相艳, 化学学报, 2016, 74, 271. doi: 10.6023/A15100687Xu, X.; Peng, S.-K.; Zhang, J.; Lu, S.-F.; Xiang, Y. Acta Chim. Sinica 2016, 74, 271. doi: 10.6023/A15100687
Guo, Z. B.; Xu, X.; Xiang, Y.; Lu, S. F.; Jiang, S. P. J. Mater. Chem. A 2015, 3, 148. doi: 10.1039/C4TA04952G
Lu, S. F.; Wang, D. L.; Jiang, S. P.; Xiang, Y.; Lu, J. L.; Zeng, J. Adv. Mater. 2010, 22, 971. doi: 10.1002/adma.200903091
Guo, Z. B.; Xiu, R. J.; Lu, S. F.; Xu, X.; Yang, S. C.; Xiang, Y. J. Mater. Chem. A 2015, 3, 8847. doi: 10.1039/C5TA00415B
Zhang, J.; Xiang, Y.; Lu, S. F.; Jiang, S. P. Adv. Sustainable Syst. 2018, 2, 1700184. doi: 10.1002/adsu.201700184
Jiao, K.; Li, X. G. Prog. Energy Combust. Sci. 2011, 37, 221. doi: 10.1016/j.pecs.2010.06.002
Li, Q. F.; Jensen, J. O.; Savinell, R. F.; Bjerrum, N. J. Prog. Polym. Sci. 2009, 34, 449. doi: 10.1016/j.progpolymsci.2008.12.003
Xu, X.; Wang, H. N.; Lu, S. F.; Guo, Z. B.; Rao, S. Y.; Xiu, R. J.; Xiang, Y. J. Power Sources 2015, 286, 458. doi: 10.1016/j.jpowsour.2015.04.028
卢善富, 徐鑫, 张劲, 相艳, 中国科学:化学, 2017, 47, 565. http://www.cnki.com.cn/Article/CJFDTotal-JBXK201705007.htmLu, S.-F.; Xu, X.; Zhang, J.; Xiang, Y. Sci. Sin. Chim. 2017, 47, 565. http://www.cnki.com.cn/Article/CJFDTotal-JBXK201705007.htm
Pan, H. Y.; Chen, S. X.; Zhang, Y. Y.; Jin, M.; Chang, Z. H.; Pu, H. T. J. Membr. Sci. 2015, 476, 87. doi: 10.1016/j.memsci.2014.11.023
Zhang, B. P.; Ni, J. P.; Xiang, X. Z.; Wang, L.; Chen, Y. M. J. Power Sources 2017, 337, 110. doi: 10.1016/j.jpowsour.2016.10.102
Yang, J. S.; Li, Q. F.; Cleemann, L. N.; Jensen, J. O.; Pan, C.; Bjerrum, N. J.; He, R. H. Adv. Energy Mater. 2013, 3, 622. doi: 10.1002/aenm.201200710
Wang, S. M.; Li, Z. H.; Lu, C. J. Colloid Interface Sci. 2015, 458, 315. doi: 10.1016/j.jcis.2015.07.056
Bo, W.; Li, H. X.; Zhang, J. J.; Song, X. J.; Hu, J. S.; Liu, C. Environ. Technol. 2016, 37, 3062. doi: 10.1080/21622515.2016.1175513
Ates, S.; Tatar-Guner, P.; Yagci, Y.; Levent Demirel, A. Des. Monomers Polym. 2012, 16, 137.
Gao, B. J.; Zhang, D. D.; Li, Y. B. J. Polym. Res. 2018, 25, 158. doi: 10.1007/s10965-018-1553-z
Zhang, J. J.; Zhang, J.; Bai, H. J.; Tan, Q. L.; Wang, H. N.; He, B. S.; Xiang, Y.; Lu, S. F. J. Membr. Sci. 2019, 572, 496. doi: 10.1016/j.memsci.2018.11.035
Qiu, J. H.; Zhang, Y. W.; Zhang, Y. T.; Zhang, H. Q.; Liu, J. D. J. Colloid Interface Sci. 2011, 354, 152. doi: 10.1016/j.jcis.2010.09.090
Beamson, G.; Briggs, D. J. Chem. Educ. 1993, 70, A25. doi: 10.1016/0003-2670(93)80419-L
Bébin, P.; Galiano, H. Adv. Polym. Technol. 2006, 25, 121. doi: 10.1002/adv.20066
Genova-Dimitrova, P.; Baradie, B.; Foscallo, D.; Poinsignon, C.; Sanchez, J. Y. J. Membr. Sci. 2001, 185, 59. doi: 10.1016/S0376-7388(00)00634-7
Iojoiu, C.; Genova-Dimitrova, P.; Maréchal, M.; Sanchez, J. Y. Electrochim. Acta 2006, 51, 4789. doi: 10.1016/j.electacta.2006.01.022
胡小莲, 唐婉莹, 何世颖, 环境科学学报, 2017, 37, 4129. doi: 10.13198/j.issn.1001-6929.2017.02.60Hu, X.-L.; Tang, W.-Y.; He, S.-Y. Acta Sci. Circumst. 2017, 37, 4129. doi: 10.13198/j.issn.1001-6929.2017.02.60
Wang, J.; Jiang, H. X.; Xu, Y. X.; Yang, J. S.; He, R. H. Appl. Surf. Sci. 2018, 452, 473. doi: 10.1016/j.apsusc.2018.05.063
Lobato, J.; Cañizares, P.; Rodrigo, M. A.; Linares, J. J.; Aguilar, J. A. J. Membr. Sci. 2007, 306, 47. doi: 10.1016/j.memsci.2007.08.028
Bose, S.; Kuila, T.; Nguyen, T. X. H.; Kim, N. H.; Lau, K.-t.; Lee, J. H. Prog. Polym. Sci. 2011, 36, 813. doi: 10.1016/j.progpolymsci.2011.01.003
He, R. H. J. Membr. Sci. 2003, 226, 169. doi: 10.1016/j.memsci.2003.09.002
Ma, Y. L.; Wainright, J. S.; Litt, M. H.; Savinell, R. F. J. Electrochem. Soc. 2004, 151, A8. doi: 10.1149/1.1630037
Tang, Q. W.; Cai, H. Y.; Yuan, S. S.; Wang, X.; Yuan, W. Q. Int. J. Hydrogen Energy 2013, 38, 1016. doi: 10.1016/j.ijhydene.2012.10.107
Yang, J. S.; Li, Q. F.; Jensen, J. O.; Pan, C.; Cleemann, L. N.; Bjerrum, N. J.; He, R. H. J. Power Sources 2012, 205, 114. doi: 10.1016/j.jpowsour.2012.01.038
Sood, R.; Donnadio, A.; Giancola, S.; Kreisz, A.; Jones, D. J.; Cavaliere, S. ACS Appl. Mater. Interfaces 2016, 8, 16897. doi: 10.1021/acsami.6b02713
Yang, J. S.; Wang, J.; Liu, C.; Gao, L. P.; Xu, Y. X.; Che, Q. T.; He, R. H. J. Membr. Sci. 2015, 493, 80. doi: 10.1016/j.memsci.2015.06.010
Wang, J.; Zheng, J. F.; Zhao, Z.; Zhang, S. B. J. Mater. Chem. 2012, 22, 706. doi: 10.1039/c2jm15131f
Inaba, M.; Kinumoto, T.; Kiriake, M.; Umebayashi, R.; Tasaka, A.; Ogumi, Z. Electrochim. Acta 2006, 51, 5746. doi: 10.1016/j.electacta.2006.03.008
Virkar, A. V.; Zhou, Y. K. J. Electrochem. Soc. 2007, 154, B540. doi: 10.1149/1.2722563
Roen, L. M.; Paik, C. H.; Jarvic, T. D. Electrochem. Solid-State Lett. 2004, 7, 19.

图 1 自交联PEI-PSF的合成示意图
Figure 1 Scheme for the synthesis of self-crosslinked PEI-PSF membrane

图 2 聚合物核磁共振氢谱. (a) PSF, (b) CMPSF-58
Figure 2 1H NMR of polymer. (a) PSF, (b) CMPSF-58

图 4 CMPSF-58和PEI-PSF-58的XPS谱图. (a) Cl 2p电子结合能, (b) N 1s电子结合能
Figure 4 XPS spectra of CMPSF-58 and PEI-PSF-58. (a) Cl 2p electron binding energy, (b) N 1s electron binding energy

图 5 PSF和PEI-PSF-58聚合物的TGA曲线(N2氛围)
Figure 5 TG curves of PSF and PEI-PSF-58 polymer (N2 atmosphere)

图 6 PEI-PSF-x浸泡浓磷酸后(a)磷酸掺杂水平和体积溶胀率及(b)单位体积磷酸含量
Figure 6 (a) Phosphoric acid doping level and volume swelling ratio and (b) phosphoric acid content per unit volume of PEI-PSF-x after doping phosphoric acid

图 7 PEI-PSF-x浸泡磷酸后的(a)应力/应变测试及(b)质子传导率随温度的变化关系
Figure 7 (a) Stress/strain test and (b) proton conductivity of PEI-PSF-x membranes after doping phosphoric acid

图 8 (a) PEI-PSF-x的电池性能, (b)不同温度对PEI-PSF-58电池性能的影响和(c) PEI-PSF-58的电池稳定性
Figure 8 (a) Cell performance of PEI-PSF-x, (b) effect of different temperatures on the PEI-PSF-58 membrane cell performance and (c) cell stability of PEI-PSF-58
表 1 PEI-PSF-x与其他PSF基高温膜关于吸酸量、质子传导率和拉伸强度的比较
Table 1. Comparison of PEI-PSF-x with other PSF membrane on acid absorption, proton conductivity and tensile strength
| PA doping sample |
PA uptake/ wt% |
σ/(mS• cm-1) @160℃ |
Tensile strength/ MPa |
Power Densitya/ (mW•cm−2)@150 ℃ |
Ref. |
| EtPSU | 168.5 | 20b | 5.1c | 204 | [38] |
| ImPSF | 132 | 34.5d | 2.4 | — | [39] |
| PSU-DMIm | ADL=13 | 38 | 7.2 | 269e | [40] |
| PSf-Im | 235.8 | 48 | 4.2 | — | [41] |
| TDPA-PSF-51 | 79.7 | 17 | 8.4 | 160 | [25] |
| TDPA-PSF-75 | 159.8 | 35 | 3.7 | 310 | [25] |
| PEI-PSF-58 | 122 | 27.9b | 26.1 | 200 | This work |
| a H2和O2下测试, b 150 ℃下测试, c 130 ℃下测试, d 140 ℃下测试, e 160℃下测试. | |||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
