Received Date:
30 April 2019 Available Online:
15 July 2019
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21773166, 21673152)
Abstract:
The promising application of surface-enhanced Raman spectroscopy (SERS) was definitely based on the high quality substrates which were restricted to the rough noble metals and colloidal nanoparticle materials. However, semiconductor has become a potential substrate for the SERS investigation due to its high stability and reproducibility. It remains significant challenges in interpreting the enhancement mechanisms. Herein, broom-like ZnO nanoparticles with novel morphology and uniform size was prepared by pyrolysis of (CH3COO)2Zn. By using p-nitrophenylthiophenol (PNTP), phenylthiophenol (TP) and p-aminophenylthiophenol (PATP) as probe molecules, the SERS effect on ZnO surfaces was systematically studied under the irradiation of excitation lines with the wavelength of 532 nm and 638 nm. The different substituents in p-position of TP allowed to change the energy levels by the electron withdrawing or donating group, it was beneficial to match the energy level gap between the probe molecules and semiconductor for triggering the photon driven charge transfer. The surface enhancement factor (EF) of broom-like ZnO nanoparticles were estimated accordingly, and the contribution of non-resonance and charge transfer to SERS effect was distinguished. The results demonstrated that the surface enhancement factor was about 10 to 35 times depending on the probe molecules and excitation wavelengths. Therefore, the different enhancement origination contributed to the different molecules on the ZnO substrate. For the TP and PATP, the charge transfer from the HOMO level of molecule to CB of ZnO was achieved by the assistance of the laser photon with the appropriate energy. Moreover, the higher energy of the photon is, the stronger the SERS enhancement effect. As for the PNTP, the photon driven charge transfer was absent due to the significant change of the HOMO and LUMO level caused by the electron withdrawing group of NO2. It revealed that the enhancement effect of PNTP molecule about 10 times was contributed by the non-resonance enhancement mechanism which was mainly due to the changes in the polarizability caused by the chemical adsorption. Comparing to the noble metal surface, the enhancement of charge transfer on ZnO was decreased with 1~2 orders of magnitude. The relatively lower rate of charge transfer in semiconductor resulted in the decrease of the charge transfer enhancement. The preliminary studies provided a novel approach for the preparation and regulation of new semiconductor SERS substrates.
Wu, D. Y.; Li, J. F.; Ren, B.; Tian, Z. Q. Chem. Soc. Rev. 2008, 37, 1025. doi: 10.1039/b707872m
[11]
Wang, Y. F.; Ruan, W. D.; Zhang, J. H.; Yang, B.; Xu, W. Q.; Zhao, B.; Lombardi, J. R. J. Raman Spectrosc. 2009, 40, 1072. doi: 10.1002/jrs.v40:8
[12]
Klingshirn, C.; Fallert, J.; Zhou, H.; Sartor, J.; Thiele, C.; Maier Flaig, F.; Schneider, D.; Kalt, H. Phys. Status Solidi B 2010, 247, 1424. doi: 10.1002/pssb.v247:6
[13]
Yang, P. D.; Yan, R. X.; Fardy, M. Nano Lett. 2010, 10, 1529. doi: 10.1021/nl100665r
[14]
Chu, S.; Wang, G. P.; Zhou, W. H.; Lin, Y. Q.; Chernyak, L.; Zhao, J. Z.; Kong, J. Y.; Li, L.; Ren, J. J.; Liu, J. L. Nat. Nanotechnol. 2011, 6, 506. doi: 10.1038/nnano.2011.97
[15]
Zhao, D.; Zhang, X. X.; Dong, H. B.; Yang, L. J.; Zeng, Q. S.; Li, J. Z.; Cai, L.; Zhang, X.; Luan, P. S.; Zhang, Q.; Tu, M.; Wang, S.; Zhou, W. Y.; Xie, S. S. Nanoscale 2013, 5, 4443. doi: 10.1039/c3nr00049d

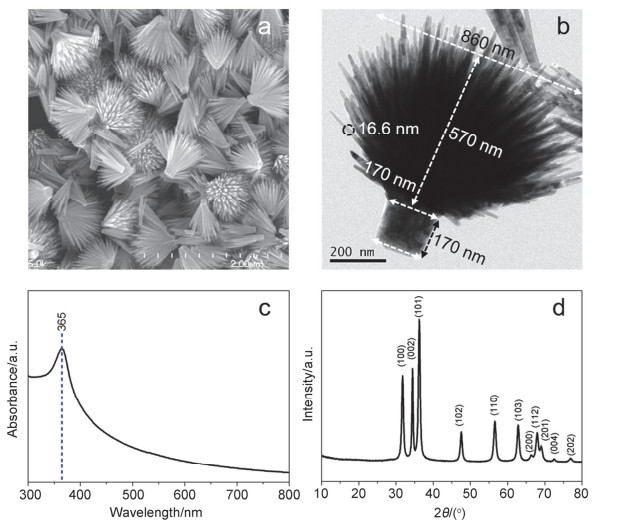
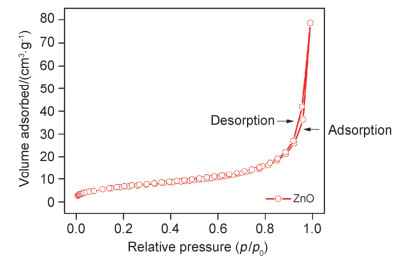
 下载:
下载:

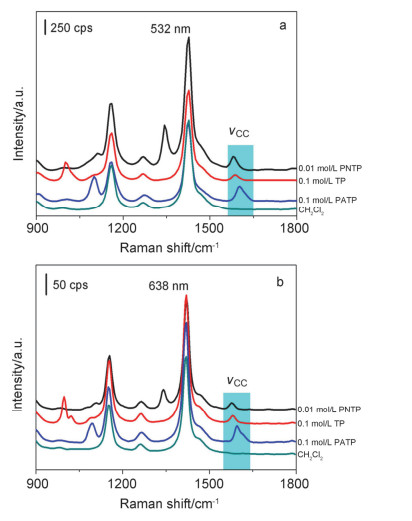

 下载:
下载:





