图 1.
远程C—H键官能化反应
Figure 1.
Remote C—H functionalization via HAT

光学活性的烷基腈类化合物广泛存在于天然产物、药物、农药、材料等领域[1], 而且可以进一步直接被转化为合成化学中应用广泛的光学活性的羧酸、酰胺、烷基胺等[2].因此, 光学活性的烷基腈类化合物的高效合成方法的建立已成为有机化学的研究热点之一.其中, C—H键的直接不对称氰基化无疑是合成手性烷基腈的最简洁高效的方式.
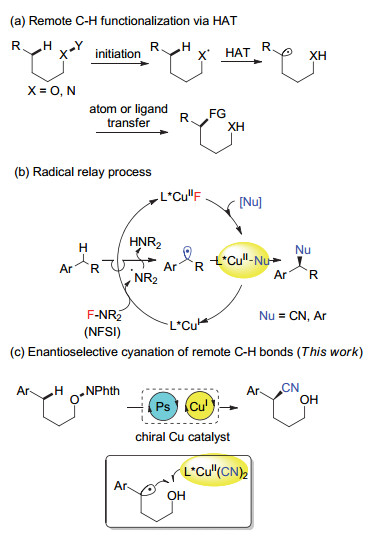
sp3 C—H键的选择性官能化反应是有机化学研究的重要领域之一, 为药物活性分子的后期修饰提供了高效工具[3].但是目前仍然存在着很多挑战, 如sp3 C—H键往往是比较惰性的, 而且底物分子中的性质相似的C—H键很多, 很难有效区别, 因此对反应的区域和立体选择性控制非常困难[4].受细胞色素P450和非血红素铁酶等酶催化反应的启发, 国内外很多课题组通过自由基攫氢(HAT)方式发展了不同类型的C—H键官能化反应[5], 尤其是通过动力学有利的1, 5-氢迁移方式, 高区域选择性地实现远端C—H键的官能化反应(图 1a)[6~9].这类反应通常涉及如下过程:生成自由基前体, 产生自由基, 区域选择性氢迁移和捕获接力的自由基.其中, 较常见的引发氢迁移的自由基包括氮自由基、氧自由基和碳自由基. 2015年, Muñiz课题组[7a]报道了碘催化的Hofmann-Löffler反应, 高效合成了含氮杂环化合物. Knowles课题组[7b]和Rovis课题组[7c]分别利用光催化氧化N—H键产生氮自由基, 通过1, 5-氢迁移实现了远程C—H键的烷基化反应.此外, 除了氮自由基驱动的氢迁移反应, 氧自由基也可以驱动这一反应.陈以昀课题组[8b]报道了首例可见光催化产生烷氧自由基, 通过1, 5-氢迁移实现C—H键烯丙基化反应.焦宁课题组[8e]利用银/过硫酸钾体系产生氧自由基, 实现了非活化烷基醇的直接远程C—H键官能化反应.朱晨课题组[8f~8h]利用高价碘试剂氧化醇获得氧自由基, 通过1, 5-氢迁移实现了δ位芳基化、氰基化等官能化反应.最近, 祝介平课题组[8k]光催化还原N—O键产生氧自由基, 发生1, 5-氢迁移后, 通过铜催化自由基接力的策略, 实现了远程C—H键的叠氮化、氰基化等反应.相比之下, 碳自由基驱动的氢迁移反应报道较少, 这是由于不同C—H键的键能相近, 氢迁移的驱动力较小.刘心元课题组[9a]利用三氟甲基自由基对烯烃加成产生的碳自由基, 促进5位活性氢(胺基α位)迁移, 实现了膦催化的胺基β位官能化反应.许鹏飞课题组[9c]报道了光催化芳基碘化物产生芳基自由基发生1, 5-氢迁移合成氧化吲哚.尽管通过1, 5-氢迁移反应可以很好控制反应的区域选择性, 但是产生的新的烷基自由基往往经历的是直接的自由基官能化反应, 其对映选择性控制非常困难, 因此相应的不对称反应几乎没有报道[8k].
我们课题组一直致力于发展自由基不对称反应, 通过发展铜催化自由基接力的策略[10], 分别以三甲基腈硅烷(TMSCN)和ArB(OH)2作为亲核试剂, 实现了苄位C—H键的不对称氰基化和芳基化反应(图 1b)[11].机理研究表明, 通过自由基攫氢产生的苄位自由基可以高选择性地被活性的手性二价铜氰物种捕获, 形成的三价铜中间体发生还原消除, 从而高效得到光学纯的苄基腈类化合物.基于这一策略, 进一步实现了一系列铜催化烯烃的不对称氰基化、芳基化和炔基化反应[12].近期, 我们还报道了光/铜共催化N-羟基邻苯二甲酰亚胺羧酸酯的不对称脱羧氰基化反应, 反应中光催化剂可以还原N—O键产生氧自由基, 继而脱羧产生苄位自由基, 实现不对称氰基化反应[13].在此基础上, 本文利用光/铜共催化体系, 通过N-烷氧基邻苯二甲酰亚胺底物的单电子还原产生氧自由基, 发生1, 5-氢迁移得到苄位自由基, 实现了高选择性的远程sp3 C—H键的不对称氰基化反应(图 1c).
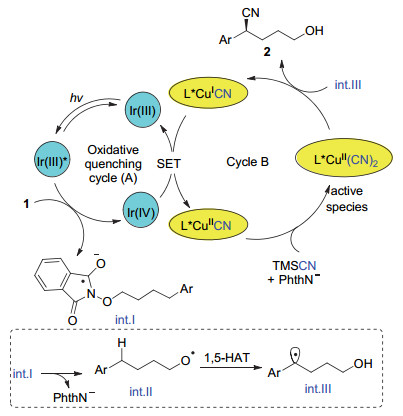
文献报道N-烷氧基邻苯二甲酰亚胺是高效产生氧自由基的活性前体之一[14, 8b].在前期工作中我们发现, Ir(ppy)3在光照下激发后可以经历氧化淬灭还原N—O键, 同时产生的Ir(ppy)3+可以氧化一价铜至二价铜, 实现苄基羧酸酯的脱羧不对称氰基化反应[13].因此, 设想通过这一体系有可能通过N—O键断裂产生烷氧自由基, 结合氧自由基的1, 5-迁移反应获得苄位自由基, 与手性铜氰物种反应, 实现氧自由基启动的远程C—H键不对称氰基化反应.于是我们以N-烷氧基邻苯二甲酰亚胺1a为模板底物, Cu(CH3CN)4BF4为催化剂和L1为配体, TMSCN为氰基来源, 在DMF溶剂中首先考察了一系列光催化剂对反应的影响, 结果如表 1所示:当使用Ir(bpy)3为光敏剂时, 在24 W的蓝光照射下, 反应可以以31%的收率和77%的ee值得到远程C—H键不对称氰基化产物(2a+2a'), 其中2a'是生成的羟基产物2a部分与体系中的三甲基硅基反应所致; 同时反应还伴随着大量烷氧基还原产物3a和环化副产物4a的生成(表 1, Entry 1).当使用其他氧化性更高的光催化剂时, 如Ir(ppy)2(dtbbpy)PF6 ([Ir]-1), Ir(dFCF3ppy)2(dtbbpy)PF6 ([Ir]-2)和Ru(bpy)3Cl2•6H2O ([Ru]), 反应效率并没有得到提升, 但是反应ee值没有受到影响(表 1, Entries 2~4).然而当使用有机光敏剂Eosin Y时, 反应无法发生(表 1, Entry 5).溶剂考察显示在二氯甲烷和三氟甲苯中反应能够表现出较优的对映选择性控制(85% ee), 令人高兴的是在二氯甲烷中反应的收率能够达到93%(表 1, Entries 6~8).对照实验显示在避光条件下, 反应无法发生, 而且铜催化剂和光催化剂都在反应中起了至关重要的作用(表 1, Entries 9~11).
 下载:
导出CSV
下载:
导出CSV
 |
||||
| Entry | Photo cat. | Solvent | 2a+2a' Yield (ee)b | 3a+4a Yieldb |
| 1 | Ir(bpy)3 | DMF | 31% (77%) | 47% |
| 2 | [Ir]-1 | DMF | 16% (77%) | 16% |
| 3 | [Ir]-2 | DMF | 30% (77%) | 32% |
| 4 | [Ru] | DMF | 8% (76%) | 11% |
| 5 | Eosin Y | DMF | 0 | 0 |
| 6 | Ir(bpy)3 | DCM | 93% (85%) | 7% |
| 7 | Ir(bpy)3 | PhCF3 | 57% (85%) | 6% |
| 8 | Ir(bpy)3 | CH3CN | 71% (79%) | 27% |
| 9 | Ir(bpy)3 | DCM | 0 | 0 |
| 10 | Ir(bpy)3 | DCM | 0 | 0 |
| 11 | — | DCM | 0 | 0 |
| a The reactions were conducted on 0.1 mmol scale with photocatalyst (1 mol%) and Cu(CH3CN)4BF4 (5 mol%) in solvent (2 mL) at room temperature, irradiated with 24 W blue LED. b Crude 1H NMR yield with CH3NO2 as an internal standard, enantiomeric excess (ee) values were determined by HPLC on a chiral stationary phase. c ln dark. d Without Cu(CH3CN)4BF4. [Ir]-1=Ir(ppy)2(dtbbpy)PF6.[Ir]-2=Ir(dFCF3ppy)2(dtbbpy)PF6.[Ru]=Ru(bpy)3Cl2" 6H2O. | ||||
通过对反应条件的系统优化, 我们确定了反应的最优条件:以N-烷氧基邻苯二甲酰亚胺1为底物, Ir(bpy)3 (1 mol%)为光催化剂, Cu(CH3CN)4BF4 (5 mol%)为催化剂, L1 (7.5 mol%)为配体, TMSCN (2 equiv.)为氰基来源, 在二氯甲烷中24 W蓝光进行光照反应.我们对底物的普适性进行了考察(表 2), 实验结果显示一系列不同取代的4-芳基-1-丁醇的邻苯二甲酰亚胺衍生物都可以顺利反应, 并以良好到优秀的产率高选择性地得到目标产物(2a~2o).各种不同的官能团(如, 卤素、酯基、醚、硼酯、三氟甲氧基等)也都能够在反应体系中兼容; 其中芳基对位无论是吸电子还是供电子取代基的底物1a~1f均能够以良好至优秀的收率(80%~93%), 良好的对映选择性(69%~90% ee)得到目标产物.值得注意的是, 对位硼酯取代的底物1g也能兼容反应, 并以91% ee对映选择性得到产物2g, 这为后续的转化反应提供了可能.同样间位不同电性取代的底物(1h~1l, 1n~1o)也能够顺利进行反应; 当芳基邻位有小位阻的取代基时(1m), 反应仍然可以正常进行, 但增大位阻不利于反应.我们也进一步考察了萘基底物, 无论是α-萘还是β-萘取代的底物, 分别以92%~90%的收率和90%~89%的ee值得到产物2p和2q.鉴于杂环在生物活性分子修饰和药物分子设计中具有重要的应用价值, 我们也考察了含杂环的底物, 其中噻吩类底物能以高达95% ee对映选择性顺利给出目标产物2r.烷基链上的偕二取代也不会影响1, 5-氢迁移反应, 反应仍然能够以85%的收率和83%的ee值获得产物2s.此外2-乙基-苯甲醇类衍生物也能顺利进行反应, 以75%的收率和80%的ee值得到产物2t.最后, 所得到的目标产物1a可以通过简单的兰尼镍氢化, 高收率地得到光学活性的5-羟基烷基酰胺类化合物5, 同时对映选择性得到保留(Eq. 1).
下载:
导出CSV
 |
 |
| a All the reactions were conducted on 0.2 mmol scale. b Isolated yields after treatment with TBAF/HOAc (3 equiv.) and enantiomeric excess (ee) values were determined by HPLC on a chiral stationary phase. c For easy separation, alcohol was converted to ester with AcCl. |
基于前期的研究工作[13], 我们推测可能的反应机理如图 2所示:光催化剂Ir(ppy)3在蓝光照下被激发, 激发态的Ir(Ⅲ)*具有很强的还原性, 还原底物N-烷氧基邻苯二甲酰亚胺1形成阴离子自由基int.Ⅰ, 进一步分解为氧自由基int.Ⅱ和邻苯二甲酰亚胺阴离子(PhthN-).随后发生1, 5-氢迁移得到苄位自由基int.Ⅲ; 同时被氧化淬灭后的光催化剂Ir(Ⅳ)可以氧化一价铜(L*CuⅠCN)形成二价铜(L*CuⅡCN), 再生光催化剂Ir(Ⅲ).二价铜中间体(L*CuⅡCN)在邻苯二甲酰亚胺阴离子的促进下, 与TMSCN反应形成高活性的L*CuⅡ(CN)2, 快速地捕捉苄基自由基int.Ⅲ, 高选择性得到远程C—H键不对称氰基化产物2, 同时再生一价铜催化剂.在整个反应中, 光催化剂的氧化淬灭循环(Cycle A)与铜催化不对称氰基化反应(Cycle B)的匹配非常关键.根据前期的脱羧氰基化反应[13], Ir(ppy)3+的还原电势是{E1/2red[IrⅣ/IrⅢ]= +0.77 V vs. SCE in MeCN}, 而手性配体配位一价铜(L*CuⅠCN)的还原电势为{E1/2red[CuⅡ/CuⅠ]=+0.36 V vs. SCE in MeCN}, 因此Ir(ppy)3+可以氧化一价铜至二价铜, 促进两个催化循环顺利进行.
本文基于铜催化自由基接力的策略, 利用光/铜共催化, 发展了δ-芳基烷基醇的远程C—H键不对称氰基化反应.反应以N-烷氧基邻苯二甲酰亚胺为底物, 通过光照产生氧自由基, 经历1, 5-氢迁移过程, 以良好到优秀的对映选择性, 区域单一地得到光学活性的δ-氰基醇类化合物.该方法条件温和, 具有较好的官能团兼容性以及底物普适性.所得到的产物中氰基可以进一步通过氢化还原为胺基, 为光学活性的δ-氰基醇和1, 6-氨基醇类化合物的合成提供了高效方法.
C—H键不对称氰基化反应操作步骤:在手套箱中, 往干燥的封管中称取底物1 (0.2 mmol, 1.0 equiv.)、Cu(CH3CN)4PF6 (0.01 mmol, 5 mol%), 手性配体(0.015 mmol, 7.5 mol%)和光催化剂Ir(ppy)3 (0.002 mmol, 1 mol%).取出后氮气保护下加入二氯甲烷(4 mL), 避光下搅拌30 min, 使配体和铜催化剂配位.然后慢慢加入TMSCN (50 μL, 0.4 mmol, 2 equiv.), 封管后在24 W蓝光照射下室温反应1~7 d.反应结束后, 用二氯甲烷(10 mL)稀释, 过硅藻土短柱, 滤液加入TBAF (3 equiv.)和HOAc (3 equiv.)搅拌5 min, 加入饱和食盐水, 用乙酸乙酯萃取, 合并有机相用无水硫酸钠干燥, 过滤, 浓缩后, 用硅胶柱层析分离得到产物2.
(a) Wang, J.; Liu, H. Chin. J. Org. Chem. 2012, 32, 1643. (王江, 柳红, 有机化学, 2012, 32, 1643.) (b) Fleming, F. F.; Yao, L.; Ravikumar, P. C.; Funk, L.; Shook, B. C. J. Med. Chem. 2010, 53, 7902.
(a) Rappoport, Z. The Chemistry of the Cyano Group, Interscience Publishers, London, 1970. (b) Larock, R. C. Comprehensive Organic Transformations: A Guide to Functional Group Preparation, 2nd ed., Wiley-VCH, Weinheim, 1999, p. 821.
Cernak, T.; Dykstra, K. D.; Tyagarajan, S.; Vachal, P.; Krska, S. W. Chem. Soc. Rev. 2016, 45, 546. doi: 10.1039/C5CS00628G
(a) Meunier, B.; de Visser, S. P.; Shaik, S. Chem. Rev. 2004, 104, 3947. (b) Ortiz de Montellano, P. R. Chem. Rev. 2010, 110, 932.
For some reviews, see: (a) Che, C.-M.; Lo, V. K.-Y.; Zhou, C.-Y.; Huang, J.-S. Chem. Soc. Rev. 2011, 40, 1950. (b) Lu, H.; Zhang, X. P. Chem. Soc. Rev. 2011, 40, 1899. (c) Huang, X.; Groves, J. T. Chem. Rev. 2018, 118, 2491. (d) Bietti, M. Angew. Chem., Int. Ed. 2018, 57, 16618. (e) Pei, P.; Zhang, F.; Yi, H.; Lei, A. Acta Chim. Sinica 2017, 75, 15. (裴朋昆, 张凡, 易红, 雷爱文, 化学学报, 2017, 75, 15.)
For some reviews, see:Stateman, L. M.; Nakafuku, K. M.; Nagib, D. A. Synthesis 2018, 50, 1569. doi: 10.1055/s-0036-1591930
(a) Martínez, C.; Muñiz, K. Angew. Chem., Int. Ed. 2015, 54, 8287. (b) Choi, G. J.; Zhu, Q.; Miller, D. C.; Gu, C. J.; Knowles, R. R. Nature 2016, 539, 268. (c) Chu, J. C. K.; Rovis, T. Nature 2016, 539, 272. (d) Chen, D.; Chu, J. C. K.; Rovis, T. J. Am. Chem. Soc. 2017, 139, 14897. (e) Wappes, E. A.; Fosu, S. C.; Chopko, T. C.; Nagib, D. A. Angew. Chem., Int. Ed. 2016, 55, 9974. (f) Liu, T.; Myers, M. C.; Yu, J.-Q. Angew. Chem., Int. Ed. 2017, 56, 306. (g) Becker, P.; Duhamel, T.; Stein, C. J.; Reiher, M.; Muñiz, K. Angew. Chem., Int. Ed. 2017, 56, 8004. (h) Li, Z.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2018, 57, 13288. (i) Jiang, H.; Studer, A. Angew. Chem., Int. Ed. 2018, 57, 1692. (j) Xia, Y.; Wang, L.; Studer, A. Angew. Chem., Int. Ed. 2018, 57, 12940. (k) Dauncey, E. M.; Morcillo, S. P.; Douglas, J. J.; Sheikh, N. S.; Leonori, D. Angew. Chem., Int. Ed. 2018, 57, 744. (l) Morcillo, S. P.; Dauncey, E. M.; Kim, J. H.; Douglas, J. J.; Sheikh, N. S.; Leonori, D. Angew. Chem., Int. Ed. 2018, 57, 12945. (m) Li, C.; Lang, K.; Lu, H.; Hu, Y.; Cui, X.; Wojtas, L.; Zhang, X. P. Angew. Chem., Int. Ed. 2018, 57, 16837. (n) Chen, H.; Guo, L.; Yu, S. Org. Lett. 2018, 20, 6255. (o) Stateman, L. M.; Wappes, E. A.; Nakafuku, K. M.; Edwards, K. M.; Nagib, D. A. Chem. Sci. 2019, 10, 2693. (p) Zhang, Z.; Stateman, L. M.; Nagib, D. A. Chem. Sci. 2019, 10, 1207. (q) Wu, K.; Wang, L.; Colón-Rodríguez, S.; Flechsig, G.-U.; Wang, T. Angew. Chem., Int. Ed. 2019, 58, 1774. (r) Bao, X.; Wang, Q.; Zhu, J. Nature Commun. 2019, 10, 768. (s) Lang, K.; Torker, S.; Wojtas, L.; Zhang, X. P. J. Am. Chem. Soc. 2019, DOI: 10.1021/jacs.9b05850.
(a) Peng, Y.; Lin, J.-S.; Li, L.; Zheng, S.-C.; Xiong, Y.-P.; Zhao, L.-J.; Tan, B.; Liu, X.-Y. Angew. Chem., Int. Ed. 2014, 53, 11890. (b) Zhang, J.; Li, Y.; Zhang, F.; Hu, C.; Chen, Y. Angew. Chem., Int. Ed. 2016, 55, 1872. (c) Wang, C. Y.; Harms, K.; Meggers, E. Angew. Chem., Int. Ed. 2016, 55, 13495. (d) Hu, A.; Guo, J.-J.; Pan, H.; Tang, H.; Gao, Z.; Zuo, Z. J. Am. Chem. Soc. 2018, 140, 1612. (e) Zhu, Y.; Huang, K.; Pan, J.; Qiu, X.; Luo, X.; Qin, Q.; Wei, J.; Wen, X.; Zhang, L.; Jiao, N. Nat. Commun. 2018, 9, 2625. (f) Wu, X.; Zhang, H.; Tang, N.; Wu, Z.; Wang, D.; Ji, M.; Xu, Y.; Wang, M.; Zhu, C. Nat. Commun. 2018, 9, 3343. (g) Wu, X.; Wang, M.; Huan, L.; Wang, Wang, D. J.; Zhu, C. Angew. Chem., Int. Ed. 2018, 57, 1640. (h) Wang, M.; Huang, L.; Zhu, C. Org. Lett. 2019, 21, 821. (i) Kim, I.; Park, B.; Kang, G.; Kim, J.; Jung, H.; Lee, H.; Baik, M.; Hong, S. Angew. Chem., Int. Ed. 2018, 57, 15517. (j) Guan, H.; Sun, S.; Mao, Y.; Chen, L.; Lu, R.; Huang, J.; Liu, L. Angew. Chem. Int. Ed. 2018, 57, 11413. (k) Bao, X.; Wang, Q.; Zhu, J. Angew. Chem. Int. Ed. 2019, 58, 2139.
(a) Yu, P.; Zheng, S.-C.; Yang, N.-Y.; Tan, B.; Liu, X.-Y. Angew. Chem., Int. Ed. 2015, 54, 4041. (b) Cui, X.; Xu, X.; Jin, L.-M.; Wojtasa, L.; Zhang, X. P. Chem. Sci. 2015, 6, 1219. (c) Chen, J.-Q.; Wei, Y.-L.; Xu, G.-Q.; Liang, Y.-M.; Xu, P.-F. Chem. Commun. 2016, 52, 6455. (d) Li, T.; Yu, P.; Lin, J.-S.; Zhi, Y.; Liu, X.-Y. Chin. J. Chem. 2016, 34, 490. (e) Li, L.; Ye, L.; Ni, S.-F.; Li, Z.-L.; Chen, S.; Du, Y.-M.; Li, X.-H.; Dang, L.; Liu, X.-Y. Org. Chem. Front. 2017, 4, 2139. (f) Yuan, W.; Zhou. Z.; Gong, L.; Meggers, E. Chem. Commun. 2017, 53, 8964. (g) Li, T.; Yu, P.; Du, Y.-M.; Lin, J.-S.; Zhi, Y.; Liu, X.-Y. J. Fluorine Chem. 2017, 203, 210. (h) Wang, N.; Ye, L.; Li, Z.-L.; Li, L.; Li, Z.; Zhang, H.-X.; Guo, Z.; Gu, Q.-S.; Liu, X.-Y. Org. Chem. Front. 2018, 5, 2810. (i) Chen, J.-Q.; Chang, R.; Lin, J.-B.; Luo, Y.-C.; Xu, P.-F. Org. Lett. 2018, 20, 2395. (j) Wang, Y.; Wen, X.; Cui, X.; Zhang, X. P. J. Am. Chem. Soc. 2018, 140, 4792. (k) Wen, X.; Wang, Y.; Zhang, X. P. Chem. Sci. 2018, 9, 5082. (l) Wu, S.; Wu, X.; Wang, D.; Zhu, C. Angew. Chem., Int. Ed. 2019, 58, 1499. (m) Chuentragool, P.; Yadagiri, D.; Morita, T.; Sarkar, S.; Parasram, M.; Wang, Y.; Gevorgyan, V. Angew. Chem. Int. Ed. 2019, 58, 1794.
Wang, F.; Chen, P.; Liu, G. Acc. Chem. Res. 2018, 51, 2036. doi: 10.1021/acs.accounts.8b00265
(a) Zhang, W.; Wang, F.; McCann, S. D.; Wang, D.; Chen, P.; Stahl, S. S.; Liu, G. Science 2016, 353, 1014. (b) Zhang, W.; Wu, L.; Chen, P.; Liu, G. Angew. Chem., Int. Ed. 2019, 58, 6425. (c) Zhang, W.; Chen, P.; Liu, G. J. Am. Chem. Soc. 2017, 139, 7709.
For cyanations, see: (a) Wang, F.; Wang, D.; Wan, X.; Wu, L.; Chen, P.; Liu, G. J. Am. Chem. Soc. 2016, 138, 15547. (b) Wang, D.; Wang, F.; Chen, P.; Lin, Z.; Liu, G. Angew. Chem., Int. Ed. 2017, 56, 2054. (c) Lu, F.-D.; Liu, D.; Zhu, L.; Lu, L.-Q.; Yang, Q.; Zhou, Q.-Q.; Wei, Y.; Lan, Y.; Xiao, W.-J. J. Am. Chem. Soc. 2019, 141, 6167. For arylations, see: (d) Wu, L.; Wang, F.; Wan, X.; Wang, D.; Chen, P.; Liu, G. J. Am. Chem. Soc. 2017, 139, 2904. (e) Wang, D.; Wu, L.; Wang, F.; Wan, X.; Chen, P.; Lin, Z.; Liu, G. J. Am. Chem. Soc. 2017, 139, 6811. For alkynylation, see: (f) Fu, L.; Zhou, S.; Wan, X.; Chen, P.; Liu, P. J. Am. Chem. Soc. 2018, 140, 10965.
Wang, D.; Zhu, N.; Chen, P.; Lin, Z.; Liu, G. J. Am. Chem. Soc. 2017, 139, 15632. doi: 10.1021/jacs.7b09802
(a) Curran, D. P.; Kim, D.; Liu, H. T.; Shen, W. J. Am. Chem. Soc. 1988, 110, 5900. (b) Kim, S.; Lee, T. A.; Song, Y. Synlett 1998, 471. (c) Zlotorzynska, M.; Sammis, G. M. Org. Lett. 2011, 13, 6264.
表 1 反应条件优化a
Table 1. Reaction condition optimization
|
|
||||
| Entry | Photo cat. | Solvent | 2a+2a' Yield (ee)b | 3a+4a Yieldb |
| 1 | Ir(bpy)3 | DMF | 31% (77%) | 47% |
| 2 | [Ir]-1 | DMF | 16% (77%) | 16% |
| 3 | [Ir]-2 | DMF | 30% (77%) | 32% |
| 4 | [Ru] | DMF | 8% (76%) | 11% |
| 5 | Eosin Y | DMF | 0 | 0 |
| 6 | Ir(bpy)3 | DCM | 93% (85%) | 7% |
| 7 | Ir(bpy)3 | PhCF3 | 57% (85%) | 6% |
| 8 | Ir(bpy)3 | CH3CN | 71% (79%) | 27% |
| 9 | Ir(bpy)3 | DCM | 0 | 0 |
| 10 | Ir(bpy)3 | DCM | 0 | 0 |
| 11 | — | DCM | 0 | 0 |
| a The reactions were conducted on 0.1 mmol scale with photocatalyst (1 mol%) and Cu(CH3CN)4BF4 (5 mol%) in solvent (2 mL) at room temperature, irradiated with 24 W blue LED. b Crude 1H NMR yield with CH3NO2 as an internal standard, enantiomeric excess (ee) values were determined by HPLC on a chiral stationary phase. c ln dark. d Without Cu(CH3CN)4BF4. [Ir]-1=Ir(ppy)2(dtbbpy)PF6.[Ir]-2=Ir(dFCF3ppy)2(dtbbpy)PF6.[Ru]=Ru(bpy)3Cl2" 6H2O. | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 底物范围a
Table 2. Substrate scope
|
|
|
|
| a All the reactions were conducted on 0.2 mmol scale. b Isolated yields after treatment with TBAF/HOAc (3 equiv.) and enantiomeric excess (ee) values were determined by HPLC on a chiral stationary phase. c For easy separation, alcohol was converted to ester with AcCl. |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们