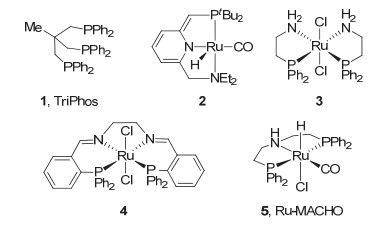
图 1.
早期酯均相催化氢化所用的代表性配体及催化剂
Figure 1.
Early representative ligands and catalysts for the hydrogenation of esters

羧酸酯还原成醇是一类非常重要的有机化学反应, 在有机合成中有着广泛的用途, 并已应用于医药、农药、香料等精细化工品的生产.酯的传统还原方法主要采用氢化锂铝、金属硼氢化物以及硼烷等负氢试剂还原, 这些方法除了价格昂贵、操作不安全等不利因素外, 往往还需要消耗当量或几倍当量的碱等来处理.这会产生大量的副产物, 导致后处理麻烦, 对环境也造成污染.工业生产上也常采用催化氢化的方法对酯进行还原, 但主要是采用非均相催化剂, 反应往往需要在高温、高压(200~300 ℃, >100 atm)的苛刻条件下才能实现.而且反应的能耗大, 副产物比较多.因此, 发展经济、高效、环境友好、节省能源和资源的羧酸酯还原方法一直受到合成化学家们的广泛关注, 也是近年来有机合成化学研究的热点和挑战.
均相催化氢化是原子经济性高、操作简单、环境友好的还原方法, 已在医药、农药、香料等的生产中得到广泛的应用.均相催化氢化研究源于20世纪30年代末化学家们尝试用一些简单的过渡金属盐如醋酸酮、氯化铑等氢化包括对苯醌等不饱和化合物[1].直到Wilkinson等[2]发现三苯基膦铑催化剂并实现烯烃在温和反应条件下的催化氢化后, 不饱和化合物的均相催化氢化才引起了化学家们的高度重视, 不对称催化氢化反应也随之得到了快速的发展[3].
酯的均相催化氢化还原最早是由Grey和Pez等[4]报道的. 1980年, 他们采用三苯基膦的钌氢催化剂如[(Ph3P)2(Ph2P)RuH2-K+•diglyme]2等在温和的反应条件下(90 ℃和60 atm)实现了酯的均相催化氢化.但该催化体系仅对三氟醋酸酯、草酸酯等高活性的酯才有较高的收率.随后的研究发现三烷基膦的钌催化剂对草酸二甲酯等也有较高的活性.如用[Ru(CO)2(OAc)2(PBu3)2]催化氢化草酸二甲酯时, 获得了高达15229的转化数, 但是反应条件相对苛刻(180 ℃和130 atm)[5].三齿膦配体TriPhos (1, 图 1)的引入显著提高了钌催化剂的活性, 草酸二甲酯可在较温和的条件下被氢化(100 ℃和70 atm)[6].此外, 邻苯二甲酸酯、苯甲酸酯和脂肪酸酯等非活化酯也可以被氢化.得到相应的芳香醇和脂肪醇(100 ℃和80 atm)[7]. 2006年, Milstein等[8]将具有吡啶骨架的P-N-N型钳形钌催化剂2(也称为Milstein催化剂)应用到芳香酯和脂肪酯的氢化还原, 并在较低的氢气压力下(115 ℃和5.3 atm)获得了很好的氢化结果(1 mol%催化剂, 86%~100%转化率).随后, 他们发现催化剂2能够将碳酸二甲酯和甲酸甲酯氢化到甲醇, 转化数高达2500[9] (110 ℃和60 atm). 2007年, Saudan等[10]报道了系列含膦和氮配位原子的双齿和四齿配体的钌催化剂, 并发现胺基膦配体的钌催化剂3和具有乙二胺骨架的四齿P-N-N-P型的钌催化剂4(也称Firmenich催化剂)对芳香酸酯和脂肪酸酯都有很高的活性.其中, 钌催化剂3对苯甲酸异丙酯的氢化转化数达到10000 (100 ℃和50 atm).钌催化剂3和4对脂肪酸酯的氢化都有较高的活性, 转化数达到2000 (100 ℃和50 atm).此外, 除末端双键和与酯基共轭的双键外, 其它类型的不饱和酯在氢化中双键均可保留.
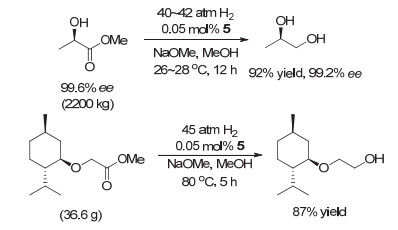
2011年, 高砂公司成功将均相催化氢化应用于(R)-1, 2-丙二醇和L-薄荷烷氧基乙醇的合成[11].他们用二乙胺连接的P-N-P型钳形钌催化剂Ru-MACHO (5, 高砂催化剂)在温和反应条件下(0.05 mol% 5, 40~42 atm, 26~28 ℃, 12 h)实现了2200 kg (R)-乳酸甲酯的催化氢化, 获得了92%的收率, 且产物的ee值在反应前后几乎保持不变(图 2).在相似的反应条件下, 他们对L-薄荷烷氧基乙酸甲酯进行了氢化, 获得87%的收率(0.05 mol% 5, 45 atm, 80 ℃, 5 h).值得一提的是, 该催化剂在室温(30 ℃)下对(R)-乳酸甲酯的氢化转化数高达4000.
上述过渡金属催化剂的出现, 实现了芳香酸酯和脂肪酸酯等非活化酯的均相催化氢化, 并在生产上得到应用.但总体而言, 已发展的酯的均相催化剂的效率仍然不高, 氢化反应的条件也较苛刻, 需要在加热或者较高压力下(>50 atm)才能得到较好的结果.正因如此, 以发展新的高效催化剂和催化体系为核心研究内容的酯的均相催化氢化在过去十多年里得到了学术界和工业界的广泛关注, 并取得了突出的研究进展.主要体现在如下几个方面: (1)高效催化剂的种类明显增加, 铁、钴、锰等丰产金属催化剂[12]的出现标志着酯的均相催化氢化更趋于向高效、绿色、可持续方向发展; (2)酯催化氢化的效率显著提高, 氢化反应的转化数最高可接近100000[13]; (3)酯氢化的底物显著拓展, 并且更加注重官能化酯氢化的化学选择性.有关酯均相催化氢化还原为醇的综述文献也相继报道[14].
酯均相催化氢化取得突破的主要原因在于新配体及其催化剂的不断发现.不同种类的配体及其催化剂具有不尽相同的催化活性和选择性, 它们的引入不仅提高了酯氢化的效率和选择性, 也扩大了酯氢化底物的范围.因此, 本论文将从配体及其催化剂的发展角度重点概述近十年来酯均相催化氢化的研究现状.特别需要指出的是, 酯的不对称催化氢化在近年来也得到了快速的发展, 本文将首次综述酯的不对称催化氢化研究进展.
如前所述, 酯均相催化氢化的配体早期主要集中在为数不多的胺基膦配体、三齿膦配体、具有二乙胺和吡啶骨架的P-N-N和P-N-P型钳形配体、以及乙二胺骨架的P-N-N-P型四齿配体.成功的催化剂主要是这些配体的钌催化剂.近年来, 用于酯氢化的配体数量发展迅速, 已接近二百多种, 主要集中在简单的二乙胺和吡啶类型的三齿钳形配体.廉价的丰产金属如铁、钴、锰的催化剂在酯氢化中得到越来越广泛的应用是这个领域发展的另一个特点.
在高砂公司用二乙胺连接的三齿P-N-P型钳形配体的钌催化剂Ru-MACHO (5)实现温和条件下脂肪酸酯的氢化并将其应用于(R)-1, 2-丙二醇和L-薄荷烷氧基乙醇的合成后[11], 此类最简单的三齿P-N-P型钳形配体及其催化剂受到了广泛的关注.除配体的种类得到快速发展以外, 中心金属已由钌金属发展到锇、铱以及铁、钴、锰等丰产金属.这是目前在酯的均相催化氢化为醇的反应中发展最快, 也是非常成功的一类三齿钳形配体及其催化剂.
2012年, Schlaf等[15]将Gusev发展的与Ru-MACHO (5)类似的三齿钳形锇催化剂6[16](图 3)应用到甘油酸三酯的模拟物己醇辛酸酯和顺-3-己烯醇己酯的氢化中.他们发现在220 ℃和54 atm H2及无溶剂的条件下, 用0.1 mol%的催化剂6a可实现相应酯的完全转化, 得到饱和醇.在优化的反应条件下, 催化剂6a可以直接将种籽油(菜籽油或大豆油)氢化为饱和脂肪醇. 2013年, Beller等[17]发现与硼烷反应后得到的Ru-MACHO类型催化剂7a(图 3)在草酸二乙酯及其类似物氢化合成乙二醇等反应中表现出很高的催化活性, 转化数和转化频率分别达到5700和360 h-1(120 ℃, 60 atm, 16 h).而采用相应的磷原子上异丙基取代的钌催化剂8a或8b(图 3)则仅能将草酸二乙酯还原为乙醇酸乙酯(100 ℃, 60 atm, 20 h).这表明在配体磷原子上引入富电子的烷基后, 相应钌催化剂的活性明显降低.同年, Ikariya等[18]发现Ru-MACHO (5)可催化系列单氟、双氟或三氟取代的α-氟代酸酯的氢化.在催化剂用量为0.005 mol%时, 对二氟乙酸乙酯的氢化可给出77%的分离收率(40 ℃, 10 atm, 23 h).在相同的反应条件下, 对三氟乙酸甲酯则得到半缩醛, 收率和选择性分别为89%和96%.对(R)-2-氟丙酸甲酯的氢化, 可以以98%的收率得到相应的手性醇, 反应前后ee值保持不变.此外, 相应的双氢钌催化剂7b表现出与Ru-MACHO (5)相当的催化活性.但是, 双氯钌催化剂7c的催化活性相对较低, 需要较长的反应时间才能给出相当的结果.
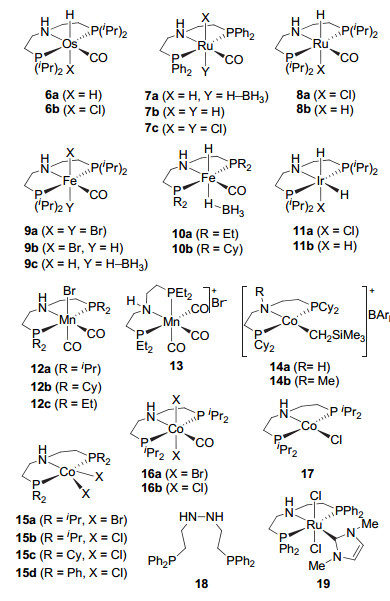
2014年, 继Milstein等[19]将具有吡啶骨架的P-N-P钳形铁催化剂引入酯的均相催化氢化后, Guan等[20]和Beller等[21]报道了铁催化酯氢化为醇的反应. Guan等[20]用硼烷活化的铁催化剂9c(图 3)对系列芳香酸酯和脂肪酸酯进行氢化(3 mol% 9c, 115 ℃, 10~15 atm, 1.5~24 h), 获得了50%~96%的收率.而相应的单氢铁催化剂9b(图 3)在添加10 mol%的叔丁醇钾时才表现出较好的催化活性.对来自椰子油的工业样品CE-1270 (73%的月桂酸甲酯, 26%的十四烷酸甲酯和少量的C10和C16烷酸酯)的氢化, 铁催化剂9c也能给出高达98.6%的收率(3 mol% 9c, 135 ℃, 50 atm, 3 h). Beller等[21]同样发现铁催化剂9c对系列脂肪酸酯、内酯以及芳香酸甲酯等具有较高活性(1 mol% 9c, 100~120 ℃, 30 atm, 6~19 h, 49%~99%收率).他们还发现当铁催化剂9c中氮原子上的氢变为甲基后, 在相同的反应条件下无催化活性.这表明铁催化剂9c中P-N-P配体上的N—H与Fe—H具有协同催化作用, 氢化反应是通过配体促进的协同机理完成的.此外, 他们还将铁催化剂9c用于阿拉泊韦(Alisporivir)的关键中间体十二肽的合成.在对氮端的甲氧羰基乙基的氢化反应中, 得到了79%的分离收率(10 mol% 9c, 120 ℃, 50 atm, 20 h).随后, Guan等[22]在系统评价脂肪酸甲酯和天然油脂的氢化时发现, 虽然铁催化剂9c对CE-1295的催化活性可以与钌催化剂7a相当(1 mol% 7a, 68 atm, 135 ℃), 但在放大实验和降低催化剂用量时催化剂9c给出较低的收率(100 g, 0.26 mol% 9c, 50 atm, 135 ℃).这可能是铁催化剂对CE-1270中含有的少量杂质敏感所致.而相应的钌催化剂5在公斤级实验中也可给出高达1860的转化数(0.05 mol% 5, 34 atm, 135 ℃, 3 h, 93%收率). Beller等[23]随后还发现当配体磷原子上的取代基为位阻较小的乙基时(催化剂10a) (图 3), 催化剂具有更高的催化活性.该铁催化剂在更温和的反应条件下(1 mol% 10a, 60 ℃, 30 atm, 6 h)对苯甲酸甲酯的氢化也有99%的收率, 而铁催化剂9c和10b在相同的条件下的收率仅为50%和30%.当反应温度降低到40 ℃, 铁催化剂10a的收率明显降低, 仅为44%;保持反应温度为60 ℃, 降低氢气压力为10 atm, 甚至2 atm时, 反应的收率又提高到82%和58%.这表明, 在较高的反应温度下有利于失去硼烷, 从而使催化剂得到活化.在优化的反应条件下, 磷原子上乙基取代的铁催化剂10a对系列芳香酯、脂肪酯和内酯给出了63%~>99%的收率(1~2 mol% 10a, 60~100 ℃, 30 atm, 6~18 h).此外, 该催化剂对非共轭酯的氢化, 双键可保留; 对共轭酯的氢化, 得到饱和的醇.与此同时, Langer等[24]也观察到同样的现象.他们用铁催化剂10a在几乎相同的反应条件下实现了苯甲酸甲酯的氢化(1 mol% 10a, 100 ℃, 50 atm, 6 h, 99%的收率).
2014年Beller等[25]还将Abdur-Rashid发展的P-N-P型铱催化剂11[26](图 3)引入酯的均相催化氢化反应中.在添加10 mol%甲醇钠的反应条件下, 铱催化剂11a对系列芳香酸酯、脂肪酸酯和内酯给出了30%~98%的收率(1~2 mol% 11a, 130 ℃, 50 atm, 18 h).在不加碱的条件下, 催化剂11b表现出很高的催化活性.接着, Beller等[27]还将他们发展的催化酮、醛和氰基化合物氢化的P-N-P型钳形锰催化剂12[28](图 3)应用于酯的氢化.他们发现将膦原子上乙基取代的P-N-P型配体与五羰基溴化锰([MnBr(CO)5])络合, 得到的是离子型锰催化剂13 (64%, 图 3)和少量中性锰催化剂12c (18%)的混合物.离子型锰催化剂13在甲苯中回流可以转化为中性的锰催化剂12c.在催化苯甲酸甲酯的氢化时, 磷原子上异丙基和环己基取代的锰催化剂12a和12b的催化活性相对较低, 而锰催化剂12c与13的催化活性相当.在添加10 mol%叔丁醇钾的反应条件下, 锰催化剂13对系列芳香酸酯、脂肪酸酯和内酯给出了57%~98%的收率(2 mol% 13, 110 ℃, 30 atm, 24 h).此外, 对含非共轭或非末端烯烃的底物, 双键保留, 主要得到不饱和醇; 而对共轭的不饱和酯, 则得到饱和醇.这是首次采用锰催化剂将酯氢化为醇.
在Milstein等[29]和de Bruin等[30]报道钴催化酯的均相催化后, Jones等[31]在2017年发现钴催化剂14[32](图 3)对芳香酸乙酯、脂肪酸乙酯氢化的收率达到67%~97% (2 mol% 14a, 120 ℃, 55 atm, 20 h), 然而对相应的甲酯氢化的转化率和收率都较低(<35%).可能原因是钴催化剂容易使甲醇脱氢产生一氧化碳, 再与钴配位使其失活.对戊内酯, 钴催化剂14a也表现出很高的催化活性, 转化数最高可达到3890 (0.01 mol% 14a, 120 ℃, 46~55 atm, 3 d).此外, 当钴催化剂14a中氮原子上的氢变为甲基(14b)后, 仍然保持相当的催化活性.这表明在钴催化剂14催化酯的氢化时配体的N—H并未参与反应. 2018年, Beller等[33]系统研究了这类二乙胺类P-N-P型钴催化剂中磷原子上取代基对酯氢化反应的影响.他们发现在50 atm H2和120 ℃以及添加20 mol%甲醇钠的反应条件下, 磷原子上苯基取代的钴催化剂15d(图 3)表现出更高的催化活性, 反应48 h可给出大于99%的转化率和99%的收率(5 mol% 15d).而钴催化剂15a~15c的反应活性较低, 转化率均小于67%.降低反应温度为100 ℃, 反应6 h钴催化剂15d仍然给出96%的收率.此外, 在该反应中, 羰基配位的催化剂16a和16b(图 3)活性也非常低, 只有一价钴催化剂17(图 3)给出较高的催化活性, 能给出97%的转化率和89%的收率(120 ℃, 50 atm, 24 h).在优化的反应条件下(5 mol% 15d, 20 mol%甲醇钠, 120 ℃, 50 atm, 6~24 h), 钴催化剂15d对系列芳香酸酯和脂肪酸酯给出了26%~99%的收率.然而, 当催化剂15d的N—H变为N—Me后所得催化剂却无催化活性.这表明配体中的N—H参与了酯的催化氢化, 氢化反应是经配体促进的双官能化机理进行的.这与Jones等[31]观察到的结果不完全一致.
2015年张绪穆等[34]在二乙胺类P-N-P钳形配体中增加了一个氮原子, 合成出含肼基的配体18(图 3).该配体与[RuCl2(p-cymene)]2或RuCl2(PPh3)3现场生成的钌催化剂在添加5 mol%的乙醇钠及50 atm H2和80 ℃的反应条件下, 对芳香酸及脂肪酸甲酯和乙酯等给出了40%~99%的收率.对苯甲酸甲酯氢化的转化数最高可达17200.但配体18是如何与钌金属配位的仍然未知. 2016年, Ogata和Kayaki等[35]注意到多数报道的Ru-MACHO类型催化剂上均有一个具有反馈π-键配位的羰基.虽然羰基的存在可以提高催化剂的稳定性, 但在氢化过程中产生的钌氢中间体的亲核活性可能受到抑制.由此, 他们用具有较强σ-供体性质的氮杂环卡宾(NHC)来替代羰基, 发展出系列含氮杂环卡宾配体的Ru-MACHO类型催化剂.他们发现, 杂环卡宾氮原子上甲基取代的钌催化剂19(图 3)具有较高的催化活性.在常压和加热50 ℃以及添加20 mol%叔丁醇钾或甲醇钠的温和反应条件下, 用2 mol%的19对系列芳香酸甲酯、脂肪酸甲酯和丁内酯进行氢化, 收率为73%~98% (5 h).
2012年, Gusev等[36]在发展了二乙胺类P-N-P型锇催化剂6[15]以后, 又用吡啶代替其中一个配位膦原子, 发展出二乙胺类P-N-N型钌催化剂20a和锇催化剂20b (图 4).当他们用叔丁醇钾处理这些催化剂后, 发现它们转变为二聚的钌催化剂21a和锇催化剂21b.在苯甲酸甲酯的氢化中, 钌催化剂20a和21a以及锇催化剂20b和21b均表现出很高的催化活性, 在50 atm H2和100 ℃的反应条件下, 用0.05 mol%的催化剂就能在1~2 h实现底物的完全转化.其中, 催化剂20a和20b需要添加1 mol%的叔丁醇钾, 催化剂21a和21b则不需要添加碱, 并且具有更高的催化活性.其中, 钌催化剂21a的转化数高达18000 (0.005 mol% 21a, 50 atm, 100 ℃, 17 h, 90%转化率).锇催化剂21b的效率稍低, 但在相同反应条件下也有8000的转化数(0.01 mol% 21b, 19 h, 80%转化率).然而对含有双键的底物, 锇催化剂21b显示出比钌催化剂21a更好的选择性.如在3-壬烯酸甲酯的氢化中, 锇催化剂21b具有高催化活性, 且底物双键保留(0.05 mol% 21b, 50 atm, 100 ℃, 6 h, 100%的转化率), 而钌催化剂21a在相同的反应条件下无催化活性.在Z-油酸甲酯的氢化中, 锇催化剂21b能给出几乎100%双键保留且构型不发生异构化的烯醇类氢化产物, 但钌催化剂21a只得到40%的双键保留(双键异构化, Z/E=20:80)和60%的双键氢化产物; 对三油酸甘油脂的氢化, 0.1 mol%的锇催化剂21b也能给出98%的转化率.由此可见, 虽然锇催化剂的活性较差, 但对非共轭不饱和酯的氢化具有很好的选择性.随后, Gusev等[37]还发现将钌催化剂20a中磷原子上的异丙基变为苯基后所得到的钌催化剂22(图 4)对酯的氢化具有更高活性.在温和的反应条件下(0.05~0.005 mol% 22, 1~10 mol%乙醇钠或甲醇钾, 50 atm, 40 ℃, 16~18 h), 钌催化剂22对苯基酸甲酯、脂肪酸甲酯等给出了94%~100%的转化率.其中, 对乙酸乙酯和己酸甲酯的氢化转化数分别达到20000和18800 (40 ℃, 16或18 h).这是当时最高的转化数.
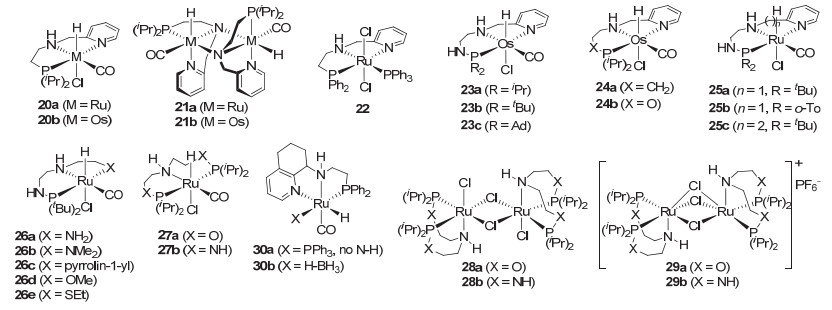
由于引入吡啶基团替代配位磷原子的成功, 且控制非共轭不饱和酯氢化中的化学选择性仍然存在挑战. Gusev等[38]对含吡啶基团的乙二胺类P-N-N三齿钳形配体进一步进行改造, 发展了在配位磷原子和碳原子之间插入NH、O和CH2的P-N-N型钳形锇催化剂23和24 (图 4).在10-十一烯酸甲酯的氢化中(0.05 mol%催化剂, 1 mol%叔丁醇钾, 50 atm, 100 ℃, 2.5 h), 插入一个NH的锇催化剂23a~23c表现出很高的催化活性和选择性(>94%的转化率, >97%的选择性), 双键重排产物<1%;增加CH2和插入O的锇催化剂24a和24b虽然也能给出大于85%的选择性, 但转化率较低, 仅达到40%和5%.值得提及的是, 他们先前发展的锇催化剂20b在相同条件下也仅给出74%的转化率和63%的选择性.此外, Milstein催化剂(2)和Firmenich催化剂3和4能给出大于70%的转化率, 但选择性较低. Milstein催化剂(2)仅得到饱和产物, Firmenich催化剂4的选择性也只有22%, 且伴随39%的双键迁移到C-9的饱和醇.高砂催化剂5能给出89%的选择性, 但转化率仅为18%.由于磷原子上叔丁基取代的锇催化剂23b更稳定, 他们用该催化剂在相同的反应条件下(0.01~0.05 mol% 23b, 0.2~2 mol%甲醇钠或碳酸钾/铯, 50 atm, 100 ℃, 1.5~24 h)评价了系列不饱和的酯, 包括共轭不饱和酯、孤立双键的不饱和多烯酯、含有环丙烷的不饱和酯的氢化.其中, 除共轭不饱和酯给出饱和的产物和相对较低的转化率外, 其余底物均给出了大于97%的转化率和大于96%的选择性; 10-十一烯酸甲酯氢化的转化数达到9800.这表明在锇催化剂20b的钳形P-N-N配体的磷原子和碳原子之间插入NH后可显著改变催化剂的催化性能和化学选择性.
2016年, Gusev[39]对上述新发展的P-N-N钳形配体进行了系统的改造, 包括改变磷原子上的取代基为邻甲苯基及在吡啶与氮原子之间增加一个CH2, 得到钳形钌催化剂25, 以及保持膦胺基部分不变, 变化吡啶基团为各种胺基、甲氧基和乙硫醇基等得到钌催化剂26 (图 4).然而这些钌催化剂在10-十一烯酸甲酯的氢化中(0.05 mol%催化剂, 1 mol%叔丁醇钾, 50 atm, 100 ℃, 2.5 h)并未超越锇催化剂23b.只有仅与锇催化剂23b具有相同P-N-N配体的钳形钌催化剂25a给出相当的催化活性和选择性(84%的转化率和96%的选择性), 但转化数最高只能达到5600;磷原子上为邻甲苯基取代基的钌催化剂25b的催化活性和选择性明显下降, 仅得到66%的转化率和75%的选择性, 且可观察到25%的双键重排到C-9位的产物.吡啶基变为胺基的钌催化剂26a和变为乙硫基的钌催化剂26e能给出69%和74%的转化率, 但选择性却很低, 分别仅为32%和37%.
2017年, Carpentier等[40]在Gusev成功发展出锇催化剂21b[36]等的启发下设计和发展了在二乙胺类P-N-P钳形配体的磷原子和碳原子间同时插入O和NH的P-N-P型钳形钌催化剂27~29(图 4).但在对苯甲酸甲酯的氢化中, 仅催化剂29b表现出较高的催化活性.在0.05 mol%催化剂用量, 50 atm H2和120 ℃及添加叔丁醇钾的反应条件下, 反应30 h可给出98%的转化率; 但对10-十一烯酸甲酯的氢化, 化学选择性非常差, 主要得到饱和氢化产物.这类催化剂虽然也能够催化芳香酸甲酯、脂肪酸酯和内酯的氢化, 但仅给出55%~92%的转化率.同年, 刘庆彬等[41]对Gusev发展的含吡啶的钌催化剂22进行改造, 设计发展了用四氢喹啉代替吡啶的钳形钌催化剂30a和30b(图 4).在用苯甲酸甲酯评价催化剂的活性时, 他们发现在添加5 mol%的硼氢化钠, 用0.05 mol%的钌催化剂30a和30b在50 atm H2和120 ℃的条件下反应3.5或4 h能给出99%的转化率.在相同的反应条件下, 催化剂30a对一系列芳香酸酯和脂肪酸酯都表现出较高的催化活性, 对4-三氟甲基苯甲酸甲酯氢化的转化数高达56000, 对乙酸乙酯的转化数则较低, 为1920.
与此同时, 将二乙胺基连接的三齿P-N-P钳形催化剂Ru-MACHO (5)改变为不含磷的三齿钳形配体及催化剂也受到关注.如2013年, Gusev等[42]将二乙胺基连接的三齿S-N-S钳形配体[43]引入到酯的氢化中发展了钌催化剂31(图 5).含有三苯基膦的钳形钌催化剂31a和31b在苯甲酸甲酯和正己酸甲酯的氢化中(0.05 mol%催化剂, 5 mol%甲醇钾, 50 atm, 40 ℃, 3 h)表现出很高的催化活性, 并且优于他们自己发展的含吡啶的钌催化剂22[36].含三苯基砷和羰基的钳形钌催化剂31c和31d虽然活性不如钌催化剂31a和31b, 但也仅略逊于含吡啶的钌催化剂22.在50 atm H2, 40~100 ℃以及0.1~0.00125 mol%催化剂用量的反应条件下, 催化剂31a对系列芳香酸甲酯和脂肪酸甲酯等给出了73%~100%的转化率.值得提及的是钌催化剂31a对己酸甲酯可给出高达9800的高转化数(100 ℃, 2 h), 明显优于Firmenich催化剂(3) (转化数1880);对乙酸乙酯在无溶剂及温和的反应条件下(40 ℃, 21 h), 钌催化剂31a可给出高达58400的高转化数.这打破了他们先前保持的高转化数记录.此外, 钌催化剂31a在乙醇钠的作用下可转化为含乙氧基的钌催化剂32, 而钌催化剂32的乙醇复合物在甲苯中加热回流可生成相应的两个氢处于fac-S-N-S构型的双氢钌催化剂33, 并产生乙酸乙酯.酯的氢化恰是其逆过程.因而, 钌催化剂31a在催化酯氢化的过程中应该经历了双氢钌催化剂33.由于相应的氮甲基取代的S-N-S钳形钌催化剂无活性, 因此钌催化剂31a中氮原子上的氢对酯的氢化应该起了非常关键的作用.
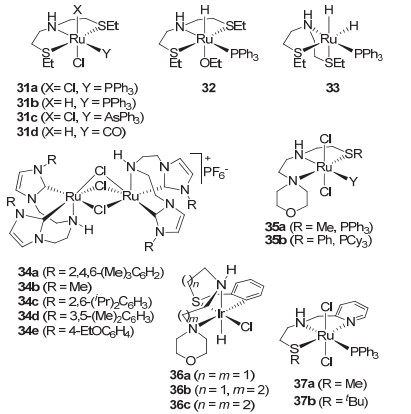
虽然卡宾配体具有强的σ-供电子性质, 且比膦配体稳定, Milstein等[44]和Song等[45]先后用卡宾配体代替Milstein催化剂2中的膦配体而发展出了含吡啶骨架的N-N-C三齿钳形配体的钌催化剂, 但这些催化剂在酯的氢化中并未表现出比含膦三齿钳形钌催化剂更高的催化活性(详细见2.1.2节). Pidko等[46]也基于Milstein催化剂2的吡啶骨架发展了相应的双卡宾C-N-C钳形钌催化剂, 同样没有表现出更高的活性.于是, Pidko等[47]在2015年将二乙基胺桥联的双卡宾配体[48]引入酯的氢化, 从而发展了以二聚体形式存在的C-N-C类型的钳形钌催化剂34(图 5).氮杂环卡宾上的取代基对反应活性影响较大, 取代基为体积较大的2, 4, 6-三甲基苯基(34a)和2, 6-二异丙基苯基(34c)时, 催化剂活性最高; 卡宾配体氮原子上取代为甲基(34b)、间位或对位取代的苯基(34d和34e)时, 催化剂的稳定性差, 活性低甚至无活性.催化剂34a在温和反应条件下(50 atm, 70 ℃)对脂肪酸酯和芳香酸酯均有很高催化氢化活性.对于己酸乙酯, 转化数高达77280 (0.00125 mol% 34a, 16 h); 即使在室温(50 atm, 20 ℃), 催化剂用量为0.025 mol%的条件下反应16 h, 34a也能给出37%的转化率.对于苯甲酸乙酯, 转化数也可达到9370 (0.01 mol% 34a).值得提及的是, 在催化剂用量为0.00125 mol%时(40 atm, 70 ℃), 34a对己酸乙酯的初始转化频率(TOF)可达283200 h-1, 反应1 h的转化数也可达到53900.这些结果表明二乙胺桥联双卡宾配体的引入显著提高了钌催化剂对酯氢化的效率, 并取得了目前三齿钳形催化剂最高的转化数.
2015年, Gordon等[49]设计发展了系列在空气中稳定的二乙胺类S-N-N型钳形配体及其钌和铱催化剂.就钌催化剂而言, 配体硫原子上甲基和苯基取代的催化剂35a和35b(图 5)具有较高的催化活性和选择性.在三氟乙酸甲酯的氢化为半缩醛的反应中, 催化剂35a和35b能给出接近2000的转化数(0.05 mol%催化剂, 25 mol%甲醇钾, 25 atm, 40 ℃, 24 h)和86%的选择性.催化活性虽然不如催化剂Ru-MACHO (5), 但其选择性却优于后者(24000的转化数和75%的选择性).与Gusev等[42]发展的S-N-S钳形钌催化剂31a相比, 催化活性和选择性相当(1660的转化数和83%的选择性).相比较而言, 具有铱碳键的单氢铱催化剂36a~36c(图 5)具有更高的活性, 在相同条件下可给出11000~11600的转化数(0.005 mol%催化剂), 选择性也高达98%.同样与P-N-P型铱催化剂11b[26](达24200的转化数, 59%的选择性)相比, 虽然活性稍差一些, 但铱催化剂36a~36c生成半缩醛产物的选择性明显优于前者. 2018年Vries等[50]用吡啶代替胺基发展了二乙胺类S-N-N型钳形配体及其钌催化剂37.研究发现该催化剂在甲苯溶剂中可以选择性地氢化肉桂酸类不饱和酯中的酯基, 得到相应的烯丙醇.在催化剂用量为0.25 mol% (37a), 添加叔丁醇钾, 30 atm H2和40 ℃的条件下反应4或16 h, 烯丙醇产物的收率接近90%, 选择性最高达到95:5.此外, 催化剂37a对苯甲酸酯、戊内酯等酯的氢化也具有很高的催化活性.如在0.05 mol%的催化剂用量, 30~60 atm H2和80 ℃反应2~24 h, 对系列酯的氢化收率为65%~99%.
此外, 2018年Zimmermann和Waser等[51]对含硫配位基的S-N-S, S-N-N以及S-N-C等类型的钳形钌催化剂包括31a和37a等进行了系统的评价, 发现31a中硫原子上乙基变为甲基的钌催化剂为最优秀的催化剂.对不同类型的酯类底物均可在20 atm H2的条件下进行氢化, 反应具有易于操作, 容易放大的优点.
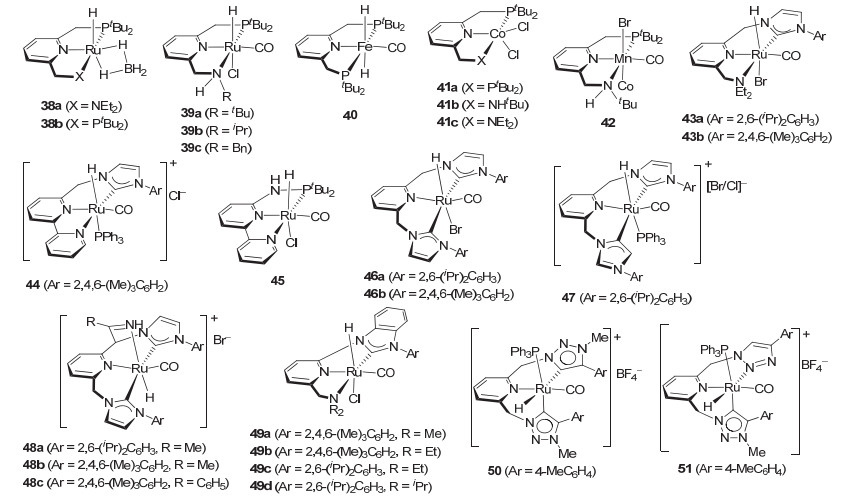
自从2006年Milstein等[8]报道第一例具有吡啶骨架的三齿P-N-P型钳形钌催化剂2催化酯的氢化反应以来, 该类三齿钳形配体及其催化剂也得到了快速的发展. 2011年, Milstein等[52]将钌催化剂2发展为含有硼烷的三齿P-N-N型钌催化剂38a以及三齿P-N-P型钌催化剂38b(图 6), 并发现在不添加碱的条件下, 钌催化剂38a的催化活性明显优于38b.如在丁酸丁酯和苯甲酸苄酯的氢化中(10 atm H2和110 ℃的条件下反应12 h, 0.5 mol%)钌催化剂38a给出98%和99%的转化率.而P-N-P型钌催化剂38b活性较低, 转化率仅为10%和17%.他们认为三齿P-N-N型钌催化剂38a效率高的主要原因是由于二乙胺基对钌金属的弱配位效应所致. 2014年, Milstein等[53]出于对金属-配体协同催化机理的兴趣而设计了含有NH的P-N-N型钳形钌催化剂39(图 6).在0.5 mol%的催化剂用量, 添加1.1 mol%的叔丁醇钾和5 atm H2, 室温条件下, 钌催化剂39c对一些脂肪酸酯和芳香酸酯以及庚内酯都表现出很高的催化活性, 反应24~48 h给出了75%~99%的收率.当用钌催化剂39a时, 需要添加更多的叔丁醇钾(5 mol%), 才能得到较好的结果.催化剂的用量减少到0.02 mol%时且在50 atm H2的条件下, 钌催化剂39a对脂肪酸酯和芳香酸酯, 以及庚内酯也能给出93%~99%的收率.
同年, Milstein等[19]将吡啶骨架的三齿P-N-P钳形配体的铁催化剂引入酯的氢化反应, 并发现铁催化剂40 (图 6)能够在温和的反应条件下(1 mol% 40, 5 mol%甲醇钠, 25 atm, 40 ℃, 1, 4-二氧六环, 16 h)催化系列三氟乙酸酯的氢化, 收率为25%~99%.对活性更高的三氟乙基三氟乙酸酯的氢化, 转化数还可达到1280 (0.05 mol% 40, 1 mol%叔丁醇钾, 10 atm, 40 ℃, 90 h, 64%收率).这是首例铁催化酯的均相催化氢化, 在此之后Guan等[20]和Beller等[21]也在同年报道了铁催化酯的均相催化氢化, 并实现了非活化酯的氢化(详细见2.1.1节).在此基础上, Milstein等[29]随后又发现吡啶骨架的三齿P-N-P和P-N-N型钳形钴催化剂也能有效催化脂肪酸酯的氢化, 但对芳香酸酯和三氟乙酸酯没有催化活性.在环己基己酸酯的氢化中, P-N-P型钴催化剂41a (图 6, 27%收率)的催化活性不如P-N-N型钴催化剂41b (85%收率)和41c (67%收率), 氮原子上含有氢的P-N-N型钴催化剂41b给出最高的催化活性(2 mol% 41b, 4 mol%三乙基硼氢化钠, 25 mol%叔丁醇钾, 50 atm, 130 ℃, 38 h).在相同的反应条件下, 催化剂41b可以氢化一系列脂肪酸酯, 给出50%~85%的收率.鉴于41b对芳香酸酯和三氟乙酸酯无催化活性, 他们认为脂肪酸酯的氢化是通过烯醇形式进行的.芳香酸酯和三氟乙酸酯不能形成烯醇式因而不能被氢化. 2017年, Milstein等[54]进一步将锰催化剂引入酯的氢化, 并发现吡啶骨架的三齿P-N-P型钳形锰催化剂42(图 6)也能够催化脂肪酸和芳香酸酯的氢化.在1 mol%催化剂, 2 mol%氢化钾以及100 ℃和20 atm H2的反应条件下, 锰催化剂42对系列脂肪酸和芳香酸酯给出了60%~99%的收率.与钴催化剂41b不同, 锰催化剂42对不能形成烯醇的芳香酸酯和三氟乙酸酯也表现出与脂肪酸酯相当的催化活性.核磁共振研究表明, 42催化酯的氢化反应经历了锰氢中间体.因此该催化剂很可能是经配体促进的协同机理实现酯的氢化.这也是继Beller等[27]首次报道锰催化酯的氢化反应后, 又一例锰催化酯氢化的文献报道.
与此同时, 基于吡啶骨架发展新的三齿钳形配体及催化剂并用于酯基氢化的研究也广受关注.如2011年, Song等[45]用强σ-供电子性质且比膦配体稳定的卡宾配体代替Milstein催化剂2中的膦配体, 发展出三齿N-N-C钳形钌催化剂43 (图 6).研究表明, 在1 mol%催化剂和添加8 mol%叔丁醇钾或KHMDS, 以及在105 ℃和5.3 atm H2的条件下反应2~3 h, 钌催化剂43a对乙酸酯和苯甲酸酯能给出90%~99%的收率.同年, Milstein等[44]在引入卡宾配体替换膦配体的同时将催化剂2中的二乙胺基换为吡啶基, 从而发展出具有联吡啶骨架类型的三齿N-N-C钳形配体的钌催化剂44 (图 6).在1 mol%催化剂用量和添加1 mol%叔丁醇钾以及在5.4 atm H2和135 ℃的条件下反应2 h, 钌催化剂44对丁酸乙酯、苯甲酸乙酯和苄酯等的氢化给出了89%~97%的收率.催化剂的用量降低到0.025 mol%时, 催化剂44对苯甲酸乙酯的氢化也可给出2840的转化数(50 atm, 12 h, 72%的转化率).此外, Milstein等[55]还将钌催化剂44用于乙交酯氢化为乙二醇的反应中(10 atm, 110 ℃, 48 h, 58%的收率), 但催化活性和效率均不如Milstein催化剂2 (12 h, 93%的收率). 2014年, Wang和Huang等[56]将他们设计和发展的具有吡啶骨架的PN3-类型的钳形钌催化剂用到酯的氢化中, 并发现钌催化剂45[57](图 6)具有较高的催化活性.在1 mol%催化剂用量, 添加8 mol%叔丁醇钾, 27 atm H2和120 ℃的条件下反应2 h, 催化剂45对苯甲酸酯和脂肪酸酯能给出96%~98%的收率.值得提及的是, 该催化剂在添加水的情况下对酯的氢化仍保留很高的催化活性. DFT计算表明该催化剂催化酯的氢化时经历了水参与的“双分子质子梭(Bimolecular proton shuttle)”的反应机制.该机制使催化剂通过一个更低的过渡态能垒活化氢分子.
2014年, Pidko等[46]设计发展了基于吡啶骨架的双卡宾类型的C-N-C钳形钌催化剂46(图 6).同时, 他们在制备该类钳形催化剂时, 还得到了异常配位的钌催化剂47, 以及在腈类溶剂中进行时得到的与腈类溶剂加合的钌催化剂48(图 6).这些含有吡啶骨架的双卡宾C-N-C钳形钌催化剂均能催化酯的氢化.在0.1~0.5 mol%催化剂用量和添加甲醇钾或叔丁醇钾的反应条件下(50 atm, 70~100 ℃, 4~16 h), 双卡宾类型的C-N-C钳形钌催化剂46~48对苯甲酸酯和脂肪酸酯的氢化均可给出几乎定量的收率. 2016年, Chianese等[58]在Song[45]发展了含单卡宾的C-N-N型钳形钌催化剂43的基础上设计合成了卡宾与吡啶直接相连的单卡宾C-N-N型钳形钌催化剂49(图 6).催化剂49b具有更高的催化活性.在添加叔丁醇钠以及6 atm H2和105 ℃的反应条件下, 0.1~0.8 mol%的催化剂49b对苯甲酸乙酯和环己基甲酸乙酯等的氢化都给出大于96%的收率.然而, 该催化剂对苯甲酸甲酯和环己基甲酸甲酯等甲酯表现出非常低的催化活性, 在相同的反应条件下收率不超过10%.值得提及的是, 当添加等当量的苄醇后, 49b可以催化壬酸甲酯的氢化, 并给出了与相应乙酯相当的催化活性.随后, Chianese等[59]将钌催化剂49中卡宾氮原子上的芳基变为位阻更大的2, 6-二异丙基苯基, 合成了49c和49d等钳形钌催化剂.由于大位阻的2, 6-二异丙基苯基的引入, 显著提高了该类C-N-N型钳形钌催化剂的活性.如胺基氮原子上乙基取代的钌催化剂49c在相同的反应条件下(6 atm H2, 105 ℃, 0.2~0.05 mol% 49c, 20 h), 对系列脂肪酸酯和芳香酸酯给出了高达2000的转化数, 明显优于卡宾氮原子上的2, 4, 6-三甲基取代的钌催化剂49a和49b. 2017年, Elsevier等[60]用三唑代替卡宾设计发展了具有吡啶骨架的C-N-C型钳形钌催化剂50(图 6).然而只有在添加20 mol%叔丁醇钾的反应条件下, 钌催化剂50 (0.75 mol%)才对苯甲酸甲酯、丁酸甲酯等脂肪酸酯表现出较高的催化活性, 并给出了53%~100%的收率(50 atm, 100 ℃).此外, 他们还得到了三唑氮配位的C-N-N型钳形钌催化剂51(图 6).该催化剂在相同的反应条件下对苯甲酸叔丁酯的氢化活性与催化剂50相当.
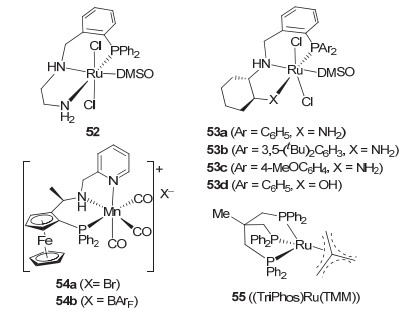
2012年Clarke等[61]在系统评价Noyori类型的双膦-钌-双胺催化剂对氟苯甲酸甲酯的氢化时, 对他们发展的含NH的三齿P-N-N和P-N-O型钳形钌催化剂52和53(图 7)也进行了评价.在催化剂用量为0.5 mol%, 叔丁醇钾做碱和100 ℃及50 atm H2的条件下反应2~16 h, 这些钌催化剂对氟苯甲酸甲酯的氢化均给出了94%~98%的收率.其中, 磷原子上苯基取代的钌催化剂52, 53a和53d的催化活性较高, 反应在2 h内结束, 并有很高的选择性.在相同的反应条件下, 钌催化剂52对系列苯环上不同取代的苯甲酸甲酯也给出了相当的氢化结果.但对含吡啶配位基的吡啶甲酸甲酯而言, 在较高氢气压力(100 atm)的条件下也只对3-位吡啶甲酸甲酯才表现出催化活性.此外, Clarke等[62]还报道了钌催化剂52对聚对苯二酸乙二酯(PET)的氢化.在110 ℃及50 atm H2的条件下反应48 h, 2.0 mol% 52可给出73%的收率.
2017年Clarke等[63]基于二茂铁骨架的手性三齿吡啶胺基膦配体发展了锰催化剂54[64](图 7), 并发现该催化剂除了对简单酮的不对称催化氢化具有很高的催化活性和对映选择性以外, 对酯的氢化也表现出较高的催化活性.在0.9 mol%的催化剂用量下和添加10 mol%叔丁醇钾以及75 ℃及50 atm H2的条件下反应16 h, 催化剂54a对不同取代的芳香酸甲酯和乙酯都给出了80%~90%的收率.对含有炔基的芳香酸甲酯氢化时, 炔基可部分保留.对丁酸丁酯, 在0.1 mol%催化剂用量下也可给出82%的转化率(90 ℃, 15 h).最近, 他们还将催化剂54a用于苯甲酸乙酯、α-取代苯甲酸酯和α-氨基酸酯等的氢化[65].当用异丙醇做溶剂时, 反应的收率为66%~96%, 转化数达到1000 (1~0.1 mol% 54a, 10~100 mol%碳酸钾, 50~110 ℃, 16 h).值得一提的是, 在氢化光学活性α-氨基酸酯和α-取代苯甲酸酯为相应的手性伯醇反应中(1 mol% 54a), 采用碳酸钾做碱, 产物的ee值与相应底物的ee值基本保持一致, 没有明显降低.只是在α-四氢萘甲酸乙酯氢化为相应的四氢萘甲醇时, ee值降低了11%.
此外, 2014年Leitner等[66]发现三齿Triphos配体(1)的钌催化剂55(图 7)对官能化酯如γ-戊内酯、衣康酸甲酯、丙交酯等表现出较高的催化活性.在1.0 mol%的催化剂用量下以及50 atm H2和140 ℃的条件下反应16~24 h, 催化剂55可在无外加碱等添加剂下直接氢化上述官能化酯, 给出95%~99%的收率.值得提及的是, 该催化剂可直接氢化羧酸, 制备相应的醇, 但需要相对较高的反应温度(140~220 ℃). 2015年, Elsevier和de Bruin等[30]还发现由四氟硼酸钴与Triphos配体(1)现场生成的钴催化剂可在80 atm H2和100 ℃的反应条件下催化苯甲酸甲酯、丁酸甲酯、γ-戊内酯以及三油酸甘油脂的氢化, 在5~10 mol%的催化剂用量下反应5~22 h, 给出47%~95%的收率.该钴催化剂同样可直接催化羧酸氢化到醇的反应.
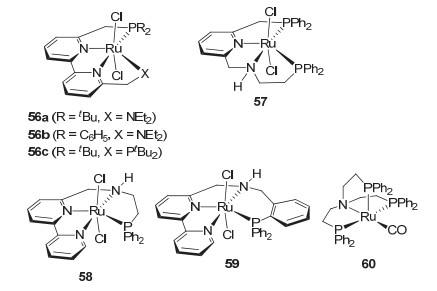
虽然2007年Saudan等[10]已报道了四齿配体的钌催化剂在酯的氢化中具有催化活性, 但含四齿配体的钌催化剂用于酯氢化的研究相对较少. 2014年, 周其林等[13]基于联吡啶骨架设计发展了四齿P-N-N-N型的钌催化剂56a和56b以及P-N-N-P型的钌催化剂56c(图 8).他们发现P-N-N-N型的钌催化剂56a和56b在氢化生物质平台分子γ-戊内酯时比相应的P-N-N-P型催化剂56c有更高的催化活性, 尤其是磷原子上叔丁基取代的钌催化剂56a活性最高.在以异丙醇为溶剂, 添加甲醇钠为碱, 0.001 mol%的催化剂用量, 室温和100 atm H2(初始)的条件下反应48 h, 钌催化剂56a给出了91%的收率, 催化剂的转化数达到91000, 转化频率也高达1896 h-1.这是目前γ-戊内酯均相催化氢化的最高转化数.此外, 在非常温和的反应条件如10 atm H2和室温的反应条件下反应16 h, 用0.1 mol%的56a对γ-戊内酯的氢化也可给出95%的收率.催化剂56a对γ-酮酸酯、丁酸二甲酯、草酸二甲酯等官能化酯, 以及简单的苯甲酸甲酯和乙酸乙酯等芳香酸酯和脂肪酸酯均有很高的活性.特别是在苯甲酸甲酯和乙酸乙酯氢化反应中分别得到91000和90000的高转化数(40 ℃, 50 atm, 48 h; 25 ℃, 100 atm, 64 h).这也是目前文献报道的酯催化氢化最好的结果. 2015年, 张绪穆等[67]基于吡啶骨架设计发展了含有NH的P-N-N-P型的钌催化剂57.他们发现催化剂57在催化剂用量为0.1~0.001 mol%, 添加叔丁醇钾做碱的反应条件下, 对系列芳香酸酯、脂肪酸酯以及官能化的酯如丁酸二甲酯等的氢化能给出80%~>99%的收率(80 ℃, 50 atm).对乙酸乙酯的氢化, 转化数可达80000 (30 h).该催化剂对长链脂肪酸甲酯也能给出高达50000的转化数.此外, 催化剂57对不饱和共轭酸酯的氢化, 双键不能保留, 得到相应的饱和醇; 而对含非末端双键的非共轭不饱和酸酯的氢化, 双键可以保留. 2016年, 张绪穆等[68]又发展了四齿P-N-N-N型的钌催化剂58和59.催化剂59的催化活性优于58, 但总体而言不如催化剂56a和57.
2018年, Schaub等[69]基于P-N-P型钳形钌催化剂Ru-MACHO (5)发展了N-3P型的钌催化剂60.他们发现该四齿类型的钌催化剂可在不添加碱和无溶剂的条件下(0.016 mol% 60, 60 atm H2, 130 ℃, 40 h)催化乙酸乙酯的氢化, 转化数达到5688.然而, 该催化剂对芳香酸酯如对苯二甲酸甲酯却显示惰性.
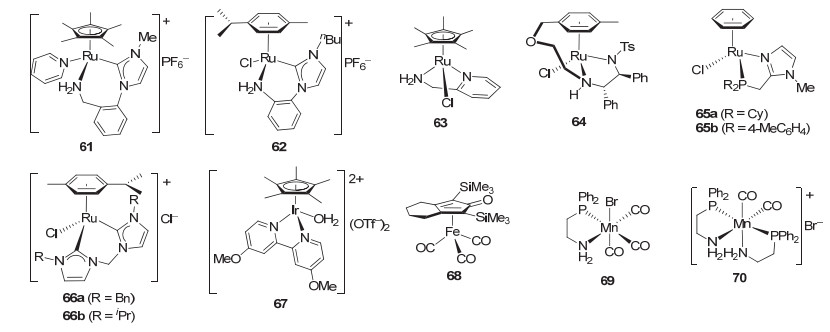
双胺配体配位的半夹心类型的金属催化剂等在简单酮的催化氢化中应用非常广泛[70].目前, 这一类型的催化剂以及类似的胺基膦配体配位的半夹心金属催化剂已在酯氢化反应中表现出很高的催化活性. 2010年, Morris等[71]首先将伯胺类型的氮-卡宾配体的茂钌催化剂61(图 9)用于酯的氢化.在50 ℃和25 atm H2以及添加叔丁醇钾的条件下反应2 h, 催化剂61 (0.067 mol%)对苯甲酸甲酯给出78%的转化率.在相同的反应条件下, 催化剂61对其它芳香酸酯和脂肪酸酯的氢化也表现出较高的催化活性, 特别是对叔戊酸甲酯氢化的转化数达到1480[72].机理研究表明该催化剂是通过配体促进的双功能机理实现酯的氢化. 2013年, Elsevier等[73]发展了系列芳胺类型的氮-卡宾配体, 并发现其钌催化剂62(图 9)对生物质平台分子丁酸甲酯表现出一定的催化活性.在80 ℃和80 atm H2的条件下反应24 h, 能给出29%的转化率(0.5 mol% 62); 但在相同条件下对草酸甲酯却没有氢化产物生成.
2011年, Ikariya等[74]在评价了系列乙基连接的胺基膦及双胺等配体后发现吡啶甲胺的茂钌催化剂63 (图 9)对苯并呋喃酮类型的内酯具有较高的催化活性.在1 mol% 63, 25 mol%叔丁醇钾, 100 ℃, 50 atm, 6~18 h的反应条件下, 该催化剂对系列α-取代或γ/δ-取代的γ-丁内酯和δ-戊内酯也都给出大于99%的转化率和80%~90%的收率.同时, 他们还发现Ts-DPEN类型的钌催化剂64(图 9)对γ-丁内酯的氢化也具有很高的催化活性[75].在添加叔丁醇钾(10 mol%)以及60 ℃和50 atm氢气压力的条件下反应48 h, 催化剂64 (2 mol%)可以以>99%的转化率将γ-丁内酯转化为1, 4-丁二醇.
2012年Beller等[76]发展了系列膦-咪唑类型双齿配体, 并发现磷原子上环己基和4-甲基苯基取代的钌催化剂65a和65b(图 9)可以有效地氢化酯.在膦-咪唑(1 mol%)与[Ru(C6H6)Cl2]2 (0.5 mol% Ru)现场生成催化剂, 添加10%叔丁醇钾, 以及在100 ℃和50 atm H2的条件下反应4.5 h, 催化剂65a和65b对苯甲酸甲酯的氢化可给出92%和94%的收率.在相同的反应条件下, 催化剂65b能够氢化芳香酸酯, 收率17%~99%.而催化剂65a对脂肪酸甲酯及内酯等的氢化效果较好, 能给出26%~99%的收率.值得提及的是, 膦-咪唑双齿配体与钌的比例为1:1时反应活性较低, 2:1时活性最高, 增加到4:1时则无活性. 2013年, Beller等[77]在此基础上又发展了亚甲基桥联的双卡宾类型的钌催化剂.与膦-咪唑类型双齿配体的钌催化剂类似.亚甲基桥联的双卡宾配体与[Ru(p-cymene)Cl2]2配位得到的钌催化剂如66a(图 9), 虽然对苯甲酸甲酯的氢化具有催化活性, 但活性较低, 只有39%的收率.当额外加入1 equiv.的双卡宾配体时, 催化活性显著提高, 反应收率提高到82% (1 mol% 66a, 30%叔丁醇钾, 100 ℃, 50 atm, 6 h).在相同的反应条件下, 催化剂66a可以氢化芳香酸、脂肪酸的甲酯和乙酯以及戊内酯等, 给出32%~92%的收率.
2016年, Goldberg等[78]报道了联吡啶的半夹心铱催化剂67(图 9)在酯氢化中的应用.在100 ℃和30 atm H2以及无溶剂和添加剂的条件下, 催化剂67可催化甲酸乙酯、乙酸甲酯和乙酸乙酯的氢化, 转化数分别达到341、305和106 (4 μmol 67, 16 h).当添加40 μmol Sc(OTf)3后, 氢化反应的转化数显著提高, 可分别达1317、346和200.催化剂67对内酯也具有催化活性, 在相同的氢化条件和无溶剂下对戊内酯和γ-丁内酯的氢化转化数可达487和200. 2016年, Pignataro等[79]将环戊二烯酮的铁催化剂68(图 9)引入酯的氢化, 发现其仅对三氟乙酸酯这样的活化酯表现出较高的催化活性.在90 ℃和70 atm H2以及添加2 mol% Me3NO、20 mol%三乙胺的条件下反应17 h, 催化剂68对三氟乙酸酯的氢化收率为60%~100%. 2017年, Pidko等[80]利用乙基桥联的胺基-膦配体发展了锰催化剂69和70(图 9), 并发现它们对酯的氢化也具有较高的催化活性.与Saudan等[10]发展的乙基桥联的胺基-膦配体的钌催化剂3不同, 他们发现只含有一个胺基膦配体的锰催化剂69比含有两个胺基膦配体的锰催化剂70的催化活性更高.对苯甲酸甲酯的氢化, 前者的收率66%, 后者的收率仅为24% (1 mol%催化剂, 10 mol%叔丁醇钾, 100 ℃, 50 atm, 20 h).当增加叔丁醇钾的用量为75 mol%时, 催化剂69也可给出98%的收率.在相同的氢化条件下, 催化剂69对系列脂肪酸酯、芳香酸酯给出了81%~98%的收率(0.2 mol% 69).对含末端双键或非共轭双键的长链脂肪酯的氢化, 双键基本保留.如在10-十一烯酸甲酯和顺-9-十八烯酸甲酯的氢化中以98%和95%的收率得到不饱和醇(0.5 mol% 69).然而对共轭的不饱和酯, 如肉桂酸甲酯, 则仅给出相应的饱和醇(99%的收率). DFT计算表明, 催化剂69在酯氢化中是通过配体促进的双官能化机理进行的.
通过消旋酯的动力学拆分和动态动力学拆分的不对称催化氢化可以合成相应的光学活性的手性伯醇.这是合成手性伯醇的高原子经济性的绿色方法.然而, 由于简单酯, 如脂肪酸酯和芳香酸酯的氢化往往需要在较高的温度和氢气压力下才能进行.目前已发展的消旋酯的不对称催化氢化合成手性伯醇仅限于内酯或在氢化反应条件下能够形成内酯的羟基酯等的氢化.
2011年, Ikariya等[74]报道了用手性双胺的半夹心茂钌催化剂71(图 10)催化消旋α-苯基取代丁内酯的不对称氢化合成相应的手性1, 4-二醇, 但对映选择性很低.采用最好的氮原子甲基取代的1, 2-环己二胺为配体的催化剂71d, 也仅得到32% ee的对映选择性.这是关于酯不对称催化氢化合成手性伯醇的首次报道.
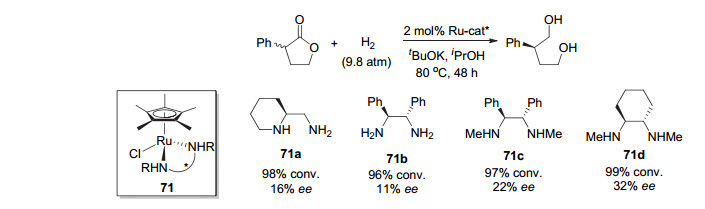
2013年, 周其林和谢建华等[81]在研究侧链含有酯基的α, α'-双取代环状脂肪酮的动态动力学拆分不对称催化氢化时, 意外发现手性螺环双膦配体SDP的钌-双膦-双胺催化剂72(图 11)能够高效催化侧链酯基的氢化, 得到含有三个连续手性中心的手性二醇, 对映选择性高达75%~99.9% ee.同年, 他们用手性螺环吡啶-胺基膦配体的铱催化剂Ir-SpiroPAP (73, 图 11)氢化δ-酮酸酯合成δ-羟基酯, 发现氢化反应很难停留在酮氢化的产物, 酯基也被氢化成醇[82].从而实现了δ-酮酸酯的不对称催化氢化合成手性二醇, 反应的对映选择性高达99.9% ee, 转化数也可达到100000.通过对反应中间体的跟踪, 他们发现反应是先氢化酮羰基, 生成δ-羟基酯. δ-羟基酯在反应条件下形成内酯中间体, 然后被氢化到二醇.该研究工作引起了Ohkuma等[83]的研究兴趣, 他们采用手性膦配体Skewphos和吡啶胺基配体和釕形成的双膦-钌-双胺催化剂74(图 11)对γ-和δ-酮酸酯进行氢化, 也得到了手性二醇, 对映选择性达到95%~99% ee.同样, 该催化剂对β-和ω-酮酸酯等很难在氢化反应条件下生成内酯的酮酸酯只能得到羰基被氢化的产物.
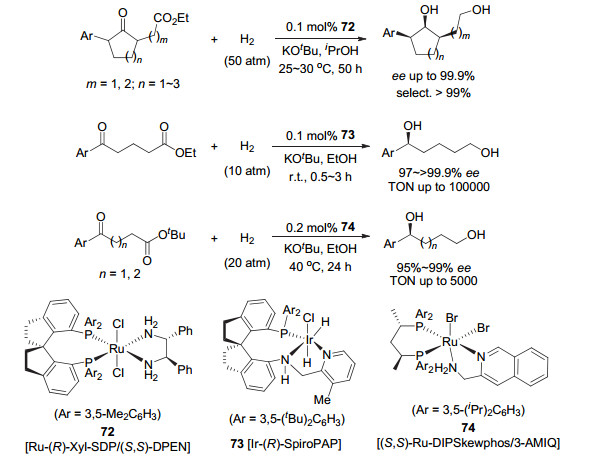
基于上述发现, 周其林和谢建华进一步深入研究了消旋酯的不对称催化氢化反应.他们在消旋脂肪醇的侧链上引入酯基, 并通过酯基的对映选择性氢化实现消旋脂肪醇的拆分, 得到相应的光学活性手性羟基酯和手性二醇[84](图 12).采用催化剂73, 消旋δ-羟基酯获得了很好的拆分效果:氢化产物手性二醇的收率为44%~50%, ee为91%~97%;回收的δ-羟基酯的收率为43%~49%, 和ee值为90%~99%.当催化剂用量降低到0.001 mol%时, 转化数达到52000的高转化数, 且氢化产物和回收的δ-羟基酯的ee值几乎保持不变.该催化剂对γ-羟基酯的拆分效果较差.经过对该氢化反应进行NMR跟踪实验发现, 酯的氢化也是通过形成内酯实现的.在此基础上, 他们最近又报道了消旋α-取代内酯的动态动力学拆分的不对称催化氢化[85].他们发现手性螺环铱催化剂73对一系列消旋α-芳基和烷基取代的戊内酯氢化均有较高的对映选择性(86%~95% ee) (图 12), 对消旋α-芳基和烷基取代的丁内酯的效果稍差, 对映选择性接近70% ee.这是首次将消旋α-取代内酯动态动力学拆分的不对称催化氢化合成手性二醇的反应对映选择性提高到大于90% ee以上.值得提及的是, 这些酯的不对称催化氢化反应已成功应用于手性药物和天然产物分子的不对称合成.
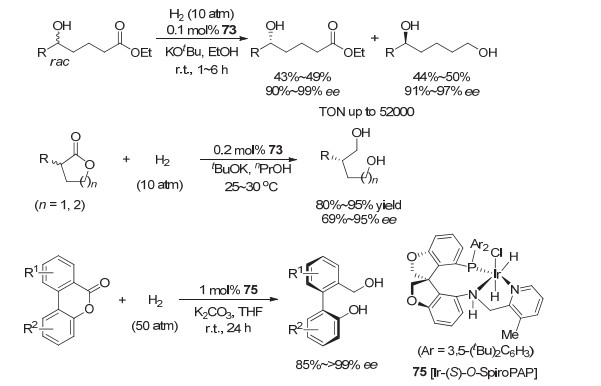
2018年, 张绪穆等[86]对手性螺环吡啶胺基膦的铱催化剂73进行修饰, 引入氧原子到螺环结构中, 发展出铱催化剂Ir-O-SpiroPSAP (75, 图 12).催化剂75在Bringmann’s内酯的不对称催化氢化中表现出较高的催化活性和很高的对映选择性.在优化的反应条件下(1 mol% 75, 5 mol% K2CO3, 50 atm H2, 室温), 催化剂对一系列Bringmann’s内酯氢化反应的对映选择性达到85%~>99% ee.通过反应中间体的分离鉴定, 他们提出反应是通过易于消旋化的轴手性阻转异构体的拆分氢化, 先形成半缩醛, 再氢化得到相应的含有联芳环骨架的手性醇.
在近十年的时间里, 酯的均相催化氢化得到了快速的发展, 已经发现近两百种均相催化剂可以催化酯的氢化.这些均相催化剂几乎都是金属配位物, 其配体主要有含二乙胺和吡啶骨架的钳形三齿配体、含联吡啶和吡啶骨架类型的四齿配体、含双胺、胺基膦、胺基咪唑、胺-卡宾、联吡啶等类型的双齿配体.催化剂的金属种类由早期的钌催化剂拓展到锇、铱等贵金属催化剂, 以及铁、钴、锰等丰产金属催化剂.虽然与简单酮的催化氢化相比, 酯的催化氢化的效率还有待提高.但是一些酯化合物的氢化已经有了高效催化剂.例如, 对于乙酸乙酯和苯甲酸甲酯等简单酯, 以及生物平台分子γ-戊内酯等的氢化, 具有联吡啶骨架的P-N-N-N型四齿配体的钌催化剂56a给出了90000或者更高的转化数[13].对于内酯或现场生成的内酯的氢化, 手性螺环吡啶胺基膦铱催化剂73的转化数达到100000[82].这些都达到或者接近了实际应用的要求.
总体而言, 酯的均相催化氢化为醇的反应多数效率不高, 只有少数几例得到了应用.其主要原因是高效催化剂的种类还有限, 特别是对不饱和酯的氢化具有化学选择性的催化剂非常稀少.丰产金属催化剂, 特别是铁催化剂虽然能够催化酯氢化反应, 但与钌催化剂相比, 效率还相差甚远.此外, 尽管内酯或现场生成的内酯不对称催化氢化合成光学活性手性二醇的研究已经取得了一些突破, 但简单的α-取代酯的动态动力学拆分不对称氢化还没有成功的例子.目前, 相应的氢化产物α-取代伯醇只能经由光学活性的酯合成.因此, 未来酯均相催化氢化的发展方向仍然是注重发展新型催化剂, 提高反应的效率和化学选择性, 以及注重发展酯的不对称催化氢化新反应.
(a) Calvin, M. J. Am. Chem. Soc. 1939, 61, 2230. (b) Iguchi, M. J. Chem. Soc. Japan 1939, 60, 1287.
(a) Young, J. F.; Osborn, J. A.; Jardine, F. H.; Wilkinson, G. J. Chem. Soc., Chem. Commun 1965, 131. (b) Osborn, J. A.; Jardine, F. H.; Young, J. F.; Wilkinson, G. J. Chem. Soc. A 1966, 1711.
(a) de Vries, J. G.; Elsevier, C. J. Eds The Handbook of Homogeneous Hydrogenation, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2007. (b) Zhang, L.; Han, Z.; Zhang, L.; Li, M.; Ding, K. Chin. J. Org. Chem. 2016, 36, 1824 (in Chinese). (张琳莉, 韩召斌, 张磊, 李明星, 丁奎岭, 有机化学, 2016, 36, 1824.) (c) Li, Y.; Wang, Z.; Liu, Q. Chin. J. Org. Chem. 2017, 37, 1978 (in Chinese). (李勇, 王征, 刘庆彬, 有机化学, 2017, 37, 1978.) (d) Chen, S.; Yang, W.; Yao, Y.; Yang, X.; Deng, Y.; Yang, D. Chin. J. Org. Chem. 2018, 38, 2534 (in Chinese). (陈姝琪, 杨文, 姚永祺, 杨新, 邓颖颍, 杨定乔, 有机化学, 2018, 38, 2534.)
(a) Grey, R. A.; Pez, G. P.; Wallo, A.; Corsi, J. J. Chem. Soc., Chem. Commun. 1980, 783. (b) Grey, R. A.; Pez, G. P.; Wallo, A. J. Am. Chem. Soc. 1981, 103, 7536.
Matteoli, U.; Menchi, G.; Bianchi, M.; Piacenti, F. J. Mol. Catal. 1988, 44, 347. doi: 10.1016/0304-5102(88)80020-8
Teunissen, H. T.; Elsevier, C. J. Chem. Commun. 1997, 667. doi: 10.1039/a700862g
Teunissen, H. T.; Elsevier, C. J. Chem. Commun. 1998, 1367.
Zhang, J.; Leitus, G.; Ben-David, Y.; Milstein, D. Angew. Chem., Int. Ed. 2006, 45, 1113. doi: 10.1002/(ISSN)1521-3773
Balaraman, E.; Gunanathan, C.; Zhang, J.; Shimon, L. J. W.; Milstein, D. Nat. Chem. 2011, 3, 609. doi: 10.1038/nchem.1089
Saudan, L. A.; Saudan, C. M.; Debieux, C.; Wyss, P. Angew. Chem., Int. Ed. 2007, 46, 7473. doi: 10.1002/(ISSN)1521-3773
Kuriyama, W.; Matsumoto, T.; Ogata, O.; Ino, Y.; Aoki, K.; Tanaka, S.; Ishida, K.; Kobayashi, T.; Sayo, N.; Saito, T. Org. Process Res. Dev. 2012, 16, 166. doi: 10.1021/op200234j
Filonenko, G. A.; van Putten, R.; Hensen, E. J. M.; Pidko, E. V. Chem. Soc. Rev. 2018, 47, 1. doi: 10.1039/C8CS90001A
Li, W.; Xie, J.-H.; Yuan, M.-L.; Zhou, Q.-L. Green Chem. 2014, 16, 4081. doi: 10.1039/C4GC00835A
(a) Pritchard, J.; Filonenko, G. A.; van Putten, R.; Hensen, E. J. M.; Pidko, E. V. Chem. Soc. Rev. 2015, 44, 3808. (b) Werkmeister, S.; Junge, K.; Beller, M. Org. Process Res. Dev. 2014, 18, 289. (c) Dub, P. A.; Ikariya, T. ACS Catal. 2012, 2, 1718. (d) Clarke, M. L. Catal. Sci. Technol. 2012, 2, 2418.
Acosta-Ramirez, A.; Bertoli, M.; Gusev, D. G.; Schlaf, M. Green Chem. 2012, 14, 1178. doi: 10.1039/c2gc15960k
Bertoli, M.; Choualeb, A.; Lough, A. J.; Moore, B.; Spasyuk, D.; Gusev, D. G. Organometallics 2011, 30, 3479. doi: 10.1021/om200437n
Ziebart, C.; Jackstell, R.; Beller, M. ChemCatChem 2013, 5, 3288. http://www.ncbi.nlm.nih.gov/pubmed/23281333
Otsuka, T.; Ishii, A.; Dub, P. A.; Ikariya, T. J. Am. Chem. Soc. 2013, 135, 9600. doi: 10.1021/ja403852e
Zell, T.; Ben-David, Y.; Milstein, D. Angew. Chem., Int. Ed. 2014, 53, 4685. doi: 10.1002/anie.201311221
Chakraborty, S.; Dai, H.; Bhattacharya, P.; Fairweather, N. T.; Gibson, M. S.; Krause, J. A.; Guan, H. J. Am. Chem. Soc. 2014, 136, 7869. doi: 10.1021/ja504034q
Werkmeister, S.; Junge, K.; Wendt, B.; Alberico, E.; Jiao, H.; Baumann, W.; Junge, H.; Gallou, F.; Beller, M. Angew. Chem. Int. Ed. 2014, 53, 8722. doi: 10.1002/anie.201402542
Fairweather, N. T.; Gibson, M. S.; Guan, H. Organometallics 2015, 34, 335. doi: 10.1021/om5011337
Elangovan, S.; Wendt, B.; Topf, C.; Bachmann, S.; Scalone, M.; Spannenberg, A.; Jiao, H.; Baumann, W.; Junge, K.; Beller, M. Adv. Catal. Synth. 2016, 358, 820. doi: 10.1002/adsc.201500930
Schneck, F.; Assmann, M.; Balmer, M.; Harms, K.; Langer, R. Organmetallics 2016, 35, 1931. doi: 10.1021/acs.organomet.6b00251
Junge, H.; Wendt, B.; Jiao, H.; Beller, M. ChemCatChem 2014, 6, 2810. doi: 10.1002/cctc.v6.10
Clarke, Z. E.; Maragh, P. T.; Dasgupta, T. P.; Gusev, D. G.; Lough, A. J.; Abdur-Rashid, K. Organometallics 2006, 25, 4113. doi: 10.1021/om060049z
Elangovan, S.; Carbe, M.; Jiao, H.; Spannenberg, A.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2016, 55, 15364. doi: 10.1002/anie.v55.49
Elangovan, S.; Topf, C.; Fischer, S.; Jiao, H.; Spannenberg, A.; Baumann, W.; Ludwig, R.; Junge, K.; Beller, M. J. Am. Chem. Soc. 2016, 138, 8809. doi: 10.1021/jacs.6b03709
Srimani, D.; Mukherjee, A.; Goldberg, A. F. G.; Leitus, G.; Diskin-Posner, Y.; Shimon, L. J. W.; Ben-David, Y.; Milstein, D. Angew. Chem. Int. Ed. 2015, 54, 12357. doi: 10.1002/anie.201502418
Korstanje T. J.; van der Vlugt, J. I.; Elsevier, C. J.; de Bruin, B. Science 2015, 350, 298. doi: 10.1126/science.aaa8938
Yuwen, J.; Chakraborty, S.; Brennessel, W. W.; Jones, W. D. ACS Catal. 2017, 7, 3735. doi: 10.1021/acscatal.7b00623
Zhang, G. Q.; Scott, B. L.; Hanson, S. K. Angew. Chem., Int. Ed. 2012, 51, 12102. doi: 10.1002/anie.201206051
Junge, K.; Wendt, B.; Cingolani, A.; Spannenberg, A.; Wei, Z.; Jiao, H.; Beller, M. Chem. Eur. J. 2018, 24, 1046. doi: 10.1002/chem.201705201
Tan, X.; Wang, Q.; Liu, Y.; Wang, F.; Lv, H.; Zhang, X. Chem. Commun. 2015, 51, 12193. doi: 10.1039/C5CC04242A
Ogata, O.; Nakayama, Y.; Nara, H.; Fujiwhara, M.; Kayaki, Y. Org. Lett. 2016, 18, 3894. doi: 10.1021/acs.orglett.6b01900
Spasyuk, D.; Smith, S.; Gusev, D. G. Angew. Chem. Int. Ed. 2012, 51, 2772. doi: 10.1002/anie.201108956
Spasyuk, D.; Gusev, D. G. Organometallics 2012, 31, 5239. doi: 10.1021/om300670r
Spasyuk, D.; Vicent, C.; Gusev, D. G. J. Am. Chem. Soc. 2015, 137, 3743. doi: 10.1021/ja512389y
Gusev, D. G. ACS Catal. 2016, 6, 6967. doi: 10.1021/acscatal.6b02324
Henrion, M.; Roisnel, T.; Couturier, J.-L.; Dubois, J.-L.; Sortais, J.-B.; Darcel, C.; Carpentier, J.-F. Mol. Catal. 2017, 432, 15. doi: 10.1016/j.mcat.2017.01.025
Wang, Z.; Chen, X.; Liu, B.; Liu, Q.-B.; Solan, G. A.; Yang, X.; Sun, W.-H. Catal. Sci. Technol. 2017, 7, 1297. doi: 10.1039/C6CY02413K
Spasyuk, D.; Smith, S.; Gusev, D. G. Angew. Chem., Int. Ed. 2013, 52, 2538. doi: 10.1002/anie.v52.9
McGuinness, D. S.; Wasserscheid, P.; Morgan, D. H.; Dixon, J. T. Organometallics 2005, 24, 552. doi: 10.1021/om049168+
Fogler, E.; Balaraman, E.; Ben-David, Y.; Leitus, G.; Shimon, L. J. W.; Milstein, D. Organometallics 2011, 30, 3826. doi: 10.1021/om200367j
Sun, Y.; Koehler, C.; Tan, R.; Annibale, V. T.; Song, D. Chem. Commun. 2011, 47, 8349. doi: 10.1039/c1cc12601f
Filonenko, G. A.; Cosimi, E.; Lefort, L.; Conley, M. P.; Coperet, C.; Lutz, M.; Hensen, E. J. M.; Pidko, E. A. ACS Catal. 2014, 4, 2667. doi: 10.1021/cs500720y
Filonenko, G. A.; Aguila, M. J. B.; Schulpen, E. N.; van Putten, R.; Wiecko, J.; Mgller, C.; Lefort, L.; Hensen, E. J. M.; Pidko, E. A. J. Am. Chem. Soc. 2015, 137, 7620. doi: 10.1021/jacs.5b04237
(a) Edworthy, I. S.; Blake, A. J.; Wilson, C.; Arnold, P. L. Organometallics 2007, 26, 3684. (b) Edworthy, I. S.; Rodden, M.; Mungur, S. A.; Davis, K. M.; Blake, A. J.; Wilson, C.; Schroöder, M.; Arnold, P. L. J. Organomet. Chem. 2005, 690, 5710. (c) Douthwaite, R. E.; Houghton, J.; Kariuki, B. M. Chem. Commun. 2004, 6, 698.
Dub, P. A.; Scott, B. L.; Gordon, J. C. Organometallics 2015, 34, 4464. doi: 10.1021/acs.organomet.5b00432
Stadler, B. M.; Puylaert, P.; Diekamp, J.; van Heck, R.; Fan, Y.; Spannenberg, A.; Hinze, S.; de Vries, J. G. Adv. Synth. Catal. 2018, 360, 1151. doi: 10.1002/adsc.v360.6
Schörgenhumer, J.; Zimmermann, A.; Waser, M. Org. Process Res. Dev. 2018, 22, 862. doi: 10.1021/acs.oprd.8b00142
Zhang, J.; Balaraman, E.; Leitus, G.; Milstein, D. Organometallics 2011, 30, 5716. doi: 10.1021/om200595m
Fogler, E.; Garg, J. A.; Hu, P.; Leitus, G.; Shimon, L. J. W.; Milstein, D. Chem. Eur. J. 2014, 20, 15727. doi: 10.1002/chem.v20.48
Espinosa-Jalapa, N. A.; Nerush, A.; Shimon, L. J. W.; Leitus, G.; Avram, L.; Ben-David, Y.; Milstein, D. Chem. Eur. J. 2017, 23, 5934. doi: 10.1002/chem.201604991
Balaraman, E.; Fogler, E.; Milstein, D. Chem. Commun. 2012, 48, 1111. doi: 10.1039/C1CC15778G
Chen, T.; Li, H.; Qu, S.; Zheng, B.; He, L.; Lai, Z.; Wang, Z.-X.; Huang, K.-W. Organometallics 2014, 33, 4152. doi: 10.1021/om500549t
He, L.-P.; Chen, T.; Gong, D.; Lai, Z.; Huang, K.-W. Organometallics 2012, 31, 5208. doi: 10.1021/om300422v
Kim, D.; Le, L.; Drance, M. J.; Jensen, K. H.; Bogdanovski, K.; Cervarich, T. N.; Barnard, M. G.; Pudalov, N. J.; Knapp, S. M. M.; Chianese, A. R. Organometallics 2016, 35, 982. doi: 10.1021/acs.organomet.6b00009
Le, L.; Liu, J-C.; He, T-Y.; Kim, D.; Lindley, E. J.; Cervarich, T. N.; Malek, J. C.; Pham, J.; Buck, M. R.; Chianese, A. R. Organometallics 2018, 37, 3286. doi: 10.1021/acs.organomet.8b00470
Sluijter, S. N.; Korstanje T. J.; van der Vlugt, J. I.; Elsevier, C. J. J. Organomet. Chem. 2017, 845, 30. doi: 10.1016/j.jorganchem.2017.01.003
Carpenter, I.; Eckelmann, S. C.; Kuntz, M. T.; Fuentes, J. A.; France, M. B.; Clarke, M. L. Dalton Trans. 2012, 41, 10136. doi: 10.1039/c2dt30973d
Fuentes, J. A.; Smith, S. M.; Scharbert, T.; Carpenter, T.; Cordes, D. B.; Slawin, A. M. Z.; Clarke, M. L. Chem. Eur J. 2015, 21, 10851. doi: 10.1002/chem.v21.30
Widegren, M. B.; Harkness, G. J.; Slawin, A. M. Z.; Cordes, D. B.; Clarke, M. L. Angew. Chem., Int. Ed. 2017, 56, 5825. doi: 10.1002/anie.201702406
Nie, H.; Zhou, G.; Wang, Q.; Chen, W.; Zhang, S. Tetrahedron:Asymmetry 2013, 24, 1567. doi: 10.1016/j.tetasy.2013.10.012
Widegren, M. B.; Clarke, M. L. Org. Lett. 2018, 20, 2654. doi: 10.1021/acs.orglett.8b00864
vom Stein, T.; Meuresch, M.; Limper, D.; Schmitz, M.; Hölscher, M.; Coetzee, J.; Cole-Hamilton, D. J.; Klankermayer, J.; Leitner, W. J. Am. Chem. Soc. 2014, 136, 13217. doi: 10.1021/ja506023f
Tan, X.; Wang, Y.; Liu, Y.; Wang, F.; Shi, L.; Lee, K.-H.; Lin, Z.; Lv, H.; Zhang, X. Org. Lett. 2015, 17, 454. doi: 10.1021/ol503456j
Wang, F.; Tan, X.; Lv, H.; Zhang, X. Chem. Asian J. 2016, 11, 2103. doi: 10.1002/asia.v11.15
Anaby, A.; Schelwies, M.; Schwaben, J.; Rominger, F.; Hashmi, A. S. K.; Schaub, T. Organometallics 2018, 37, 2193. doi: 10.1021/acs.organomet.8b00353
(a) Xie, J.-H.; Bao, D.-H.; Zhou, Q.-L. Synthesis 2015, 47, 460. (b) Zhao, B.; Han, Z.; Ding, K. Angew. Chem. Int. Ed. 2013, 52, 4744.
O, W. W. N.; Lough, A. J.; Morris, R. H. Chem. Commun. 2010, 46, 8240. doi: 10.1039/c0cc02664f
O, W. W. N.; Morris, R. H. ACS Catal. 2013, 3, 32. doi: 10.1021/cs300619q
Jansen, E.; Jongboed, L. S.; Tromp, D. S.; Lutz, M.; de Bruin, B.; Elsevier, C. J. ChemSusChem 2013, 6, 1737. doi: 10.1002/cssc.201300363
Ito, M.; Ootsuka, T.; Watari, R.; Shiibashi, A.; Himizu, A.; Ikariya, T. J. Am. Chem. Soc. 2011, 133, 4240. doi: 10.1021/ja1117254
Touge, T.; Hakamata, T.; Nara, H.; Kobayashi, T.; Sayo, N.; Saito, T.; Kayaki, Y.; Ikariya, T. J. Am. Chem. Soc. 2011, 133, 14960. doi: 10.1021/ja207283t
Junge, K.; Wendt, B.; Westerhaus, F. A.; Spannenberg, A.; Jiao, H.; Beller, M. Chem. Eur. J. 2012, 18, 9011. doi: 10.1002/chem.v18.29
Westerhaus, F. A.; Wendt, B.; Dumrath, A.; Wienhöfer, G.; Junge, K.; Beller, M. ChemSusChem 2013, 6, 1001. doi: 10.1002/cssc.v6.6
Brewster, T. P.; Rezayee, N. M.; Culakova, Z.; Sanford M. S.; Goldberg, K. I. ACS Catal. 2016, 6, 3113. doi: 10.1021/acscatal.6b00263
Gajewski, P.; Gonzalez-de-Castro, A.; M.; Renom-Carrasco, M.; Piarulli, U.; Gennari, C.; de Vries, J. G.; Lefort, L.; Pignataro, L. ChemCatChem 2016, 8, 3431. doi: 10.1002/cctc.201600972
van Putten, R.; Uslamin, E. A.; Garbe, M.; Liu, C.; Gonza-lez-de-Castro, A.; Lutz, M.; Junge, K.; Hensen, E. J. M.; Beller, M.; Lefort, L.; Pidko, E. A. Angew. Chem., Int. Ed. 2017, 56, 7531. doi: 10.1002/anie.201701365
Liu, C.; Xie, J.-H.; Li, Y.-L.; Chen, J.-Q.; Zhou, Q.-L. Angew. Chem. Int. Ed. 2013, 52, 593. doi: 10.1002/anie.201207561
Yang, X.-H.; Xie, J.-H.; Liu, W.-P.; Zhou, Q.-L. Angew. Chem. Int. Ed. 2013, 52, 7833. doi: 10.1002/anie.v52.30
Arai, N.; Namba, T.; Kawaguchi, K.; Masumoto, Y.; Ohkuma, T. Angew. Chem. Int. Ed. 2018, 57, 1386. doi: 10.1002/anie.201711363
Yang, X.-H.; Wang, K.; Zhu, S.-F.; Xie, J.-H.; Zhou, Q.-L. J. Am. Chem. Soc. 2014, 136, 17426. doi: 10.1021/ja510990v
Yang, X.-H.; Yue, H.-T.; Yu, N.; Li, Y.-P.; Xie, J.-H.; Zhou, Q.-L. Chem. Sci. 2017, 8, 1811. doi: 10.1039/C6SC04609F
Chen, G.-Q.; Lin, B.-J.; Huang, J.-M.; Zhao, L.-Y.; Chen, Q.-S.; Jia, S.-P.; Yin, Q.; Zhang, X. J. Am. Chem. Soc. 2018, 140, 8064. doi: 10.1021/jacs.8b03642

图 1 早期酯均相催化氢化所用的代表性配体及催化剂
Figure 1 Early representative ligands and catalysts for the hydrogenation of esters

图 2 高砂公司合成(R)-1, 2-丙二醇和L-薄荷烷氧基乙醇
Figure 2 Takasago's processes for synthesising (R)-1, 2-propanediol and 2-(l-menthoxy)ethanol

图 3 二乙基胺类P-N-P型钳形配体及催化剂
Figure 3 Diethylamine P-N-P type of pincer ligands and catalysts

图 4 修饰改造的二乙基胺类含磷钳形配体及催化剂
Figure 4 Pincer ligands and catalysts derived from diethylamine

图 5 不含磷的二乙基胺类钳形配体及催化剂
Figure 5 Phosphine-free pincer ligands and catalysts derived from diethylamine

图 10 Ikariya报道的消旋α-苯基γ-丁内酯的不对称催化氢化
Figure 10 Asymmetric hydrogenation of racemic α-phenyl γ-lactone reported by Ikariya

图 11 酮酸酯的不对称催化氢化合成手性二醇
Figure 11 Asymmetric hydrogenation of ketoesters to chiral diols

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:




