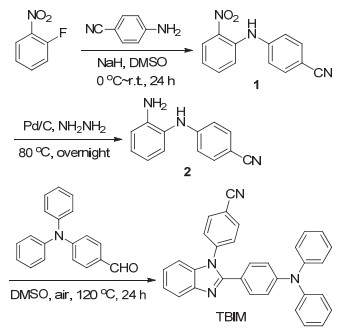
图式1.
化合物TBIM的合成路线
Scheme 1.
Synthetic route to compound TBIM

力致荧光变色是一种十分重要的固体光学现象, 具有该性能的材料可在受力条件下呈现两种甚至多种色彩的荧光颜色变化或荧光强度变化[1].受力条件下晶相至无定形相[2]以及晶相至晶相[3]或无定形相至晶相的转变[4]被认为是引起化合物产生力致荧光变色活性的原因, 其中晶相至无定形相的转变是最普遍存在的类型.利用在力刺激下荧光性能变化的动态可逆性, 此类分子可作为一种智能材料广泛地应用于传感[5]、加密纸张[6]或墨水[7]、记忆芯片[8]、光信息记录介质等[9]方面, 已成为近年来材料化学领域研究的热点之一.固体的力致荧光变色性能主要取决于其结晶态的发射性质, 而这与分子构象、堆积方式以及分子间相互作用关系密切.
另一种与结晶态性质密切相关的固体光学现象是摩擦发光, 它首次于1605年被培根发现.当在黑暗中碾压糖块时, 糖块会发出微弱的光.相较于力致荧光变色体系, 纯粹由有机小分子构成的摩擦发光晶体并不是很多[10].由于大部分具有摩擦发光性能的晶体拥有非中心对称空间群, 压电效应被广泛认为是引起晶体产生摩擦发光现象的原因.然而, 少数一些具有中心对称空间群的有机小分子晶体也呈现出良好的摩擦发光性能[11].由于压电晶体必须具有非中心对称空间群, 显然压电效应无法对后者做出合理的解释.因此, 探究具有中心对称空间群的有机小分子晶体产生摩擦发光活性的原因是值得关注的课题.
同质多晶体在单分子水平上具有完全相同的分子结构, 但在超分子水平上具有不同的分子构象、分子堆积方式以及分子间相互作用, 是研究晶体微观生长与其宏观性能之间构效关系的良好载体.本文通过芳香亲核取代、还原以及空气氛围下DMSO中的氧化环合反应, 将作为给电子基的三苯胺与作为吸电子基的4-氰基苯基分别引入苯并咪唑的C(2)与N(1)位, 所得化合物的石油醚/二氯甲烷混合液经挥发获得两种晶体TBIMB与TBIMG, 前者具有深蓝色荧光, 后者具有绿色荧光.两种晶体在研磨下其发射波长分别发生红移与蓝移, 均呈现出力致荧光变色活性, 但前者的变色性能是可逆的, 而后者的是单向的.此外, 尽管两种晶体都具有中心对称空间群, 但它们依然呈现良好的摩擦发光性能, 发光波长与晶态下的荧光发射波长几乎一致.本文结合晶体学解析与量子化学理论计算, 从分子构象、堆积方式以及分子间相互作用的角度出发, 对两种晶体在结晶态发射波长、力致荧光变色可逆性以及摩擦发光性能等方面的相似性与差异性进行了详细深入的分析与探讨, 从而揭示了两种晶体产生上述性能异同的根本原因, 为多晶体构效关系的研究提供可借鉴的方法.
目标分子TBIM的合成可以通过三步反应并以较好的收率而方便地实现: 2-氟硝基苯首先在NaH的存在下与4-氰基苯胺发生芳香亲核取代反应得到中间体1, 其在钯催化下被水合肼还原成为中间体2.在空气气氛下, 用DMSO做氧化剂, 中间体2与4-(二苯胺基)苯甲醛发生氧化环化反应生成4-(2-(4-(二苯胺基)苯基)-1H-苯并咪唑基苯腈(TBIM).目标分子以及中间体的结构经1H NMR、13C NMR以及HRMS确证.
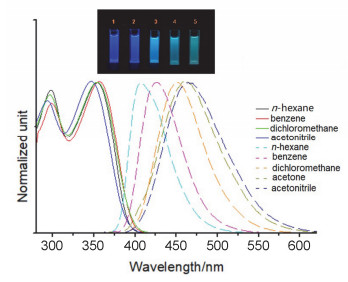
首先对TBIM在二氯甲烷溶液中的摩尔吸光系数以及荧光量子产率进行了测量.该分子的摩尔吸光系数为3.88×104 mol-1•cm-1, 表明分子受激后发生的主要是π-π*跃迁, 荧光量子产率为0.572.从TBIM在不同溶剂中的吸收与发射光谱可见(图 1), 该分子的吸收光谱对溶剂极性的变化不敏感, 吸收谱的峰型与波长在不同溶剂中基本保持不变; 发射光谱对溶剂极性的变化较为敏感, 随着溶剂极性的增大, 发射波长也相应地出现红移, 产生一定程度的正向溶剂效应.但当溶剂的极性超过二氯甲烷的极性后, 进一步增大溶剂的极性, 溶液发射波长的红移变得非常小.由此说明, 分子受激后确实发生了分子内电荷转移, 但激发态电荷分离的程度并不十分的显著, 即便在很高极性的溶剂中分子受到的溶剂稳定化作用也不足够大, 无法产生大幅的发射红移与荧光淬灭.此外, 纵然在极小溶剂效应的正己烷中, TBIM的吸收与发射光谱的重叠面积也较小, Stockes位达到60 nm, 这暗示着激发态在回落到基态前发生了较为明显的几何弛豫.由于分子上存在多个σ键, 它们所连接的基团在分子受激后可以发生不同程度的自由转动并相应地消耗激发能, 从而导致发射相较于吸收出现较大幅度的波长红移.
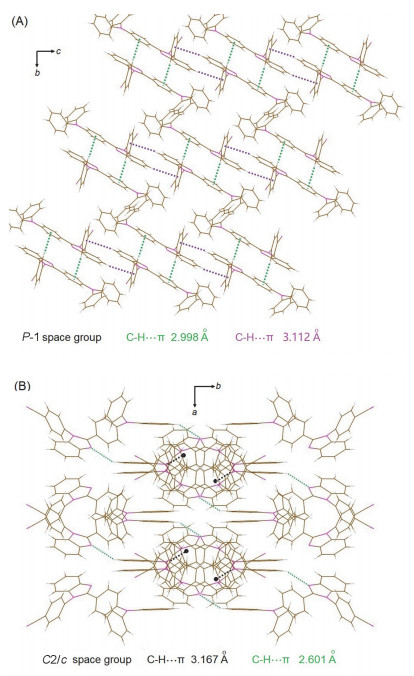
将TBIM化合物溶解于正己烷与二氯甲烷(1:1, V/V)的混合溶剂中, 所得溶液在空气中挥发可以获得两种具有不同荧光颜色的晶体.在溶液中优先生长出的是具有深蓝色荧光的晶体TBIMB, 随后生长出的是具有绿色荧光的晶体TBIMG. X-射线单晶衍射表明, 前者属于单斜晶系C2/c空间群, 晶体中含有与TBIMB分子为1:1摩尔比的二氯甲烷溶剂分子, 而后者属于三斜晶系P-1空间群, 两者均为中心对称空间群.我们推测, 两种晶体的生长是溶液极性依赖的, 溶液的极性在培养初期较高, 此时优先长出TBIMB晶体; 由于二氯甲烷的挥发速率快于正己烷, 随着时间的推移, 溶液的极性逐渐减小, 从而利于TBIMG晶体的析出.
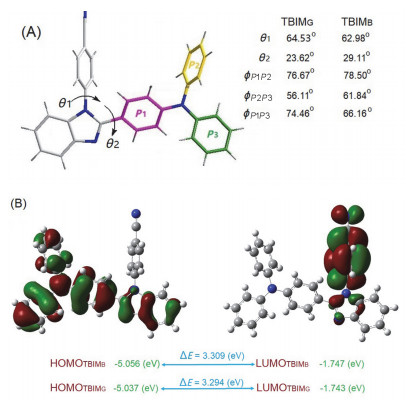
对两种晶体中分子的构象进行分析可以发现(图 2A), 两者在氰基苯基与咪唑环之间(θ1)和三苯胺上P1苯基与咪唑环之间(θ2)的扭角, 以及三苯胺上三个苯环彼此之间的二面角(ϕ)方面的差别并不十分显著.依据晶体中的分子构象, 我们采用B3LYP密度泛函方法在6-31G*水平下对其进行了HOMO与LUMO能级的计算(图 2B).结果表明在两种晶体中, 分子前线轨道能级间的能差十分接近, 不足以导致两种晶体发射波长的差异达到70 nm.事实上, 与气态或溶液态中分子间彼此处于孤立状态的情况大为不同, 晶体中存在大量的分子间相互作用, 仅凭分子构象往往不能获得关于分子能量的准确信息, 必须考虑它们彼此间相互作用的情况[12].
最近, 池振国小组[13]采用飞秒瞬态时间分辨发射光谱证实了含有三苯胺与三苯磷的酮化合物的两种晶体, 其不同波长的荧光分别源于分子LE(局部发射)激发态与TICT(扭转分子内电荷转移)激发态的发射.在蓝色荧光(473 nm)晶体中, 三苯胺上的三个苯基均与相邻分子间存在C—H···π相互作用; 绿色荧光(504 nm)晶体中, 只有两个三苯胺上的苯基与相邻分子间存在C—H···π相互作用.因此, 前者中三苯胺上不与羰基共轭的两个苯基受到分子间作用力的束缚较强而无法发生显著的构象扭转, 分子受激后只能形成LE激发态并发射短波荧光; 后者中的这两个苯基受到分子间作用力的束缚较弱而能够产生较大幅度的构象扭转, 分子受激后可形成TICT激发态并发射长波荧光.
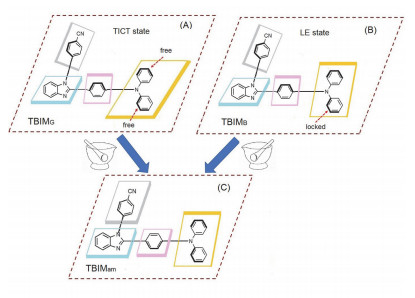
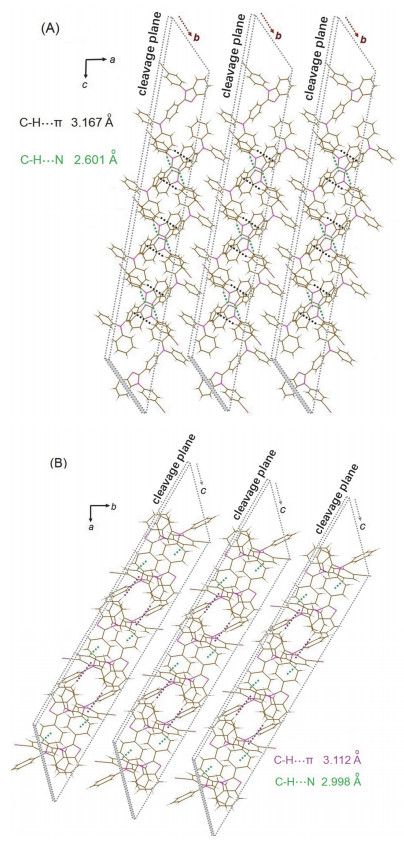
由于缺少飞秒瞬态时间分辨发射光谱仪, 尚无法开展相关的实验证实工作, 但鉴于本文中的化合物与上述报道的分子在结构上存在相似性, 推测TBIMB与TBIMG晶体发射波长的巨大差异也极有可能源于两种晶体中的分子在受激后处于不同的激发态.晶体学分析表明, 这两种晶体中均不存在π-π相互作用, 这使得两种晶体呈现出强烈的固体荧光.在TBIMG晶体中(图 3A), 相邻分子主要通过两种较强的C—H···π相互作用而进行堆积, 一种存在于一个分子的氰基苯基与另一个分子的苯并咪唑上的苯环之间(3.112 Å ), 另一种存在于一个分子的氰基苯基与另一个分子中三苯胺上的P1苯环之间(2.998 Å ).在TBIMB晶体中(图 3B), 一个分子上的咪唑双键N原子与另一个分子三苯胺上的P3苯环之间存在氢键(2.601 Å );此外, 一个分子上的氰基苯基与另一个分子三苯胺上的P1苯环之间形成C—H···π相互作用(3.167 Å ).由于在TBIMB晶体中, P3苯环受到分子间相互作用的约束而使之无法与P2苯环一起通过C—N键绕着P1苯环转动, 分子受激后只能形成LE激发态并发射短波荧光.相反, 由于P2苯环与P3苯环均不与相邻分子间存在相互作用, TBIMG晶体中的分子受激后这两个苯环可以一起通过C—N键绕着P1苯环转动, 由此可形成TICT激发态并发射长波荧光(图 4).
此外, 我们发现TBIMG晶体对不同角度入射日光的吸收有所差别, 从而沿着不同的宏观晶轴方向进行观察时, 晶体会呈现出不同的颜色(图 5).当沿着晶体厚度最大方向观察时, 晶体呈现为黄绿色; 沿着其厚度最小方向观察时, 晶体仍然呈现为黄绿色; 而沿着其厚度较小方向观察时, 晶体却是完全无色的.由此可见, 这种色泽的不同并非晶体厚度差别引起, 应当归咎于晶体的二向色性.因此, 大尺寸的TBIMG晶体可以开发成为偏振片.这种二向色性在TBIMB晶体上没有观察到.
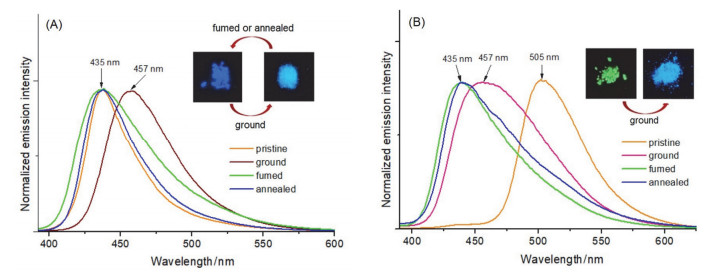
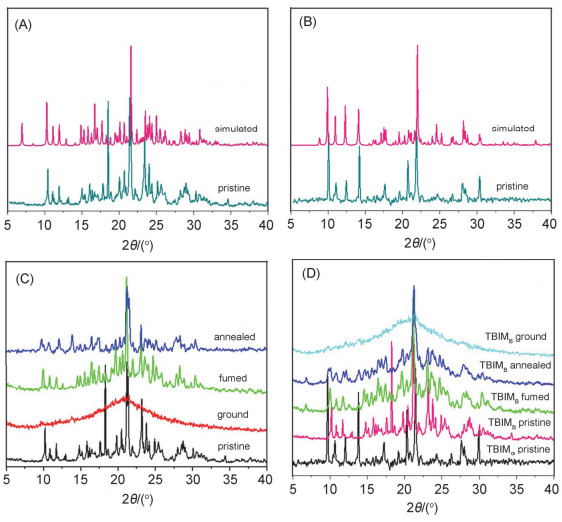
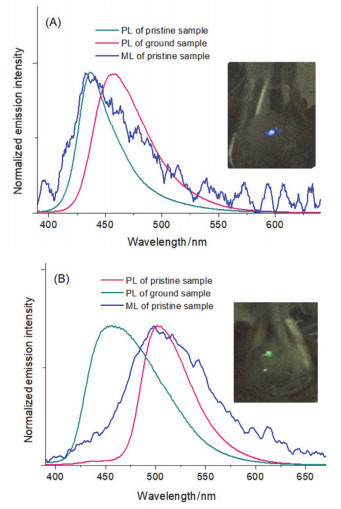
对两种晶体在研磨前的发射光谱进行了测量(图 6A, 6B), TBIMB与TBIMG的发射波长分别为435 nm和505 nm.两种晶体在研磨后的荧光发射波长发生相反方向的位移:前者红移至457 nm, 而后者则蓝移至455 nm, 这表明两种晶体都具有力致荧光变色活性.我们对两种晶体在研磨前后的样品进行了X-射线粉末衍射测定(PXRD).衍射图形表明(图 7A, 7B), 研磨前的多晶粉末具有与其单晶完全相同的分子堆积结构, 单晶中获得的晶体结构信息可以用来揭示多晶粉末中的分子间相互作用以及分子构象.粉末衍射图表明(图 7C, 7D), TBIMB与TBIMG两种晶体在研磨后发生了由结晶态向无定形态的转变, 其力致荧光变色活性应是由固体的形貌变化所引起.当使用良溶剂(例如二氯甲烷、氯仿、四氢呋喃、丙酮、乙腈等)进行熏蒸或在烘箱中于120 ℃下保温热处理一定时间后, 研磨后的TBIMB粉末可以完全恢复成研磨前的晶相结构, 而TBIMG粉末在相同处理条件下无法再次恢复成其研磨前的晶相结构, 却呈现出与TBIMB晶体几乎一致的衍射峰图形, 且这种在退火条件下由TBIMG向TBIMB转变的过程不受热处理温度与时间的影响.这表明两种晶体在固体条件下的转变是单向的, 它们之间的相互转化只能在溶液中实现.
尽管两种晶体具有不同的分子构象与堆积模式, 但受力后晶格受到了彻底破坏, 在无定形状态下分子的构象趋于一致, 因而两个研磨后的样品呈现出几乎完全一致的发射特征.此时, 氰基苯基、苯并咪唑核以及三苯胺上的P1苯环这三者可能均处于同一平面, 且三苯胺上P2与P3苯环所在平面与苯并咪唑核之间也可能处于同一平面(图 4C).前者类型的构象平面化加强了给电子与吸电子基团间的共轭, 使得分子的发射波长红移.对于TBIMB晶体, 研磨后分子主要产生这种类型的几何变化.对于TBIMG晶体, 由于其荧光来源于P2与P3苯环所在平面与P1苯环之间的大幅扭转, 尽管研磨后分子也产生前者类型的构象平面化, 但后者类型的构象平面化对其荧光的影响更为强烈, 因而TBIMG晶体在研磨后产生发射蓝移.
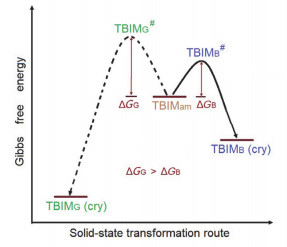
造成TBIMG晶体具有不可逆力致荧光变色活性的原因很有可能在于其晶体中存在更为密集的分子间相互作用, 从而导致研磨后的无定形样品欲恢复成原有晶相结构时需要的活化能较高, 尽管更为密集的分子间相互作用使得晶体在热力学上具有更大的晶格能和更高的稳定性.因此, 在退火(熔点之下的热处理)与溶剂熏蒸(动态的溶解-挥发过程)这些动力学晶化条件下, 研磨后的样品分子将选择性地翻越较小的活化能垒, 优势地转变成TBIMB晶体并呈现出该晶体的发射特征(图 8).
此外, TBIMB与TBIMG的晶体在黑暗处被用力摩擦时会分别发出清晰的蓝色与绿色的闪光, 这说明两种晶体均具有摩擦发光活性.对两者的摩擦发光(ML)光谱进行了测量(图 9A, 9B).结果表明, 两种晶体的ML发射波长与荧光(FL)发射波长几乎完全一致, 这说明机械力激发的不是无定形状态下而是晶相状态下的分子, 这些受激分子应当处于晶体原生表面以及晶体受力发生折断或解理而形成的新表面.由于两种晶体均属中心对称空间群, 压电效应导致它们具有摩擦发光活性的可能性可以排除.
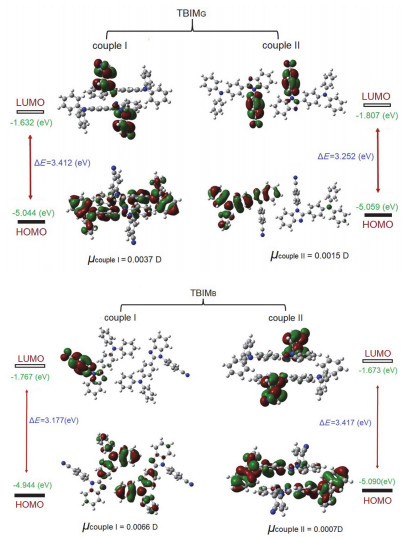
最近, 李振课题组[11b]对一个具有中心对称空间群的摩擦发光晶体中存在强相互作用的分子对进行了理论计算, 结果表明分子对的净偶极矩并非接近于零, 而是具有相当大的数值.因而该晶体在受力碎裂后, 碎裂面上的分子对之间可能产生放电效应, 从而激发这些表面上的分子使之产生光辐射.这种效应有别于压电现象.类似地, 采用李振等提出的方法, 对TBIMB与TBIMG晶体中存在较强相互作用的分子对进行DFT/B3LYP/6-31G*水平下的计算.结果显示(图 10A, 10B), 在TBIMG晶体的分子对Ⅰ中(C—H···π 2.998 Å )以及在TBIMB晶体的分子对Ⅱ中(C—H···π 3.167 Å ), 前线轨道上的电子云均分布在分子对中的两个分子上, 并发生一定程度的分子内电荷转移; 而在TBIMG晶体的分子对Ⅱ中(C—H···π 3.112 Å )以及在TBIMB晶体的分子对Ⅰ中(C—H···N 2.601 Å ), HOMO或LUMO轨道上的电子云分别选择性地仅分布于分子对中的一个分子上, 并发生一定程度的分子间电荷转移.此外, 计算表明所有分子对的净偶极矩均非常接近于零.显然, 既非压电效应也非上述李振等提出的机理可以合理解释TBIMB与TBIMG晶体的摩擦发光行为.
晶体学研究表明, 在两种晶体中, 分子间相互作用将分子堆积成链状排列(图 11A, 11B), 相邻的分子链之间没有显著的相互作用存在, 因此晶体在受到外力作用时会很容易发生解理而形成许多新的解理面, 晶体的脆性较高.我们推测, 这些解理面在外力作用下发生彼此间的相对运动而导致内生摩擦放电, 由此激发解理面上的分子使之受激后释放出光子.一些报道也支持上述的推测[14].由于两种晶体产生的摩擦发光波长明显不同, 可以排除这种发光来源于施力物体与晶体之间的表面摩擦放电(两者间摩擦产生的静电火花).此外, 有研究表明[15], 摩擦发光的强度与可产生内生摩擦放电的晶体表面积成正比.对于这两种晶体来说, 链状分子堆积所导致的晶体脆性有助于其受力后发生解理从而提高这种表面积, 晶体的摩擦发光活性得到了增强.由于晶体产生摩擦发光效应时仅处于解理状态而非晶格完全破坏状态, 机械力激发与光致激发的分子处于相同的受激状态, 因而晶体的摩擦发光波长与晶体的荧光发射波长几乎完全一致.
通过芳香亲核取代、还原、氧化闭环反应, 以较好的总收率获得了含有氰基苯基与三苯胺基的苯并咪唑化合物TBIM, 其溶液经缓慢挥发先后生成两种晶体TBIMB与TBIMG, 两者的发射波长差异达到70 nm.量子化学理论计算表明, 波长的显著差异并非源于两者在晶态下的构象差别, 而更可能由两者的分子堆积模式以及分子间相互作用的不同所造成.两种晶体在研磨后均产生力致荧光变色效应, TBIMB发生发射红移而TBIMG发生发射蓝移, 且前者的变色效应是可逆的而后者则是不可逆的.两种晶体之间的相互转变只能在溶液中实现, 在固态下只能发生TBIMG向TBIMB的单向转变, 其原因很可能在于前者晶体中更密集的分子间相互作用导致的较高活化能使TBIMG分子从无定形态向结晶态的转变在动力学上无法实现.此外, 两种晶体在施力下均可观察到清晰的摩擦发光现象, 发光波长与两者的荧光发射波长几乎完全一致.晶体学分析、理论计算以及实验测量均表明, 压电效应、具有较高净偶极矩的分子对之间的放电激发以及施力物体与晶体之间的表面摩擦放电均不是TBIMB与TBIMG晶体出现摩擦发光的原因.特殊的链状分子堆积所导致的晶体高度脆性以及受力后晶体解理面间的相对运动所引起的内生摩擦放电引发解理面上的分子受激很有可能是两种晶体具有摩擦发光活性的重要原因.
所用试剂均为分析纯且部分试剂在使用前进行了重结晶或蒸馏纯化.熔点在OptiMelt自动显微熔点仪上测定. 1H NMR (400 MHz)与13C NMR (100 MHz)谱图用DMSO-d6或CDCl3作为溶剂、在Bruker Avance Ⅱ DMX 400型液体核磁共振仪上测定.吸收光谱在Shimadzu UV 3600型UV-Vis-NIR光谱仪上测试, 发射光谱在Perkin-Elmer LS55型光谱仪上进行测试.溶液中的荧光量子产率采用稀溶液比较法, 用硫酸奎宁的0.1 mol/L硫酸溶液作为参照(其荧光量子产率为0.55).摩擦发光光谱在海洋光学flame微型光纤光谱仪上测定.粉末衍射在DX2700型衍射仪上、以铜Kα射线为入射源, 扫描电压40 kV、扫描电流40 mA、扫描速率0.3 (°)/min进行测定.动态差示热扫描在Waters TA Q20型仪器上完成.高分辨质谱的测定在Waters TOFMS GCT Premier上完成.单晶衍射在Bruker Gemini Ultra CCD衍射仪上以石墨单色化的钼射线(λ=0.71073 Å )或铜射线(λ=1.54178 Å )为入射源完成测量.
0 ℃冰浴下, 4-氰基苯胺(12 mmol)的无水DMSO溶液(10 mL)缓慢地滴入2-氟硝基苯(10 mmol)与氢化钠(15 mmol, 60 wt%, 矿物油吸附)的无水DMSO溶液(15 mL), 所得的混合物立刻变成深红色并在冰浴下继续保温30 min, 然后在室温下搅拌24 h.反应液倒入水中, 搅拌后有大量橙红色沉淀生成, 静置后过滤.所得固体用水和乙醇洗涤数次, 在硅胶柱色谱上用二氯甲烷/石油醚(1:3, V/V)混合液进行洗脱分离.流出液在旋转蒸发仪上除去溶剂后得到深黄色粉末状固体2.11 g, 收率88%. m.p. 182.3~183.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 6.98 (dt, J1=2.0 Hz, J2=8.8 Hz, 1H), 7.33 (dt, J1=2.0 Hz, J2=8.8 Hz, 2H), 7.47~7.52 (m, 2H), 7.65 (dt, J1=2.0 Hz, J2=8.8 Hz, 2H), 8.22 (dd, J1=2.0 Hz, J2=8.8 Hz, 1H), 9.43 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 106.68, 117.35, 118.78, 120.23, 121.21, 126.91, 133.86, 135.63, 135.69, 139.50, 143.78. HRMS (ESI) m/z calcd. for [C13H9N3O2+Na+] 262.0592, found 262.0587.
室温下, 将水合肼(80 wt%, 18 mL)缓慢滴入含有中间体1 (10 mmol)与钯碳(10 wt%, 0.5 mmol)的无水乙醇溶液中(15 mL), 所得混合液在80 ℃下回流过夜.反应液冷至室温后过滤, 滤饼用无水乙醇洗涤数次, 合并滤液.滤液倒入水中, 用乙酸乙酯萃取三次(15 mL×3), 合并的萃取液经无水硫酸钠干燥后过滤.滤液在旋转蒸发仪上除去溶剂后, 在硅胶柱色谱上用二氯甲烷/石油醚(1:1, V/V)混合液进行洗脱分离.流出液在旋转蒸发仪上除去溶剂后得到浅灰色粉末状固体1.98 g, 收率95%. m.p. 159.4~161.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 3.77 (br, s, 2H), 5.67 (s, 1H), 6.65 (dt, J1=2.0 Hz, J2=8.8 Hz, 2H), 6.78 (dt, J1=1.2 Hz, J2=7.6 Hz, 1H), 6.82 (d, J=7.6 Hz, 1H), 7.10 (d, J=7.6 Hz, 2H), 7.42 (dt, J1=2.0 Hz, J2=8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 100.61, 113.99, 116.41, 119.19, 120.14, 125.19, 127.03, 127.77, 133.77, 143.00, 149.58. HRMS (ESI) m/z calcd. for [C13H11N3+H+] 210.1031, found 210.1042.
室温下, 4-(二苯基氨基)苯甲醛(12 mmol)的无水DMSO溶液(15 mL)加入到中间体2 (10 mmol)的无水DMSO(15 mL)溶液中, 所得混合液在敞口条件下加热至120 ℃搅拌24 h.反应结束后将反应液冷至室温, 并将反应液倒入大量水中, 用二氯甲烷萃取三次(15 mL× 3), 合并的萃取液经无水硫酸钠干燥后过滤.滤液在旋转蒸发仪上除去溶剂后, 在硅胶柱色谱上用乙酸乙酯/石油醚(1:3, V/V)混合液进行洗脱分离.流出液在旋转蒸发仪上除去溶剂后得到浅黄色粉末状固体3.28 g, 收率71%. m.p. 216.8~218.2 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 6.85 (d, J=8.8 Hz, 2H), 7.09 (d, J=7.2 Hz, 4H), 7.14 (t, J=7.2 Hz, 2H), 7.22~7.31 (m, 2H), 7.33~7.39 (m 7H), 7.68 (d, J=8.8 Hz, 2H), 7.78 (d, J=8.0 Hz, 1H), 8.09 (d, J=8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 110.62, 111.83, 118.68, 119.64, 120.65, 122.17, 123.46, 123.75, 124.74, 125.76, 129.02, 130.25, 130.76, 134.66, 136.97, 141.21, 143.26, 146.77, 149.06, 152.08; HRMS (ESI) m/z calcd. for [C32H22N4+H+] 463.1923, found 463.1928.
晶胞参数由全角度最小二乘法修正确定, 经SHELXS-97软件由直接法获得初步结构, 通过全矩阵最小二乘法对非氢原子的坐标以及热力学各向异性参数进行精修, 所有氢原子设定在理想的平衡位置并约束在其所处的母原子上. TBIMB•CH2Cl2 (CCDC 1945883)和TBIMG (CCDC 1945888)的晶体学数据已在剑桥晶体学数据中心进行了登记, 并可通过网址www.ccdc.cam. ac.uk/data_ request/ cif免费获取.
化合物的核磁谱图以及晶体学数据见支持信息(Supporting information).
(a) Peng, B. Y.; Xu, S. D.; Chi, Z. G.; Zhang, X. Q.; Zhang, Y.; Xu, J. R. Prog. Chem. 2013, 25, 1805(in Chinese). (彭邦银, 许适当, 池振国, 张锡奇, 张艺, 许家瑞, 化学进展, 2013, 25, 1805.)(b) Di, B. H.; Chen, Y. L. Chin. Chem. Lett. 2018, 29, 245; (c) Yuan, Y.; Yuan, W.; Chen, Y. L. Sci. China Mater. 2016, 59, 507; (d) Yuan, W.; Yuan, Y.; Chen, Y. L. Acta Polym. Sinica 2016, 11, 1495(in Chinese). (袁伟, 袁媛, 陈于蓝, 高分子学报, 2016, 11, 1495.) (e) Yang, J.; Chi, Z.; Zhu, W.; Tang, B. Z.; Li, Z. Sci. China Chem. 2019, 62, 1090; (f) Li, Q.; Li, Z. Adv. Sci. 2017, 4, 1600484. http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201311002.htm
(a) Dong, Y. Q.; Lam, J. W. Y.; Tan, B. Z. J. Phys. Chem. Lett. 2015, 6, 3429; (b) Chi, Z.; Zhang, X.; Xu, B.; Zhou, X.; Ma, C.; Zhang, Y.; Liu, S.; Xu, J. Chem. Soc. Rev. 2012, 41, 3878; (c) Wang, C.; Li, Z. Mater. Chem. Front. 2017, 1, 2174; (d) Yang, Z.; Chi, Z.; Mao, Zhang, Z. Y.; Liu, S.; Zhao, J. M.; Aldred, P.; Chi, Z. Mater. Chem. Front. 2018, 2, 861; (e) Varughese, S. J. Mater. Chem. C 2014, 2, 3499; (f) Sagara, Y.; Yamane, S.; Mitani, M.; Weder, C.; Kato, T. Adv. Mater. 2016, 28, 1073; (g) Bian, G. F.; Huang, H.; Zhan, L. L.; Lü, X. J.; Cao, F.; Zhang, C.; Zhang, Y. J. Acta Phys.-Chim. Sin. 2016, 32, 589(in Chinese). (边高峰, 黄华, 占玲玲, 吕晓静, 曹枫, 张诚, 张玉建, 物理化学学报, 2016, 32, 589.) (h) Sun, J. B.; Zhang, G. H.; Jia, X. Y.; Xue, P. C.; Jia, J. H.; Lu, R. Acta Chim. Sinica 2016, 74, 165(in Chinese). (孙静波, 张恭贺, 贾小宇, 薛鹏冲, 贾俊辉, 卢然, 化学学报, 2016, 74, 165); (i) Ouyang, M.; Yu, C. H.; Zhang, Y. J.; Hu, B.; Lü, X. J.; Sun, J. W.; Zhang, C. Acta Phys.-Chim. Sin. 2012, 28, 2944(in Chinese). (欧阳密, 俞春辉, 张玉建, 胡彬, 吕晓静, 孙璟玮, 张诚, 物理化学学报, 2012, 28, 2944.) http://www.cnki.com.cn/Article/CJFDTotal-WLHX201602025.htm
(a) Seki, T.; Takamatsu, Y.; Ito, H. J. Am. Chem. Soc. 2016, 138, 6252; (b) Jin, M.; Seki, T.; Ito, H. J. Am. Chem. Soc. 2017, 139, 7452; (c) Yagai, S.; Okamura, S.; Nakano, Y.; Yamauchi, M.; Kishikawa, K.; Karatsu, T.; Kitamura, A.; Ueno, A.; Kuzuhara, D.; Yamada, H.; Seki, T.; Ito, H. Nat. Commun. 2014, 5, 4013. https://www.ncbi.nlm.nih.gov/pubmed/27070308
(a) Zheng, K.; Zheng, Y.; Peng, L.; Xiang, Y.; Tong, A. J. J. Phys. Chem. C 2017, 121, 21610; (b) Lü, Y.; Liu, Y.; Ye, X.; Liu, G. F.; Tao, X. T. CrystEngComm 2015, 17, 526; (c) Chung, K.; Kwon, M. S.; Leung, B. M.; Kim, J. S. ACS Cent. Sci. 2015, 1, 94.
(a) Shi, P.; Duan, Y.; Wei, W.; Xu, Z.; Li, Z.; Han, T. J. Mater. Chem. C 2018, 6, 2476; (b) Feng, C.; Wang, K.; Xu, Y.; Liu, L.; Zou, B.; Lu, P. Chem. Commun. 2016, 52, 3836; (c) Wang, L.; Wang, K.; Zou, B.; Ye, K.; Zhang, H.; Wang, Y. Adv. Mater. 2015, 27, 2918; (d) Xie, W.-Z.; Zheng, H.-C.; Zheng, Y.-S. J. Mater. Chem. C 2017, 5, 10462.
(a) Hirata, S.; Watanabe, T. Adv. Mater. 2006, 18, 2725; (b) Lim, S. J.; An, B. K.; Jung, S. D.; Chung, M. A.; Park, S. Y. Angew. Chem., Int. Ed. 2004, 43, 6346; (c) Olson, C. E.; Previte, M. J. R.; Fourkas, J. T. Nat. Mater. 2002, 1, 225; (d) Irie, M.; Fukaminato, T.; Sasaki, T.; Tamai, N.; Kawai, T. Nature 2002, 420, 759.
(a) Kishimura, A.; Yamashita, T.; Yamaguchi, K.; Aida, T. Nat. Mater. 2005, 4, 546; (b) Zhu, X.; Liu, R.; Li, Y.; Huang, H.; Wang, Q.; Wang, D.; Zhu, X.; Liu, S.; Zhu, H. Chem. Commun. 2014, 50, 12951; (c) Qi, Q.; Liu, Y.; Fang, X.; Zhang, Y.; Chen, P.; Wang, Y.; Yang, B.; Xu, B.; Tian, W.; Zhang, S. X. RSC Adv. 2013, 3, 7996; (d) Kumar, P.; Dwivedi, J.; Gupta, B. K. J. Mater. Chem. C 2014, 2, 10468; (e) Lu, X.-L.; Xia, M. J. Mater. Chem. C 2016, 4, 9350.
(a) Yuan, W. Z.; Tan, Y.; Gong, Y.; Lu, P.; Lam, J. W. Y.; Shen, X. Y.; Feng, C.; Sung, H. Y.; Lu, Y.; Williams, I. D.; Sun, J. Z.; Zhang, Y.; Tang, B. Z. Adv. Mater. 2013, 25, 2837; (b) Li, C.; Tang, X.; Zhang, L.; Li, C.; Liu, Z.; Bo, Z.; Dong, Y. Q.; Tian, Y.-H.; Dong, Y.; Tang, B. Z. Adv. Optical Mater. 2015, 3, 1184; (c) Naeem, K. C.; Subhakumari, A.; Varughese, S.; Nair, V. C. J. Mater. Chem. C 2015, 3, 10225; (d) Sun, J.; Han, J.; Liu, Y.; Duan, Y.; Han, T.; Yuan, J. J. Mater. Chem. C 2016, 4, 8276; (e) Xue, P.; Yang, Z.; Chen, P. J. Mater. Chem. C 2018, 6, 4994.
(a) Sagara, Y.; Kato, T. Angew. Chem. Int. Ed. 2011, 50, 9128; (b) Yoon, S.-J.; Chung, J. W.; Gierschner, J.; Kim, K. S.; Choi, M.-G.; Kim, D.; Park, S. Y. J. Am. Chem. Soc. 2010, 132, 13675; (c) Sun, H.; Liu, S.; Lin, W.; Zhang, K. Y.; Lü, W.; Huang, W.; Huo, F.; Yang, H.; Jenkins, G.; Zhao, Q.; Huang, W. Nat. Commun. 2014, 5, 3601; (d) Zhang, K. Y.; Liu, S.; Zhao, Q.; Huang, W. Coord. Chem. Rev. 2016, 319, 180; (e) Chen, X.; Sun, G.; Zhang, T.; Liu, S.; Zhao, Q.; Huang, W. Adv. Mater. 2016, 28, 7137; (f) Zhao, Q.; Xu, W.; Sun, H.; Yang, J.; Zhang, K. Y.; Liu, S.; Ma, Y.; Huang, W. Adv. Opt. Mater. 2016, 4, 1167; (g) Lin, W.; Zhao, Q.; Sun, H.; Zhang, K. Y.; Yang, H.; Yu, Q.; Zhou, X.; Guo, S.; Liu, S.; Huang, W. Adv. Opt. Mater. 2015, 3, 368; (h) Han, J.; Sun, J.; Li, Y.; Duan, Y.; Han, T. J. Mater. Chem. C 2016, 4, 9287.
(a) Xu, B.; Li, W.; He, J.; Wu, S.; Zhu, Q.; Yang, Z.; Wu, Y.-C.; Zhang, Y.; Jin, C.; Lu, P.-Y.; Chi, Z.; Liu, S.; Xu, J.; Bryce, M. R. Chem. Sci. 2016, 7, 5307; (b) Xu, S.; Liu, T.; Mu, Y.; Wang, Y.-F.; Chi, Z.; Lo, C.-C.; Liu, S.; Zhang, Y.; Lien, A.; Xu, J. Angew. Chem. 2015, 127, 888; (c) Yang, J.; Ren, Z.; Xie, Z.; Liu, Y.; Wang, C.; Xie, Y.; Peng, Q.; Xu, B.; Tian, W.; Zhang, F.; Chi, Z.; Li, Q.; Li, Z. Angew. Chem. Int. Ed. 2017, 56, 880; (d) Neen, K. K.; Sudhakar, P.; Dipak, K.; Thilagar, P. Chem. Commun. 2017, 53, 3641; (e) Xu, B.; He, J.; Mu, Y.; Zhu, Q.; Wu, S.; Wang, Y.; Zhang, Y.; Jin, C.; Lo, C.; Chi, Z.; Lien, A.; Liu, S.; Xu, J. Chem. Sci. 2015, 6, 3236; (f) Nakayama, H.; Nishida, J.; Takada, N.; Sato, H.; Yamashita, Y. Chem. Mater. 2012, 24, 671; (g) Nishida, J.; Ohura, H.; Kita, Y.; Hasegawa, H.; Kawase, T.; Takada, N.; Sato, H.; Sei, Y.; Yamashita, Y. J. Org. Chem. 2016, 81, 433; (h) Liu, M. L.; Wu, Q.; Shi, H. F.; An, Z. F.; Huang, W. Acta Chim. Sinica 2018, 76, 246(in Chinese). (刘明丽, 吴琪, 史慧芳, 安众福, 黄维, 化学学报, 2018, 76, 246.)
(a) Wang, C.; Xu, B.; Li, M.; Chi, Z.; Xie, Y.; Li, Q.; Li, Z. Mater. Horiz. 2016, 3, 220; (b) Liu, F.; Tu, J.; Wang, X.; Wang, J.; Gong, Y.; Han, M.; Dang, X.; Liao, Q.; Peng, Q.; Li, Q.; Li, Z. Chem. Commun. 2018, 54, 5598.
(a) Yang, J.; Ren, Z.; Chen, B.; Fang, M.; Zhao, Z.; Tang, B. Z.; Peng, Q.; Li, Z. J. Mater. Chem. C 2017, 5, 9242; (b) Yang, J.; Ren, Z.; Xie, Z.; Liu, Y.; Wang, C.; Xie, Y.; Peng, Q.; Xu, B.; Tian, W.; Zhang, F.; Chi, Z.; Li, Q.; Li, Z. Angew. Chem. Int. Ed. 2017, 56, 880.
Xie, Z.; Yu, T.; Chen, J.; Ubba, E.; Wang, L.; Mao, Z.; Su, T.; Zhang, Y.; Aldred, M. P.; Chi, Z. Chem. Sci. 2018, 9, 5787. doi: 10.1039/C8SC01703D
Hardy, G. E.; Baldwin, J. C.; Zink, J. I.; Kaska, W. C.; Liu, P.; Dubois, L. J. Am. Chem. Soc. 1977, 99, 3552. doi: 10.1021/ja00453a002
Zink, J. I.; Chandra, B. P. J. Phys. Chem. 1982, 86, 5. doi: 10.1021/j100390a003

图 1 TBIM在不同极性溶剂中的吸收光谱(实线)与发射光谱(虚线) (插图: 365 nm紫外光照下溶液的荧光照片: 1—正己烷; 2—苯; 3—二氯甲烷; 4—丙酮; 5—乙腈)
Figure 1 Absorption (solid lines) and emission spectra (dash lines) of TBIM in different solvents (Inserted: photos of solutions under 365 nm UV irradiation: 1—n-hexane; 2—benzene; 3—dichloromethane; 4—acetone; 5—acetonitrile)

图 2 (A) TBIMB与TBIMG晶体中分子的构象以及(B)两者中的分子在该构象下的HOMO-LUMO能级与能差
Figure 2 (A) Molecular conformations in TBIMB and TBIMG crystals; (B) HOMO-LUMO levels and energy gaps of the double molecules under such conformations

图 3 TBIMG (A)与TBIMB (B)晶体中分子的堆积(为了清楚起见TBIMB晶体中的溶剂分子已抽取)
Figure 3 Molecular packings in TBIMG (A) and TBIMB (B) crystals (solvent molecules in TBIMB crystal are squeezed in order to present clear view)

图 4 TBIMG (A)与TBIMB (B)晶体中以及两者无定形状态下(C)分子激发态构象示意图
Figure 4 Schematic diagram of excited-state geometry for molecules in TBIMG (A) and TBIMB (B) crystals and in their amorphous phase (C)

图 6 TBIMG (A)和TBIMB (B)在不同固体状态下的发射光谱
Figure 6 Solid-state emission spectra of TBIMG (A) and TBIMB (B) under different conditions

图 7 TBIMB (A, C)和TBIMG (B, D)在不同固体状态下的PXRD图形
Figure 7 PXRD patterns of TBIMB (A, C) and TBIMG (B, D) under different solid-state conditions

图 8 TBIMG和TBIMB在不同固体条件下转化的能量示意图
Figure 8 Schematic diagram of potential energy during transformations of TBIMG and TBIMB under different solid-state conditions

图 9 TBIMG (A)和TBIMB (B)晶体的摩擦发光光谱、研磨前后的光致发射光谱(插图:黑暗处两种晶体摩擦发光的照片)
Figure 9 ML and PL spectra of TBIMG (A)和TBIMB (B) (Inserted: ML photos of the two polymorphs)

图 10 晶体TBIMG和TBIMB中分子对的前线轨道能级、能级差以及偶极矩
Figure 10 HOMO-LUMO levels, energy gaps and dipole moments of molecular couples in TBIMG and TBIMB crystals

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:

