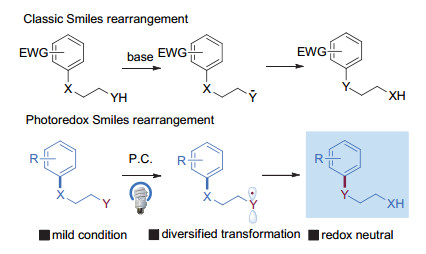
图 1.
经典Smiles重排与光催化Smiles重排反应
Figure 1.
Generalized Smiles rearrangement and photoredox Smiles rearrangement

Smiles重排是一种分子内芳基迁移反应, 是合成复杂、高附加值芳、杂环化合物的重要手段, 在过去数十年来在有机合成、药物化学领域有着广泛的应用[1].该类型反应最早可追溯到1894年由Henrique发现, 并在20世纪30年代由Smiles教授[2]发展并进行了系统的研究.经典的Smiles重排反应是分子内芳基亲核取代反应, 该反应的能垒较高, 需要在芳环上引入吸电子取代基来降低反应的能垒, 这也导致传统Smiles反应对底物的电性较为敏感.此外必要时需要使用较为剧烈的反应条件如加热、强碱活化产生活泼的亲核试剂才能使得反应顺利进行, 这进一步限制了该反应的底物适用性.自20世纪70年代Speckamp等首次报道了单电子转移Smiles过程以后, 自由基Smiles反应得到了一定程度的发展[3].这些新方法一定程度上弥补了传统Smiles重排的底物局限性, 但依旧要额外加入当量的化学氧化剂.
可见光催化氧化还原反应是一类利用光能, 在温和的条件下发生单电子转移的高效的合成模式, 近年来, 随着该领域的迅速发展, 受到了化学家们的广泛关注.过去五年中, 这一催化模式被逐渐应用到Smiles重排中, 使得这一经典反应过程重新焕发出新的活力, 一系列光催化Smiles重排反应被报道(图 1).反应条件温和, 不仅避免了大量使用昂贵氧化剂而且利用绿色的可见光能作为能源, 更重要的是较传统的Smiles反应而言光催化的该过程可以有效地提高官能团兼容性.本文将重点按促进重排反应的自由基类型进行分类并简要介绍.
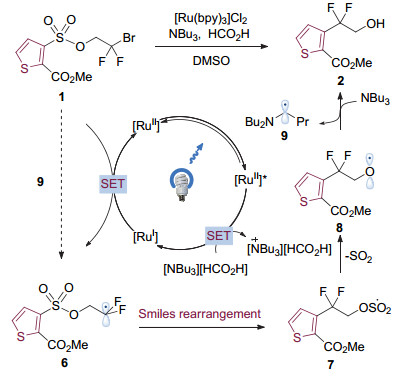
2015年, Stephenson课题组[4]报道了可见光催化的杂环芳基磺酸酯1的Smiles重排反应(图 2), 在Ru(bpy)3催化下, 高效得到了杂环苄位二氟取代的醇2.与传统热驱动Smiles重排相比, 反应条件温和, 不需要加入强碱, 底物适用性广. Stephenson随后将该类型转化应用于二氟取代的螺环结构的构建中, 实现了ORL-1拮抗剂彻块4的高效合成[5], 显示出可见光催化Smiles重排巨大的应用潜力.

在提出的反应机理中(图 3), 作者认为催化剂[Ru(bpy)3]Cl2·6H2O被光激发后生成激发态的二价钌中间体(E1/2(M*/M-)=+0.77 V vs. SCE), 该中间体能够被体系中加入的三丁基胺还原淬灭, 生成强还原性的一价钌配合物(E1/2(M/M-)=-1.33 V vs. SCE), 随后与底物中的氟烷基溴发生单电子转移(Single-Electron Transfer, SET), 本身被氧化成为二价钌配合物, 完成光催化剂的循环, 同时生成碳自由基物种6.中间体6发生Smiles重排并脱除一分子二氧化硫, 得到烷氧自由基8.中间体8被体系中的胺还原成为最终产品2.进一步研究表明, 中间体8被胺还原后产生的氮α位碳自由基9也具有一定的还原性, 能够还原底物中较为缺电子的碳溴键, 因此作者认为体系中可能同时存在自由基链式过程[6].
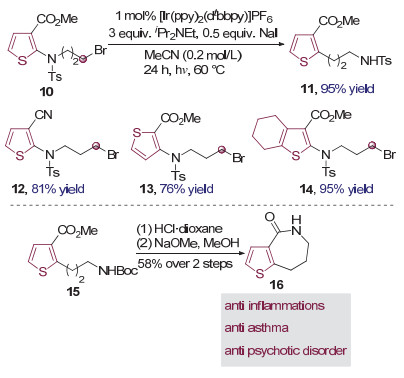
2016年, 张兆国、万均等[7]使用还原能力更强的光催化剂来还原取代的芳香磺酰胺分子内的碳溴键来产生碳自由基, 引发Smiles重排反应.由于Smiles重排中常用的芳香磺酰胺/磺酸酯制备较为繁琐, 与此相比, 如何利用更易获得的芳香胺类化合物引起了研究者们的注意. 2018年, Stephenson课题组[8]使用烷基侧链上有溴原子取代的芳胺类化合物10作为底物, 通过向体系中加入碘化钠现场生成碘代Smiles重排前体, 利用光催化还原碳碘键的方式产生强亲核性的伯碳自由基进攻缺电子芳环, 实现了可见光催化的芳香胺Smiles重排反应, 得到了杂芳基取代的酰胺化合物11(图 4).由于条件温和, 反应具有优秀的底物适用性, 芳环具有不同取代基的底物均可以顺利的发生反应.值得注意的是, 产品15可通过两步简单合成操作转化为具有多种生物活性的重要化合物tetrahydrothienoazepinone 16(图 4), 显示出了该转化方法较高的应用价值.
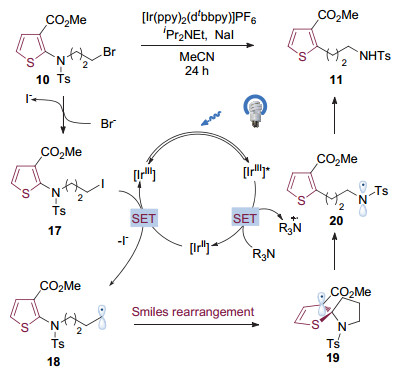
在反应机理方面, 作者认为[Ir(ppy)2(dtbbpy)]PF6在光照下被激发成为激发态的三价铱配合物(图 5), 其被体系中的二异丙基乙基胺还原成为强还原性的二价铱配合物, 与现场生成的碘代芳烃17发生单电子转移, 自身被氧化成为三价铱配合物, 完成了光催化剂的循环.同时产生的碳自由基中间体18通过ipso加成-消除完成Smiles重排, 得到芳基迁移的氮自由基中间体20, 中间体20从体系中的还原剂攫取氢原子, 生成杂芳基取代的脂肪酰胺类化合物11.
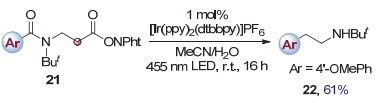
由于铱类光催化剂较宽的氧化还原电势窗口, 碳卤键以外的其它官能团也能够被还原. 2018年, Reiser等[9]使用酰胺化合物21作为底物, 通过光催化还原分子中Nphth官能团中的羰基引起氮氧键均裂, 进而脱除二氧化碳产生碳自由基引发重排, 得到了脂肪胺化合物22 (图 6).
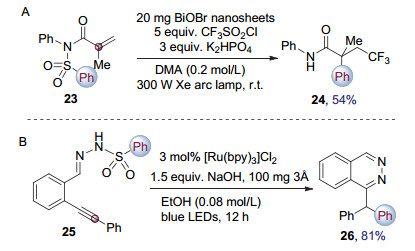
除了还原分子内缺电子官能团产生碳自由基, 通过光催化产生自由基片段对不饱和键进行加成后新生成的碳自由基也能够有效诱导重排发生, 从而生成不饱和键的双官能团化产物. 2015年, 张兵课题组[10]使用BiOBr无机半导体纳米薄片作为光催化剂, 在可见光照射下还原三氟甲磺酰氯为三氟甲基自由基, 随后对化合物23的双键进行加成, 生成的碳自由基引发Smiles重排, 在脱除一分子二氧化硫后, 得到三氟甲基取代的酰胺类化合物24 (图 7A). 2016年, Brachet和Belmont等[11]报道了在可见光催化下, 炔基取代的芳基磺酰腙25能够发生碳碳三键的氢胺化反应, 生成的烯基自由基能够进一步加成到邻近的苯环上, 导致芳基重排并放出二氧化硫, 生成取代的酞嗪类化合物26, 实现了可见光催化的氢胺化-Smiles重排串联反应.值得一提的是该串联反应导致芳基形式上发生了1, 8迁移(图 7B). 2017年, 唐石和盛瑞隆等[12]将三氟甲基拓展为全氟烷基化试剂, 通过光催化Smiles重排得到了一系列全氟烷基取代的酰胺类化合物.同年, 李毅等[13]利用可见光诱导的包括Smiles重排在内的自由基串联反应实现了复杂并环结构的构建.
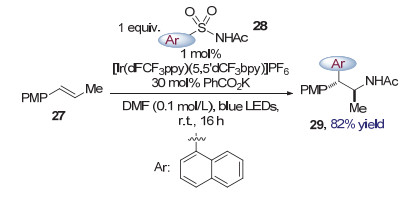
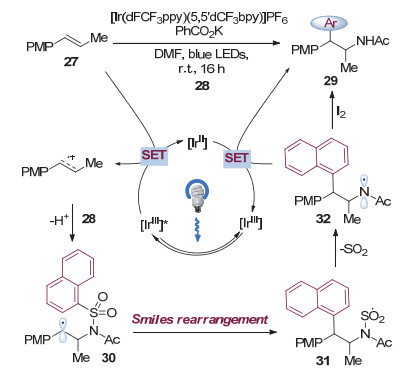
2018年, Stephenson等[14]使用富电子烯烃27作为底物, 磺酰胺28作为官能团化试剂, 通过光催化诱导的Smiles重排实现了烯烃的双官能团化, 得到了酰胺类化合物29 (图 8).与前述转化方法不同, Stephenson通过氧化底物烯烃来诱导重排反应的发生, 产生碳自由基的过程实际上从自由基加成转变为了双键的单电子氧化反应.该策略的优势在于无需产生较活泼的氮自由基, 从而避免了双键α位攫氢副产物的产生[15].在提出的反应机理中, 作者认为光催化剂[Ir(dFCF3ppy)(5, 5'-dCF3-bpy)]PF6在光照下被激发, 得到激发态的配合物, 随后与富电子烯烃27发生单电子转移生成自由基正离子和二价铱配合物.磺酰胺28对自由基正离子亲核进攻, 生成碳自由基物种30, 引发Smiles重排后脱去一分子二氧化硫得到氮自由基物种32 (图 9). 32被二价铱配合物还原进而质子化, 生成产物29.而二价铱配合物则被氧化为三价铱配合物, 完成光催化剂的循环.
同年, 朱晨等[16]也报道了使用烯烃作为底物通过光催化诱导的Smiles重排反应高效地实现烯烃二氟甲基化的转化方法(图 10), 得到了具有二氟官能团的芳香烃类化合物34.该方法条件温和, 活化以及非活化的烯烃都能够顺利转化, 且不同电性的取代基兼容性良好(图 10).产品可以通过简单的合成操作转化为其他具有高附加价值的化合物.
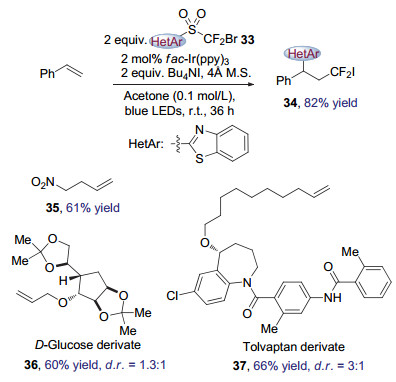
该转化的优势在于, 作者通过对双官能团化试剂33进行了设计, 将预迁移的杂芳基(自由基受体)和溴代二氟烷烃(自由基供体)通过砜基结合在一起, 从而使底物摆脱了预先装入磺酰基的限制.鉴于存在大量商品化的烯烃, 这一创新极大地拓展了底物的适用范围.
作者提出的反应机理如图 11所示.首先在光照下, 铱催化剂被激发为激发态, 与双官能团化试剂33发生单电子转移, 被氧化淬灭为四价铱配合物, 生成碳自由基物种38.四价铱配合物被体系中的碘负离子还原为三价铱配合物, 完成光催化剂的循环的同时生成单质碘.中间体38随后对苯乙烯加成, 引发自由基重排并脱去一分子二氧化硫生成碳自由基物种41.中间体41被单质碘捕获, 得到烯烃双官能团化的产物34.
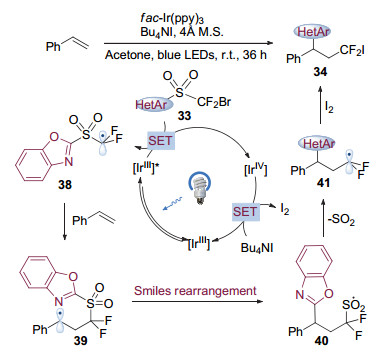
2019年, Greaney课题组[17]也利用不同底物通过可见光引发的Smiles重排得到了烯烃的二氟烷基芳基化产物.值得注意的是, 体系中没有加入光催化剂, 作者认为体系中加入的还原剂和烷基化试剂形成的电子供受体(Electron Donor-Acceptor, EDA)复合物是反应得以进行的关键.
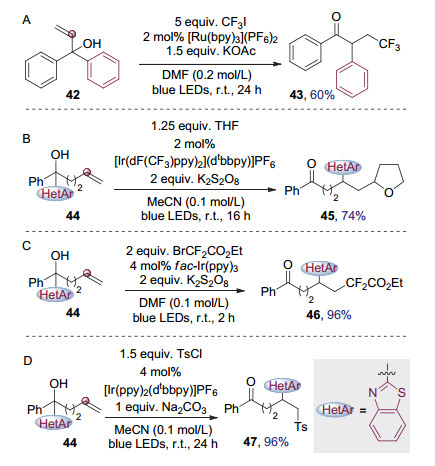
磺酰胺类化合物由于能脱除一分子二氧化硫促进平衡移动, 因而是Smiles重排的理想底物, 同时化学家们也对其它类型的底物进行了探索. 2015年, 朱成建课题组[18]使用1, 1-二芳基取代的烯丙醇42作为底物, 通过可见光催化的Smiles重排得到了烯烃的芳基三氟甲基化产物43 (图 12A).值得注意的是, 该转化中芳基进行了1, 2迁移, 通过改变不同的烷基化试剂, 还能够实现二芳基取代烯丙醇类化合物的其它芳基官能团化反应[19]. 2017年, 谷利军等[20]使用羟基α位有杂芳基取代的叔醇44作为底物, 醚类化合物作为烷基化试剂, 通过加成产生碳自由基诱导Smiles重排发生, 得到具有醚官能团的酮类化合物45(图 12B).同年, 朱成建课题组[21]使用高炔丙醇, 通过可见光催化的Smiles重排实现了炔基的芳基二氟烷基化反应. 2018年, 朱晨等[22]使用激发态光催化剂还原二氟溴乙酸酯中缺电子的碳溴键来产生二氟烷基碳自由基, 通过其与烯基叔醇44进行加成诱导杂芳基迁移, 从而实现了可见光催化Smiles重排介导的烯烃杂芳基二氟烷基化反应, 得到了具有二氟官能团的酮类化合物46(图 12C).进一步研究发现, 将二氟溴乙酸酯替换为磺酰氯类化合物则可以实现可见光催化Smiles重排介导的烯烃杂芳基磺化反应, 得到结构中含有芳基磺酰官能团的酮类化合物47[23] (图 12D).
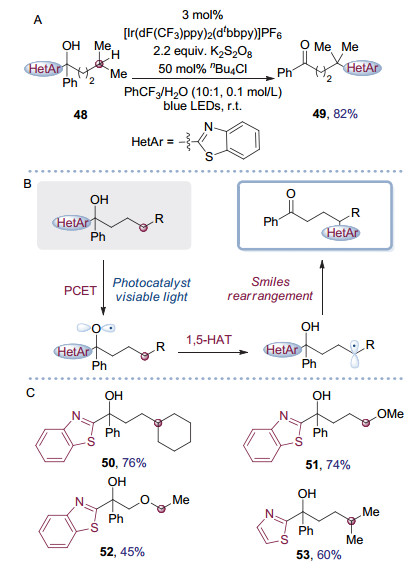
与碳卤键和碳碳双键相比, 碳氢键极性低, 键强高, 难以活化, 在化学反应中通常被认为是惰性的.但由于具有较高的原子经济性以及导向非常规的合成切断方法, 碳氢键的选择性活化官能团化一直以来都是有机化学界的研究热点. 2018年, 朱晨课题组[24]使用羟基α位有杂环取代的叔醇48为底物, 利用常见的过硫酸盐作为氧化剂, 通过可见光催化产生碳自由基诱导Smiles重排反应, 实现了叔醇δ远端C(sp3)—H键活化, 得到了羰基δ位有杂芳环取代的酮49(图 13A).在提出的反应机理中(图 13B), 作者认为叔醇在可见光催化下发生质子协同的电子转移(Proton-Coupled Electron-Transfer, PCET), 产生烷氧自由基, 随即发生选择性1, 5氢迁移反应(1, 5-Hydrogen Atom Transfer, 1, 5-HAT)生成亲核性的碳自由基, 该碳自由基物种进而发生Smiles重排及氧化, 得到具有苯甲酰官能团的化合物.降低位阻以及共轭结构的形成是重排驱动力的重要来源.反应具有较高的效率以及官能团兼容性, 能够以较高的收率合成多种具有不同官能团的酮类化合物(图 13C).并且由于具有羰基和苯并噻唑结构, 产物易于通过简单合成操作转化为具有其它官能团化合物[25], 体现了这一转化方法高的应用潜力.
与碳氢键相似, 碳碳键也是大量存在于分子当中的化学键, 选择性的碳碳键活化官能团化同样具有非常重要的意义. 2017年, Nevado课题组[26]报道了一种条件温和的基于可见光催化Smiles重排实现脂肪胺54氮γ位C—C键选择性活化烷基化的方法(图 14), 得到芳香胺55.
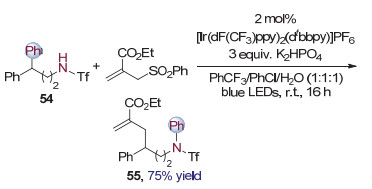
作者认为氮自由基是反应的关键中间体(图 15).首先, 光催化剂[Ir(dF(CF3)ppy)2(dtbbpy)]PF6在光照下被激发, 生成激发态的三价铱配合物, 其与去质子化后的酰胺氮负离子发生单电子转移, 本身被还原成二价铱配合物, 同时得到氮自由基56.氮自由基对γ位苯环进行ipso加成, 发生Smiles重排, 实现了氮γ位碳碳键的选择性活化.生成的碳自由基58与体系中的缺电子双键化合物发生Michael加成, 生成的碳自由基物种59进而消除得到烷基化的产物55.而消除产生的苯砜基自由基将二价铱配合物氧化为三价铱配合物, 完成光催化剂的循环.
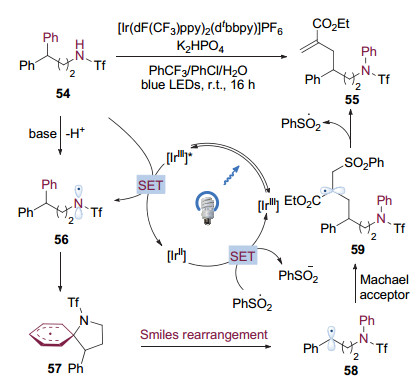
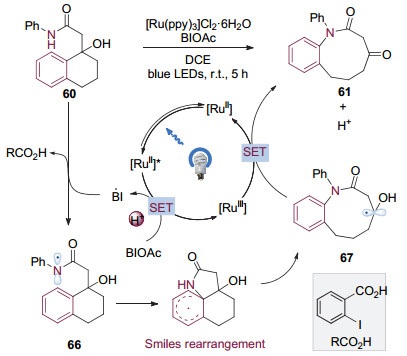
2018年, 刘心元等[27]发现, 在可见光催化条件下, 加入BIOAc来协助氧化特定结构的酰胺60可以得到内酰胺化合物61.进一步研究完善了这种通过可见光催化的Smiles重排来活化C(sp2)—C(sp3)键从而合成中环内酰胺的方法(图 16).该转化方法能够通过调整底物环系的大小合成9至11元环的内酰胺, 同时条件温和, 具有较好的官能团兼容性, 富电子的杂芳基也能够顺利的发生迁移.
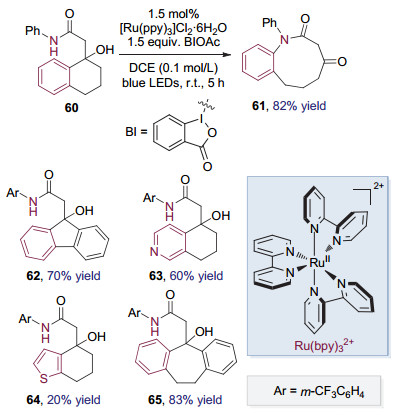
机理研究表明反应始于酸协助下氧化剂BIOAc对激发态二价钌配合物的氧化淬灭, 产生BI自由基和三价钌配合物(图 17). BI自由基在体系中酸的协助下氧化底物酰胺生成氮自由基物种66, 本身被还原成酸.氮自由基与苯环发生ipso加成, 导致Smiles重排, 生成具有内酰胺结构的氧α位碳自由基67, 67与体系中的三价钌配合物发生单电子转移后去质子化, 本身被氧化为具有羰基官能团的内酰胺61, 另外生成二价钌配合物完成光催化剂的循环.
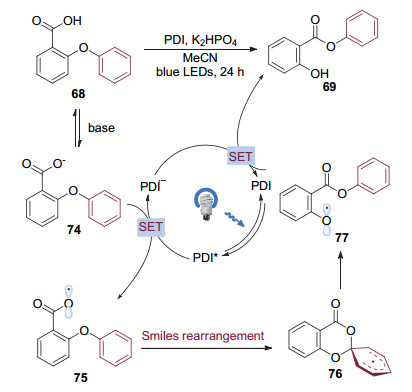
氧自由基由于能量上比碳自由基和氮自由基要更高, 产生较为困难, 因而在该重排反应中应用较少. 2017年, 曹小平和李洋等[28]在使用芳基醚68作为底物, 意图通过可见光催化的放氢反应构建二苯并呋喃结构时, 却意外发现底物发生了芳基1, 5迁移, 生成了芳香羧酸苯酚酯69.进一步的研究完善了这一可见光催化Smiles重排介导的芳基醚转化成酯的反应(图 18A).该转化条件温和, 具有较好的官能团兼容性.与离子型Smiles重排不同的是, 不论迁移芳环上连有给电子取代基还是吸电子取代基, 反应都能以很高的效率进行(图 18B).值得注意的是, 利用商业可得的原料73, 通过两步简单的合成转化, 就能够以92%的收率克级规模制备用于治疗呼吸系统炎症的药物Guacetisal(图 18C).从而显示出这一类转化模式较大的应用潜力.
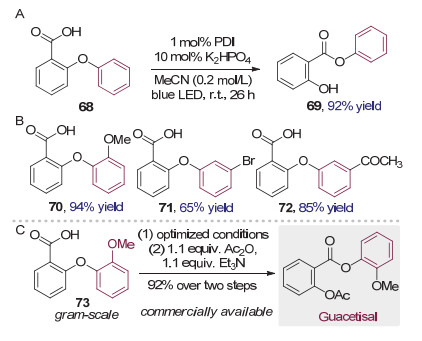
经过细致的机理研究, 作者认为氧自由基是反应历经的重要中间体, 而反应的驱动力来源于芳酯更强的碳氧键解离能.首先有机光催化剂PDI在光照下被激发, 生成强氧化性的激发态的催化剂, 与羧酸负离子74发生单电子转移, 生成氧自由基物种75, 本身被还原成自由基负离子.氧自由基75对邻近苯环进行ipso加成, 发生Smiles重排, 生成芳基迁移的氧自由基物种77.中间体77被体系中的催化剂自由基负离子还原后质子化, 得到芳酯69, 同时再生光催化剂, 完成循环(图 19).
同年, Gonzalez-Gomez等[29]使用不同的碱, 也报道了可见光催化Smiles重排介导的醚转化为酯的反应.
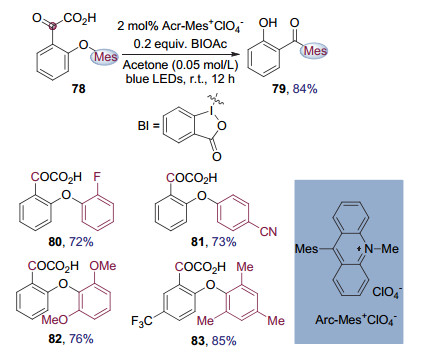
陈以昀课题组[30]发现将光催化剂Arc-Mes+ClO4-与高价碘试剂联用, 能够在可见光照射下氧化芳基酮酸类化合物78生成酰基自由基, 从而诱导芳基迁移, 得到二苯甲酮类化合物79.最近, 他们报道了这一可见光催化的酰基自由基Smiles重排反应(图 20).该转化能够构建广泛存在于具有生物活性分子中的二苯甲酮结构.与离子型Smiles重排相比, 条件温和, 并且迁移芳环上连有给电子取代基或者吸电子取代基的底物都能够被顺利转化.
综上所述, 通过可见光催化策略产生碳、氮和氧自由基诱导重排发生, 能够以温和的条件, 好的官能团兼容性实现多种底物的定向芳基化反应, 构建一系列芳环或芳杂环取代的高附加价值产品.这一类反应兼具新型催化模式和经典反应体系的优点, 令可见光催化Smiles重排显示出巨大的合成潜力以及广阔的应用前景.此外, 发展新型光催化剂, 实现其他种类原子介导的重排, 进一步拓展反应类型将成为未来的研究热点.
(a) Chen, Z.-M.; Zhang, X.-M.; Tu, Y.-Q. Chem. Soc. Rev. 2015, 44, 5220; (b) Allart-Simon, I.; Gérard, S.; Sapi, J. Molecules 2016, 21, 878; (c) Holden, C. M.; Greaney, M. F. Chem. Eur. J. 2017, 23, 8992; (d) Lin, S.-B.; He, X.-R.; Meng, J.-P.; Gu, H.-N.; Zhang, P.-Z.; Wu, J. Chin. J. Org. Chem. 2017, 37, 1864. (蔺松波, 何兴瑞, 孟金鹏, 顾海宁, 张培志, 吴军, 有机化学, 2017, 37, 1864.)
(a) Warren, L. A.; Smiles, S. J. Chem. Soc. 1930, 1327; (b) Warren, L. A.; Smiles, S. J. Chem. Soc. 1930, 956; (c) Levi, A.; Warren, L. A.; Smiles, S. J. Chem. Soc. 1933, 1490.
(a) Kong, W.; Merino, E.; Nevado, C. Angew. Chem., Int. Ed. 2014, 53, 5078; (b) Thaharn, W.; Soorukram, D.; Kuhakarn, C.; Tuchinda, P.; Reutrakul, V.; Pohmakotr, M. Angew. Chem., Int. Ed. 2014, 53, 2212; (c) Fuentes, N.; Kong, W.; Fernández-Sánchez, L.; Merino, E.; Nevado, C. J. Am. Chem. Soc. 2015, 137, 964; (d) Wu, X.; Zhu, C. Chin. J. Chem. 2019, 37, 171.
Douglas, J. J.; Albright, H.; Sevrin, M. J.; Cole, K. P.; Stephenson, C. R. J. Angew. Chem., Int. Ed. 2015, 54, 14898. doi: 10.1002/anie.201507369
Benito Collado, A. B.; Diaz Buezo, N.; Jimenez-Aguado, A. M.; Lafuente Blanco, C.; Martinez-Grau, M. A.; Pedregal-Tercero, C.; Toledo Escribano, M. A. U.S. 8232289 B2, 2011.
Douglas, J. J.; Sevrin, M. J.; Cole, K. P.; Stephenson, C. R. J. Org. Process Res. Dev. 2016, 20, 1148. doi: 10.1021/acs.oprd.6b00126
Li, Y.; Hu, B.; Dong, W.; Xie, X.; Wan, J.; Zhang, Z. J. Org. Chem. 2016, 81, 7036. doi: 10.1021/acs.joc.6b00735
Alpers, D.; Cole, K. P.; Stephenson, C. R. J. Angew. Chem., Int. Ed. 2018, 57, 12167. doi: 10.1002/anie.201806659
Faderl, C.; Budde, S.; Kachkovskyi, G.; Rackl, D.; Reiser, O. J. Org. Chem. 2018, 83, 12192. doi: 10.1021/acs.joc.8b01538
Liu, C.; Zhang, B. RSC Adv. 2015, 5, 61199. doi: 10.1039/C5RA08996D
Brachet, E.; Marzo, L.; Selkti, M.; König, B.; Belmont, P. Chem. Sci. 2016, 7, 5002. doi: 10.1039/C6SC01095D
Tang, S.; Yuan, L.; Deng, Y.-L.; Li, Z.-Z.; Wang, L.-N.; Huang, G.-X.; Sheng, R.-L. Tetrahedron Lett. 2017, 58, 329. doi: 10.1016/j.tetlet.2016.12.027
Huang, H.; Li, Y. J. Org. Chem. 2017, 82, 4449. doi: 10.1021/acs.joc.7b00350
Monos, T. M.; McAtee, R. C.; Stephenson, C. R. J. Science 2018, 361, 1369.
Zard, S. Z. Chem. Soc. Rev. 2008, 37, 1603. doi: 10.1039/b613443m
Yu, J.; Wu, Z.; Zhu, C. Angew. Chem., Int. Ed. 2018, 57, 17156. doi: 10.1002/anie.201811346
Whalley, D. M.; Duong, H. A.; Greaney, M. F. Chem. Eur. J. 2019, 25, 1927. doi: 10.1002/chem.201805712
Xu, P.; Hu, K.; Gu, Z.; Cheng, Y.; Zhu, C. Chem. Commun. 2015, 51, 7222. doi: 10.1039/C5CC01189B
(a) Huang, H.-L.; Yan, H.; Yang, C.; Xia, W. Chem. Commun. 2015, 51, 4910; (b) Li, Y.; Liu, B.; Ouyang, X.-H.; Song, R.-J.; Li, J.-H. Org. Chem. Front. 2015, 2, 1457; (c) Cai, S.; Tian, Y.; Zhang, J.; Liu, Z.; Lu, M.; Weng, W.; Huang, M. Adv. Synth. Catal. 2018, 360, 4084; (d) Lu, M.; Qin, H.; Lin, Z.; Huang, M.; Weng, W.; Cai, S. Org. Lett. 2018, 20, 7611; (e) Wang, H.; Xu, Q.; Yu, S. Org. Chem. Front. 2018, 5, 2224; (f) Wang, Q.-L.; Chen, Z.; Zhou, C.-S.; Xiong, B.-Q.; Zhang, P.-L.; Yang, C.-A.; Liu, Y.; Zhou, Q. Tetrahedron Lett. 2018, 59, 4551; (g) Yin, Y.; Weng, W.-Z.; Sun, J.-G.; Zhang, B. Org. Biomol. Chem. 2018, 16, 2356; (h) Wei, X.-J.; Noël, T. J. Org. Chem. 2018, 83, 11377.
Gu, L.; Gao, Y.; Ai, X.; Jin, C.; He, Y.; Li, G.; Yuan, M. Chem. Commun. 2017, 53, 12946. doi: 10.1039/C7CC06484E
周能能, 胥攀, 李伟鹏, 成义祥, 朱成建, 化学学报, 2017, 75, 60. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345727.shtmlZhou, N.-N.; Xu, P.; Li, W.-P.; Cheng, Y.-X.; Zhu, C.-J. Acta Chim. Sinica 2017, 75, 60. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract345727.shtml
Yu, J.; Wang, D.; Xu, Y.; Wu, Z.; Zhu, C. Adv. Synth. Catal. 2018, 360, 744. doi: 10.1002/adsc.201701229
Tang, N.; Yang, S.; Wu, X.; Zhu, C. Tetrahedron 2019, 75, 1639. doi: 10.1016/j.tet.2018.12.003
Wu, X.; Wang, M.; Huan, L.; Wang, D.; Wang, J.; Zhu, C. Angew. Chem. 2018, 130, 1656. doi: 10.1002/ange.201709025
Dondoni, A.; Marra, A. Chem. Rev. 2004, 104, 2557. doi: 10.1021/cr020079l
Shu, W.; Genoux, A.; Li, Z.; Nevado, C. Angew. Chem., Int. Ed. 2017, 56, 10521. doi: 10.1002/anie.201704068
Wang, N.; Gu, Q.-S.; Li, Z.-L.; Li, Z.; Guo, Y.-L.; Guo, Z.; Liu, X.-Y. Angew. Chem., Int. Ed. 2018, 57, 14225. doi: 10.1002/anie.201808890
Wang, S.-F.; Cao, X.-P.; Li, Y. Angew. Chem., Int. Ed. 2017, 56, 13809. doi: 10.1002/anie.201706597
Gonzalez-Gomez, J. C.; Ramirez, N. P.; Lana-Villarreal, T.; Bonete, P. Org. Biomol. Chem. 2017, 15, 9680. doi: 10.1039/C7OB02579C
Li, J.; Liu, Z.; Wu, S.; Chen, Y. Org. Lett. 2019, 21, 2077. doi: 10.1021/acs.orglett.9b00353

图 1 经典Smiles重排与光催化Smiles重排反应
Figure 1 Generalized Smiles rearrangement and photoredox Smiles rearrangement

图 2 可见光催化的杂环芳基磺酸酯的Smiles重排反应
Figure 2 The visible light mediated Smiles rearrangement of aromatic sulfonates

图 3 可见光促进的杂环芳基磺酸酯的Smiles重排反应机理
Figure 3 Proposed mechanism for the visible light mediated Smiles rearrangement of aromatic sulfonates

图 4 可见光催化的杂环芳胺的Smiles重排反应
Figure 4 The visible light mediated Smiles rearrangement of aromatic amines

图 5 可见光催化的杂环芳胺的Smiles重排反应机理
Figure 5 Proposed mechanism for the visible-light mediated Smiles rearrangement of aromatic amines

图 6 通过可见光催化还原羰基官能团引发Smiles重排反应
Figure 6 Smiles rearrangement mediated by reduction of carbonyl through visible-light photoredox catalysis

图 7 通过可见光催化不饱和键官能团化诱导Smiles重排
Figure 7 Smiles rearrangement mediated by functionalization of unsaturated C—C bond through visible-light photoredox catalysis

图 8 可见光催化Smiles重排介导的烯烃胺芳基化反应
Figure 8 Alkene aminoarylation through visible-light photocatalytic Smiles rearrangement

图 9 可见光催化Smiles重排介导的烯烃胺芳基化反应机理
Figure 9 Proposed mechanism for alkene aminoarylation through visible-light photocatalytic Smiles rearrangement

图 10 可见光催化Smiles重排介导的烯烃二氟甲基化反应
Figure 10 Alkene difluoromethylation through visible-light photocatalytic Smiles rearrangement

图 11 可见光催化Smiles重排介导的烯烃二氟甲基化反应机理
Figure 11 Proposed mechanism for alkene difluoromethylation through visible-light photocatalytic Smiles rearrangement

图 12 烯基醇类底物作为可见光催化Smiles重排前体实现烯烃的双官能团化
Figure 12 Bifunctionalization of alkenes through visible-light photocatalytic Smiles rearrangement of vinyl alcohol

图 13 光催化Smiles重排介导的叔醇δ远端C(sp3)—H键活化
Figure 13 Remote C(sp3)—H bonds activation of tertiary alcohol through photoredox catalyzed Smiles rearrangement

图 14 基于可见光催化Smiles重排的脂肪胺氮γ位C—C键活化官能团化
Figure 14 γ-Functionalizations of amines through visible-light photocatalytic Smiles rearrangement

图 15 脂肪胺氮γ位C—C键活化官能团化反应机理
Figure 15 Proposed mechanism for γ-functionalization of amines

图 16 由可见光催化Smiles重排介导的中环内酰胺合成
Figure 16 Synthesis of medium-sized lactams mediated by visible-light photocatalytic Smiles rearrangement

图 18 可见光催化Smiles重排介导的芳基醚转化成酯的反应
Figure 18 Transformation from aryl ethers to esters through visible-light photocatalytic Smiles rearrangement

图 19 可见光催化Smiles重排介导的芳基醚转化成酯的反应机理
Figure 19 Proposed mechanism for transformation from aryl ethers to esters through visible-light photocatalytic Smiles rearrangement

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:

