State Key Laboratory of Bio-organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032
b.
Institute of Clinical Science, Zhongshan Hospital, Shanghai Medical School, Fudan University, Shanghai 200032
Received Date:
25 June 2019 Available Online:
15 October 2019
Fund Project:
Project supported by the Funds from the National Natural Science Foundation of China (Nos. 21432012, 21621002), the Chinese Academy of Sciences (Strategic Priority Research Program, No. XDB20020200), the Youth Innovation Promotion Association (No. 2017300) and the K.C. Wong Education Foundation
Abstract:
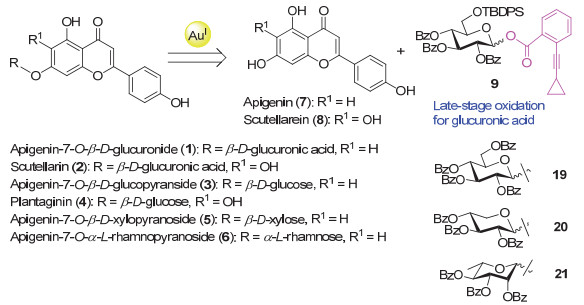
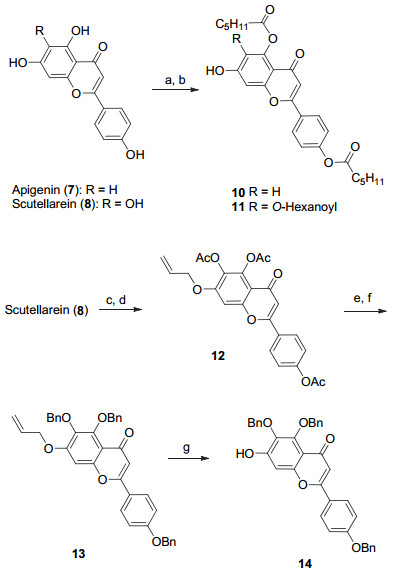
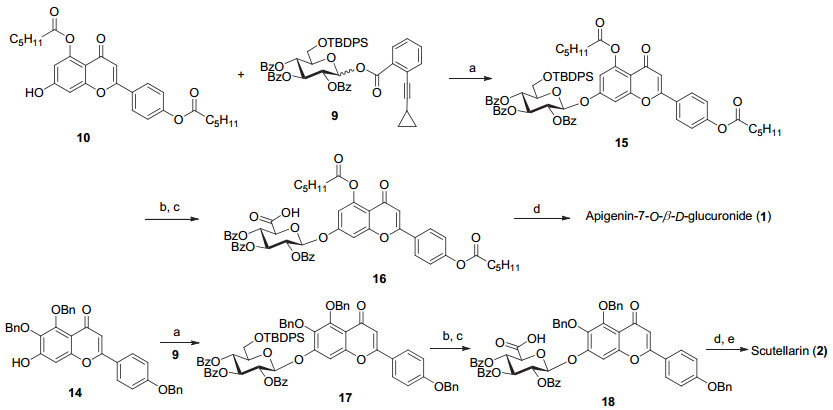
Apigenin-7-O-β-D-glucuronide (1) and scutellarin (scutellarein-7-O-β-D-glucuronide, 2) are two major flavone glucuronide components occurring in breviscapines, which are prepared from the traditional Chinese herb Erigeron breviscapus. These two flavone glycosides show potent anti-oxidative, anti-inflammatory and neuroprotective activities in various evaluations. Synthesis of these natural glycosides in an efficiently manner would facilitate studies on their structure activity relationships. As a persistent effort on the chemical syntheses of the diverse glycoconjugates from traditional Chinese herbs in our group, we report herein the synthesis of these two representative flavone O-glucuronides. It is known that the solubility of flavone compounds is rather low and this property would greatly hinder their glycosylation reactions. In order to increase the solubility of the flavone derivatives in the glycosylation solvents, hexanoyl and benzyl groups were selected as the permanent protecting groups for the hydroxyl groups of apigenin (7) and scutellarein (8). The construction of the phenolic O-glucuronide is known to be a difficult task, especially the glycosylation of the poorly nucleophilic 7-hydroxyl group which locates at the para-position of the flavone carbonyl group. We achieved the glycosylation of the flavone 7-OH with 2, 3, 4-tri-O-benzoyl-6-O-TBDPS-glucopyranosyl ortho-alkynylbenzoate (9) under the catalysis of Ph3PAuNTf2 (0.2 equiv., 4 Å MS, CH2Cl2, r.t., 5 h) in excellent yields. After that, the 6-O-TBDPS groups were removed, and the requisite glucuronides were then elaborated by oxidation of the resulting 6-OH under the conditions of DAIB/TEMPO (CH2Cl2/H2O, V:V=2:1, r.t.) in good yields. After global deprotection, the desired products apigenin-7-O-β-D-glucuronide (1) and scutellarin (2) were obtained in overall yields of 36% (5 steps) and 7% (9 steps), respectively, from the starting flavone aglycones. Following the same strategy, four naturally occurring flavone-7-O-glycosides, namely apigetrin (3), plantaginin (4), apigenin 7-O-β-D-xylopyranoside (5) and apigenin 7-O-α-L-rhamnopyranoside (6), were smoothly synthesized in 4~7 steps with the overall yields of 61%, 13%, 58% and 61%, respectively.
(a) Cui, J. M.; Wu, S. Nat. Prod. Res. Dev. 2003, 15, 255. (b) Ma, Y. H.; Luo, G. A.; Wang, Y. M. Chin. Tradit. Pat. Med. 2004, 1, 63. (c) Wang, J.; Zhang, L.; Liu, B.; Wang, Q.; Chen, Y.; Wang, Z.; Zhou, J.; Xiao, W.; Zheng, C.; Wang, Y. J. Ethnopharmacol. 2018, 224, 429.
[2]
(a) Yue, J. M.; Lin, Z. W.; Wang, D. Z. Phytochemistry1994, 36, 717. (b) Xia, H. J.; Qiu, F.; Zhu, S.; Zhang, T. Y.; Qu, G. X.; Yao, X. S. Biol. Pharm. Bull. 2007, 30, 1308.
(a) Wu, W. H.; Chen, T. Y.; Lu, R. W.; Chen, S. T.; Chang, C. C. Phytochemistry2012, 83, 110. (b) Chen, V.; Staub, R. E.; Baggett, S.; Chimmani, R.; Tagliaferri, M.; Cohen, I.; Shtivelman, E. PLoS One2012, 7, e30107.
[5]
(a) Huang, X. W.; Xu, Y.; Sui, X.; Lin, H.; Xu, J. M.; Han, D.; Ye, D. D.; Lv, G. F.; Liu, Y. X.; Qu, X. B.; Duan, M. H. Oncol. Lett. 2019, 17, 5581. (b) Li, H. M.; Gu, T.; Wu, W. Y.; Yu, S. P.; Fan, T. Y.; Zhong, Y.; Li, N. G. Med. Chem. 2018, 14, 1.
[6]
(a) Sherbeiny, E.; Ansari, E. Planta Med. 1976, 29, 129. (b) Homberg, H.; Geiger, H. Phytochemistry1980, 19, 2443. (c) Smirnova, L. P.; GlyzinA, V. I.; Patudin, A. V.; Bankovskii, A. I. Chem. Nat. Compd. 1974, 10, 687. (d) Shabrawy, M. O. A.; Hosni, H. A.; Garf, I. A.; Marzouk, M. M.; Kawashty, S. A.; Saleh, N. A. M. Biochem. Syst. Ecol. 2014, 56, 125.
[7]
(a) Jacobsson, M.; Malmberg, J.; Ellervik, U. Carbohydr. Res. 2006, 341, 1266. (b) Sun, J. S.; Laval, S.; Yu, B. Synthesis2014, 46, 1030. (c) Li, Y.; Yang, W. Z.; Ma, Y.; Sun, J. S.; Shan, L.; Zhang, W. D.; Yu, B. Synlett2011, 915. (d) Yang, W. Z.; Sun, J. S.; Yang, Z.; Han, W.; Zhang, W. D.; Yu, B. Tetrahedron Lett. 2012, 53, 2773. (e) Hu, Y.; Tu, Y. H.; Liu, D. Y.; Liao, J. X.; Sun, J. S. Org. Biomol. Chem. 2016, 14, 4842. (f) Liao, J. X.; Fan, N. L.; Liu, H.; Tu, Y. H.; Sun, J. S. Org. Biomol. Chem. 2016, 14, 1221. (g) Wang, Y.; Liu, M.; Liu, L.; Xia, J. H.; Du, Y. G.; Sun, J. S. J. Org. Chem. 2018, 83, 4111.
[8]
(a) Farkas, L.; Mezey-Vandor, G.; Nogradi, M. Chem. Ber. 1971, 104, 2681. (b) Farkas, L.; Mezey-Vandor, G.; Nogradi, M. Chem. Ber. 1974, 107, 3874. (c) Synthesis for 1: Li, P. H.; Zhang, Z. P.; Zhang, W.; Yang, Z. X. CN 104761599, 2015. (d) Synthesis of 2: (i) Nagashima, S.; Hirotani, M.; Yoshikawa, T. Phytochemistry2000, 53, 533; (ii) Li, P. H.; Zhang, W.; Yang, Z. X.; Zhang, X. B.; Wang, J.; Zhu, H. B.; Chen, J. X.; Bai, Y. Y. EP 2840088, 2015. (e) Synthesis of 3: (i) Nakaoki, N. Yakugaku Zasshi1940, 60, 502. (ii) Oyama, K. I.; Kondo, T. Tetrahedron2004, 60, 2025; (iii) Liu, J. D.; Chen, L.; Cai, S. L.; Wang, Q. Carbohydr. Res. 2012, 357, 41; (iv) Zheng, Z. W.; Han, Z. Y.; Cai, L.; Zhou, D. D.; Chavis, B. R.; Li, C. S.; Sui, Q.; Jiang, K. Y.; Gao, Q. Tetrahedron Lett. 2018, 59, 4442. (f) Synthesis of 4: Li, N. G.; Shen, M. Z.; Wang, Z. J.; Tang, Y. P.; Shi, Z. H.; Fu, Y. F.; Shi, Q. P.; Tang, H.; Duan, J. A. Bioorg. Med. Chem. Lett. 2013, 23, 102.
[9]
(a) Li, Y.; Yang, Y.; Yu, B. Tetrahedron Lett. 2008, 49, 3604. (b) Li, Y.; Yang, X.; Liu, Y.; Zhu, C.; Yang, Y.; Yu, B. Chem.-Eur. J. 2010, 16, 1871. (c) Zhu, Y.; Yu, B. Angew. Chem., Int. Ed. 2011, 50, 8329; (d) Tang, Y.; Li, J.; Zhu, Y.; Li, Y.; Yu, B. J. Am. Chem. Soc. 2013, 135, 18396. (e) Li, W.; Yu, B. Chem. Soc. Rev. 2018, 47, 7954. (f) Yu, B. Acc. Chem. Res. 2018, 51, 507.
[10]
(a) Zhu, D.; Yu, B. Chin. J. Chem. 2018, 36, 681. (b) Li, J.; Yu, B. Angew. Chem., Int. Ed. 2015, 54, 6618. (c) Bai, Y.; Shen, X.; Li, Y.; Dai, M. J. Am. Chem. Soc. 2016, 138, 10838. (d) Wang, B.; Liu, Y.; Jiao, R.; Feng, Y.; Li, Q.; Chen, C.; Liu, L.; He, G.; Chen, G. J. Am. Chem. Soc. 2016, 138, 3926. (e) Nicolaou, K. C.; Cai, Q.; Sun, H.; Qin, B.; Zhu, S. J. Am. Chem. Soc. 2016, 138, 3118. (f) Nie, S. Y.; Li, W.; Yu, B. J. Am. Chem. Soc. 2014, 136, 4157. (g) Zhang, X.; Zhou, Y.; Zuo, J.; Yu, B. Nat. Commun. 2015, 6, 5879. (h) Shen, R. Z.; Cao, X.; Yu. B. Acta Chim. Sinica2018, 76, 278. (沈仁增, 曹鑫, 俞飚, 化学学报, 2018, 76, 278.)
[11]
(a) Yang, W. Z.; Sun, J. S.; Lu, W. X.; Li, Y.; Shan, L.; Han, W.; Zhang, W. D.; Yu, B. J. Org. Chem. 2010, 75, 6879. (b) Yang, W. Z.; Li R. Y.; Han, W.; Zhang, W. D.; Sun, J. S. Chin. J. Org. Chem. 2012, 32, 1067. (杨为准, 李荣耀, 韩伟, 张卫东, 孙建松, 有机化学, 2012, 32, 1067.)
[12]
Liu, X.; Wen, G. E.; Liu, J. C.; Liao, J. X.; Sun, J. S. Carbohydr. Res. 2019, 475, 69. doi: 10.1016/j.carres.2019.02.005
[13]
Karst, N.; Jean-Claude, J. J. Chem. Soc., Perkin Trans. 12000, 16, 2709.
[14]
Yu, J.; Sun, J. S.; Niu, Y. M.; Li, R. Y.; Liao, J. X.; Zhang, F. Y.; Yu, B. Chem. Sci. 2013, 4, 3899. doi: 10.1039/c3sc51479j
[15]
(a) Zulueta, M. M. L.; Lin, S. Y.; Lin, Y. T.; Huang, C. J.; Wang, C. C.; Ku, C. C.; Shi, Z.; Chyan, C. L.; Irene, D.; Lim, L. H.; Tsai, T. I.; Hu, Y. P.; Arco, S. D.; Wong, C. H.; Hung, S. C. J. Am. Chem. Soc. 2012, 134, 8988. (b) Chang, C. H.; Lico, L. S.; Huang, T. Y.; Lin, S. Y.; Chang, C. L.; Arco, S. D.; Hung, S. C. Angew. Chem., Int. Ed. 2014, 53, 9876.
[16]
Li, M.; Han, X. W.; Yu, B. J. Org. Chem. 2003, 68, 6842. doi: 10.1021/jo034553e
[17]
Gao, Q.; Lian, G. Y.; Lin, F. Carbohydr. Res. 2008, 344, 511.

 下载:
下载:






 下载:
下载:




