图 1.
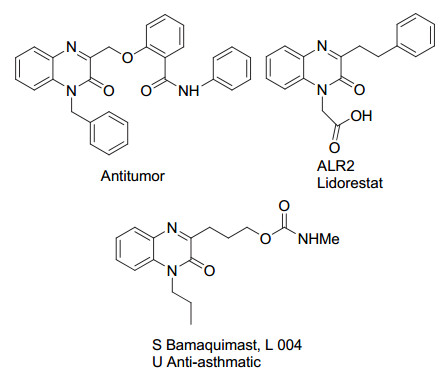
具有药物活性的喹喔啉酮类化合物
Figure 1.
Pharmaceutically active quinoxalinones

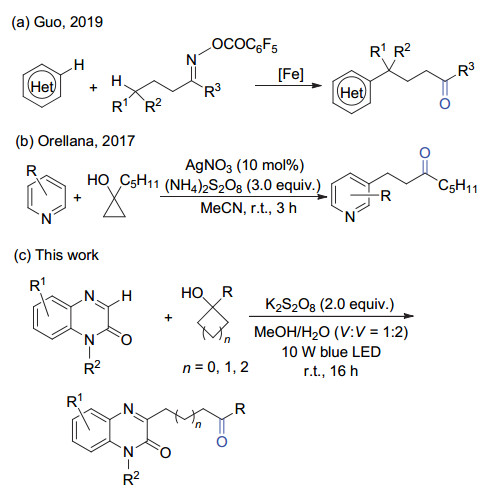
喹喔啉酮类化合物是一类广泛存在于具有生物活性的天然产物及药物分子中的特殊结构单元[1].因此, 喹喔啉酮类化合物的合成与修饰引起了化学家们的广泛关注[2].早在1971年, Minisci等[3]就报道了自由基的羟烷基化反应, 首次实现了对杂环化合物的修饰.随后, 一系列通过自由基取代的方法修饰杂环化合物方法被报道, 各类官能团可以通过该方法方便地引入到杂环化合物中[4].研究表明通过直接的C(sp2)—H键官能团化实现对喹喔啉酮的修饰无疑是一种简洁、高效的策略.然而, 如何实现高选择性的喹喔啉酮的官能团化仍具有很大的挑战性.近些年来, 传统过渡金属催化及自由基介导喹喔啉酮的C(3)位的官能团化取得了很大的进展[5], 一系列C(3)-芳基化[6]、膦化[7]、氨基化[8]、酰基化[9]及氟烷基化[10]反应相继被报道.然而, 喹喔啉酮的C(3)-烷基化反应[11], 特别是带有官能团的烷基化反应却不多见.最近, 我们课题组[12]通过亚胺自由基引发的C—C键裂解反应, 以环酮肟酯为烷基源, 在铁催化下成功实现了喹喔啉酮的C(3)-氰烷基化反应.此外, 我们课题组[13]还利用亚胺自由基引发的1, 5-氢迁移策略, 以开链酮肟酯为烷基源, 实现了喹喔啉酮的C(3)-酮烷基化反应.以上反应中, 我们在喹喔啉酮的C(3)位引入烷基的同时还引入了氰基或羰基官能团, 为进一步实现喹喔啉酮的修饰提供了机会.从图 1可以看到, 很多具有生物活性的喹喔啉酮分子中均含有不同链长的官能团化烷基片段[14].基于此, 我们将进一步开展喹喔啉酮的C(3)-烷基化反应研究.
环烷醇是一类非常重要的有机合成中间体.近些年研究表明, 环烷醇可作为潜在的酮烷基化试剂参与多种化学转化, 实现将酮烷基片段引入到各种分子骨架中.通常, 在单电子氧化条件下, 环烷醇通过烷氧自由基驱动的β-键均裂, 生成含有羰基官能团的碳自由基, 继而发生后续的加成或取代反应, 形成碳-碳或碳-杂键[15].目前, 环烷醇的氧化开环反应体系中常用的氧化剂有醋酸碘苯、过硫酸盐和过氧化叔丁醇等[15].其中, 过硫酸盐作为氧化剂的反应引起了我们的关注.过硫酸盐作为一种廉价、环境友好、易操作的强氧化剂, 在有机合成中具有广泛的应用[16].过硫酸盐在加热、光照或过渡金属还原条件下均可分解产生硫酸根自由基负离子(SO4.-).该自由基负离子具有很强的单电子氧化性, 可以氧化各种中性分子或负离子产生相应的自由基活性中间体[17]. 2015年, 我们课题组报道了过硫酸盐促进的环烷醇的开环-炔基化反应, 合成了一系列炔酮化合物[15d].在类似体系下, 我们实现了环烷醇与N-苯基丙烯酰胺的自由基环化反应, 成功地将酮烷基片段引入到了羟吲哚分子骨架中[15g].
基于上述基础, 本工作实现了无过渡金属催化下可见光促进的张力环烷醇对于喹喔啉酮类化合物的C(3)-酮烷基化反应.该反应体系不需要加入任何过渡金属催化剂, 以过硫酸钾为氧化剂, 在室温条件下仅蓝光照射16 h后便可得到最终产物(Scheme 1).
以喹喔啉酮(1a)和1-苯基环丁醇(2a)作为模板底物去探索最优的反应条件(表 1).根据以往的经验, 首先以2.0 equiv.过硫酸钾为氧化剂, 以丙酮/水(V:V=1:1)的混合溶液为反应介质, 将化合物1a和2a加热至50 ℃反应. 16 h后, 我们成功地分离到了C(3)-酮烷基化的产物3a, 然而, 仅有11%的收率(表 1, Entry 1).反应过程中, 我们观察到了较浓的环丁醇开环氢化的产物, 且喹喔啉酮1a完全消失.考虑到喹喔啉酮在氧化升温的条件下分解, 接下来在常温而其他条件不变的情况下尝试反应.室温下该反应也能顺利进行, 但产率仍不够理想(17%, 表 1, Entry 2).文献报道可见光也可促进过硫酸盐分解产生活性氧化性物种(SO4.-)[18], 因此我们将该反应置于蓝光下照射16 h.幸运的是, 我们发现产率上升至36%(表 1, Entry 3).随后, 在光照、室温反应的前提下, 我们对反应溶剂进行了筛选(详细的溶剂筛选情况见Supporting Information).研究发现, 使用水与质子性溶剂的混合液作为反应介质, 有利于该反应的进行(表 1, Entries 4~8).尤其是, 当采用MeOH/H2O (V:V=1:1)作溶剂时, 可得到74%的产率.即使采用纯水作溶剂, 也可以以13%的收率得到目标产物(表 1, Entry 9).然而, 采用MeOH作为唯一溶剂时, 仅观察到痕量的产物(表 1, Entry 10).可见, 水的存在对于该反应至关重要, 可能是因为水有利于过硫酸盐的溶解.接着, 我们对甲醇和水的比例进行了调节(表 1, Entries 5, 11).研究表明, 提升水的比例更有助于产物的生成, 当甲醇与水的比例为1:2时, 反应产率上升至89%.在此基础上, 对一些常见的无机氧化剂和有机氧化剂进行了筛选(表 1, Entries 12~15).发现K2S2O8收率最佳, (NH4)2S2O8和Na2S2O8作氧化剂也能给出很好的收率, 过氧叔丁醇(TBHP)和PhI(OAc)2对该反应不起作用.最后, 控制实验表明氧化剂和光照对于该反应均是必须的(表 1, Entries 16, 17).
 下载:
导出CSV
下载:
导出CSV
 | |||
| Entry | Oxidant | Solvent (V:V) | Yieldb/% |
| 1 | K2S2O8 | Acetone/H2O (1:1) | 11c |
| 2 | K2S2O8 | Acetone/H2O (1:1) | 17d |
| 3 | K2S2O8 | Acetone/H2O (1:1) | 36 |
| 4 | K2S2O8 | EtOH/H2O (1:1) | 61 |
| 5 | K2S2O8 | MeOH/H2O (1:1) | 74 |
| 6 | K2S2O8 | DMSO/H2O (1:1) | 37 |
| 7 | K2S2O8 | DMF/H2O (1:1) | 28 |
| 8 | K2S2O8 | DCE/H2O (1:1) | 15 |
| 9 | K2S2O8 | H2O | 13 |
| 10 | K2S2O8 | MeOH | Trace |
| 11 | K2S2O8 | MeOH/H2O (1:2) | 89 |
| 12 | (NH4)2S2O8 | MeOH/H2O (1:2) | 85 |
| 13 | Na2S2O8 | MeOH/H2O (1:2) | 86 |
| 14 | TBHP | MeOH/H2O (1:2) | Trace |
| 15 | PhI(OAc)2 | MeOH/H2O (1:2) | Trace |
| 16 17 |
— K2S2O8 |
MeOH/H2O (1:2) MeOH/H2O (1:2) |
Trace NRd, e |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (0.3 mmol, 1.5 equiv.), solvent (2 mL), oxidant (2.0 equiv.), 10 W Blue LED under N2 at room temperature for 16 h. b Yield of isolated product. c At 50 ℃, without light irradiation. d At room temperature, without light irradiation. e NR=no reaction. | |||
在最佳条件下, 首先对环烷醇的适用范围进行了考察.从表 2中可以看出, 对于1-芳基环丁醇而言, 苯环上带有吸电子或给电子基时, 均能以较好的产率得到相应的C(3)-酮烷基化产物3b~3f. 1-(β-萘基)环丁醇(2g)也能以60%的收率生成目标产物3g.噻吩基环烷醇也能很好地适用于该反应体系, 最终以60%的产率生成产物3h.而当四元环醇的环上对位带有苯基时, 目标产物3i的收率降至35%, 原料环醇回收率为45%, 考虑到采用的溶剂CH3OH为质子性溶剂, 所以我们在反应中观察到了其开环后氢化的副产物.为了在喹喔啉酮骨架上引入不同链长的烷基片段, 接下来考察了三元环醇的开环反应.实验结果表明, 苯环上的取代基无论是给电子基(OMe, Me)还是吸电子基(Cl, Br, CF3), 均能以很好的收率给出目标产物3k~3o.溴原子的保留为产物的进一步修饰提供了很好的机会.同样地, 我们将苯环取代基变为萘环, 产物3p的收率为65%. 1-环己基环丙醇在当前反应条件下也可以生成产物3q, 但产率只有28%.对于1-苄基环丙醇而言, 同样可以成功地参与到该反应当中, 以55%的收率得到产物3r.苄基的苯环上的取代基如tBu和Br对反应效率没有明显的影响(3s, 3t). 1-苯氧甲基环丙醇也可以与喹喔啉酮以52%的收率得到产物3u. 1-苄基环丙醇2r~2t作为烷基化试剂时, 产率普遍偏低(40%~55%), 主要是因为原料在反应过程当中转化率较低.值得关注的是, 环张力较小的1-苯基环戊醇在该反应条件下同样可以给出相应的产物3v, 然而产率只有15%.反应中, 分离到了13%的开环-氢化的副产物(3vꞌ).
最后, 研究了各种取代喹喔啉酮的反应普适性(表 3).结果表明, 苯环上带有供电子或吸电子基团的各种喹喔啉酮均能有效地参与该反应, 得到相应的产物4a~4i, 产率适中.包括氟、氯、溴和酯一系列官能团都具有良好的耐受性.即使像硝基这种强吸电子取代基也可以得到38%的产物.除甲基外, 氮原子上含有其他保护基如正丙基、苄基和酯基类的喹喔啉酮也适用于该反应, 以59%~77%的产率得到相应的产物4j~4l. N未保护的喹喔啉酮也可成功地以57%的收率给出目标产物. 1-苯基环丁醇与取代的喹喔啉酮也能顺利反应, 给出相应的产物4n, 4o.
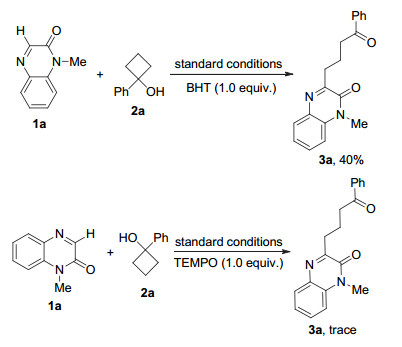
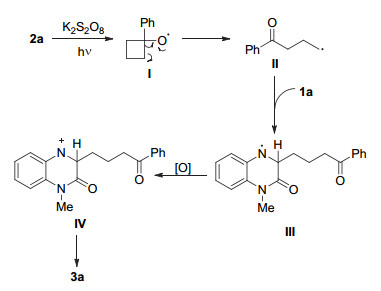
最后, 为了更好地了解反应机理, 我们进行了一些控制实验(Scheme 2).当将1.0 equiv.的自由基抑制剂2, 6-二叔丁基-4-甲基苯酚(BHT)加入到1a和2a的反应当中后, 产物3a的收率降至40%.而当将1.0 equiv.的2, 2, 6, 6-四甲基哌啶-氮-氧化物(TEMPO)加入到反应当时, 只得到痕量的产物3a.上述实验结果表明, 该反应很可能经历了自由基历程.根据上述结果和先前的报道, 我们提出了该反应可能的机理, 如Scheme 3所示.首先, 在K2S2O8氧化下, 环烷醇2a经单电子氧化产生烷氧自由基中间体Ⅰ.随后, 自由基中间体Ⅰ经历开环(β-键均裂)后生成γ-羰基的烷基自由基Ⅱ.自由基Ⅱ被喹喔啉酮1a捕获, 生成自由基中间体Ⅲ.随后中间体Ⅲ经过单电子氧化并失去H+生成最终产物3a.
发展了一种新型的可见光促进的以张力环烷醇为烷基源的喹喔啉酮的C(3)-酮烷基化反应.通过该反应, 成功地将不同链长的含羰基官能团的烷基片段引入到了一系列喹喔啉酮分子中.该反应在室温条件下进行, 使用低毒、廉价的过硫酸盐为氧化剂, 且反应可以在水相中进行, 为喹喔啉酮的烷基化提供了更加绿色、环保、简便的方法.
向干燥的石英反应管中依次加入氧化剂K2S2O8 (2.0 equiv., 0.4 mmol)、喹喔啉酮(1.0 equiv., 0.2 mmol)和环烷醇(1.5 equiv., 0.3 mmol).随后置换氮气, 在氮气条件下加入1.3 mL的H2O和0.7 mL的MeOH, 将反应管密封后, 在蓝光照射, 室温条件下搅拌16 h.反应完毕后, 向反应混合液中加入乙酸乙酯稀释, 有机相用水(三次)和饱和食盐水(一次)洗涤, 并用无水Na2SO4干燥、浓缩后得粗产物.经柱层析(石油醚/乙酸乙酯, V: V=4:1)分离得到目标产物.
Ries, U. J.; Priepke, H. W. M.; Hauel, N. H.; Handschuh, S.; Mihm, G.; Stassen, J. M.; Wienen, W.; Nar, H. Bioorg. Med. Chem. Lett. 2003, 13, 2297. doi: 10.1016/S0960-894X(03)00443-8
(a) Carta, A.; Piras, S.; Loriga, G.; Paglietti, G. Mini-Rev. Med. Chem. 2006, 6, 1179. (b) Li, X.; Yang, K.-H.; Li, W.-L.; Xu, W.-F. Drugs Future 2006, 31, 979. (c) Hussain, S.; Parveen, S.; Hao, X.; Zhang, S.-Z.; Wang, W.; Qin, X.-Y.; Yang, Y.-C.; Chen, X.; Zhu, S.-J.; Zhu, C.-J.; Ma, B. Eur. J. Med. Chem. 2014, 80, 383.
Buratti, W.; Gardini, G. P.; Minisci, F. Tetrahedron 1971, 27, 3655. doi: 10.1016/S0040-4020(01)97776-2
For review, see: (a) Proctor, R. S. J.; Phipps, R. J. Angew. Chem., Int. Ed. 2019, DOI: 10.1002/anie.201900977.For selected examples, see: (b) Huff, C. A.; Cohen, R. D.; Dykstra, K. D.; Streckfuss, E.; DiRocco, D. A.; Krska, S. W. J. Org. Chem. 2016, 81, 6980. (c) Wu, X.-X.; Zhang, H.; Tang, N.-N.; Wu, Z.; Wang, D.-P.; Ji, M.-S.; Xu, Y.; Wang, M.; Zhu, C. Nat. Commun. 2018, 9, 3343. (d) Wu, X.-X.; Wang, M.-Y.; Huan, L.-T.; Wang, D.-P.; Wang, J.-W.; Zhu, C. Angew. Chem., Int. Ed. 2018, 57, 1640.
(a) Jiao, J.; Murakami, K.; Itami, K. ACS Catal. 2016, 6, 610. (b) Legnani, L.; Cerai, G. P.; Morandi, B. ACS Catal. 2016, 6, 8162. (c) Zhou, Z.; Ma, Z.; Behnke, N. E.; Gao, H.; Kurti, L. J. Am. Chem. Soc. 2017, 139, 115. (d) Wang, P.; Li, G. C.; Jain, P.; Farmer, M. E.; He, J.; Shen, P. X.; Yu, J. Q. J. Am. Chem. Soc. 2016, 138, 14092.
(a) Yin, K.; Zhang, R. Org. Lett. 2017, 19, 1530. (b) Paul, S.; Ha, J. H.; Park, G. E.; Lee, Y. R. Adv. Synth. Catal. 2017, 359, 1515. (c) Carr r, A.; Brion, J.-D.; Messaoudi, S.; Alami, M. Org. Lett. 2013, 15, 5606. (d) Wang, L.-L.; Bao, P.-L.; Liu, W.-W.; Liu, S.-T.; Hu, C.-S.; Yue, H.-L.; Yang, D.-S.; Wei, W. Chin. J. Org. Chem. 2018, 38, 3189. (王雷雷, 鲍鹏丽, 刘维伟, 刘思彤, 胡昌松, 岳会兰, 杨道山, 魏伟, 有机化学, 2018, 38, 3189.)
Gao, M.; Li, Y.; Xie, L.; Chauvin, R.; Cui, X. Org. Biomol. Chem. 2016, 52, 2846. https://www.ncbi.nlm.nih.gov/pubmed/26779573
Li, Y.; Gao, M.; Wang, L.; Cui, X. Org. Biomol. Chem. 2016, 14, 8428. doi: 10.1039/C6OB01283C
(a) Yuan, J.-W.; Fu, J.-H.; Liu, S.-N.; Xiao, Y.-M.; Mao, P.; Qu, L.-B. Org. Biomol. Chem. 2018, 16, 3203. (b) Xie, L.-Y.; Peng, S.; Fan, T.-G.; Liu, Y.-F.; Sun, M.; Jiang, L.-L.; Wang, X.-X.; Cao, Z.; He, W.-M. Sci. China, Chem. 2019, 62, 460.
Hong, G.-F.; Yuan, J.-W.; Fu, J.-H.; Pan, G.-Y.; Wang, Z.-W.; Yang, L.-R.; Xiao, Y.-M.; Mao, P.; Zhang, X.-M. Org. Chem. Front. 2019, 6, 1173. doi: 10.1039/C9QO00105K
(a) Yuan, J.-W.; Fu, J.-H.; Yin, J.-H.; Dong, Z.-H.; Xiao, Y.-M.; Mao, P.; Qu, L.-B. Org. Chem. Front. 2018, 5, 2820. (b) Fu, J.-H.; Yuan, J.-W.; Zhang, Y.; Xiao, Y.-M.; Mao, P.; Diao, X.-Q.; Qu, L.-B. Org. Chem. Front. 2018, 5, 3382.
Yang, L.; Gao, P.; Duan, X.-H.; Gu, Y.-R.; Guo, L.-N. Org. Lett. 2018, 20, 1034. doi: 10.1021/acs.orglett.7b03984
Gu, Y.-R.; Duan, X.-H.; Chen, L.; Ma, Z.-Y.; Gao, P.; Guo, L.-N. Org. Lett. 2019, 21, 917. doi: 10.1021/acs.orglett.8b03865
(a) Liu, R.; Huang, Z.-H.; Murray, M. G.; Guo, X.-Y.; Liu, G. J. Med. Chem. 2011, 54, 5747. (b) Qin, X.-Y.; Hao, X.; Han, H.; Zhu, S.-J.; Yang, Y.-C.; Wu, B.-B.; Hussain, S.; Parveen, S.; Jing, C.-J.; Ma, B.; Zhu, C.-J. J. Med. Chem. 2015, 58, 1254.
For review, see: (a) Wu, X.-X.; Zhu, C. Chem. Select. 2017, 2, 10678. For selected examples, see: (b) Ren, R.-G.; Zhao, H.-J.; Huan, L.-T.; Zhu, C. Angew. Chem., Int. Ed. 2015, 54, 12692. (c) Zhao, H.-J.; Fan, X.-F.; Yu, J.-J.; Zhu, C. J. Am. Chem. Soc. 2015, 137, 3490. (d) Wang, S.; Guo, L.-N.; Wang, H.; Duan, X.-H. Org. Lett. 2015, 17, 4798. (e) Jia, K.; Zhang, F.; Huang, H.; Chen, Y. J. Am. Chem. Soc. 2016, 138, 1514. (f) Huan, L.-T.; Zhu, C. Org. Chem. Front. 2016, 3, 1467. (g) Guo, L.-N.; Deng, Z.-Q.; Wu, Y.; Hu, J. RSC Adv. 2016, 6, 27000. (h) Ren, R.-G.; Wu, Z.; Xu, Y.; Zhu, C. Angew. Chem., Int. Ed. 2016, 55, 2866. (i) Nikolaev, A.; Legault, C. Y.; Zhang, M.-H.; Orellana, A. Org. Lett. 2018, 20, 796. (j) Zhao, R.; Yao, Y.; Zhu, D.; Chang, D.-H.; Liu, Y.; Shi, L. Org. Lett. 2018, 20, 1228.
For selected reviews, see: (a) Zhao, J.-F.; Duan, X.-H.; Guo, L.-N. Chin. J. Org. Chem. 2017, 37, 2498. (赵景峰, 段新华, 郭丽娜, 有机化学, 2017, 37, 2498.) (b) Mandal, S.; Bera, T.; Dubey, G.; Saha, J.; Laha, J. K. ACS Catal. 2018, 8, 5085. (c) Sathyamoorthi, S.; Banerjee, S. Chem. Select. 2017, 2, 10678.
For selected examples, see: (a) Minisci, F.; Citterio, A.; Giordano, C. Acc. Chem. Res. 1983, 16, 27. (b) Chinchilla, R.; Najera, C.; Yus, M. Chem. Rev. 2004, 104, 2667. (c) Yin, F.; Wang, X.-S. Org. Lett. 2014, 16, 1128. (d) Wei, W.; Wen, J.-W.; Yang, D.-S.; Du, J.; You, J.-M.; Wang, H. Green Chem. 2014, 16, 2988. (e) Li, Y.-M.; Shen, Y.-H.; Chang, K.-J.; Yang, S.-D. Tetrahedron 2014, 70, 1991. (f) Laha, J. K.; Patel, K. V.; Tummalapalli, K. S. S.; Dayal, N. Chem. Commun. 2016, 52, 10245.
(a) Devan, S.; Shah, B.-A. Chem. Commun. 2016, 52, 1490. (b) Zhang, Y.-Q.; Teuscher, K. B.; Ji, H.-T. Chem. Sci. 2016, 7, 2111. (c) Zhao, Y.-T.; Huang, B.-B.; Yang, C.; Xia, W.-J. Org. Lett. 2016, 18, 3326. (d) Meyer, A. U.; Alexander, W.; K nig, B. Angew. Chem., Int. Ed. 2017, 56, 409.
表 1 反应条件的优化a
Table 1. Optimization of reaction condition
| |||
| Entry | Oxidant | Solvent (V:V) | Yieldb/% |
| 1 | K2S2O8 | Acetone/H2O (1:1) | 11c |
| 2 | K2S2O8 | Acetone/H2O (1:1) | 17d |
| 3 | K2S2O8 | Acetone/H2O (1:1) | 36 |
| 4 | K2S2O8 | EtOH/H2O (1:1) | 61 |
| 5 | K2S2O8 | MeOH/H2O (1:1) | 74 |
| 6 | K2S2O8 | DMSO/H2O (1:1) | 37 |
| 7 | K2S2O8 | DMF/H2O (1:1) | 28 |
| 8 | K2S2O8 | DCE/H2O (1:1) | 15 |
| 9 | K2S2O8 | H2O | 13 |
| 10 | K2S2O8 | MeOH | Trace |
| 11 | K2S2O8 | MeOH/H2O (1:2) | 89 |
| 12 | (NH4)2S2O8 | MeOH/H2O (1:2) | 85 |
| 13 | Na2S2O8 | MeOH/H2O (1:2) | 86 |
| 14 | TBHP | MeOH/H2O (1:2) | Trace |
| 15 | PhI(OAc)2 | MeOH/H2O (1:2) | Trace |
| 16 17 |
— K2S2O8 |
MeOH/H2O (1:2) MeOH/H2O (1:2) |
Trace NRd, e |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (0.3 mmol, 1.5 equiv.), solvent (2 mL), oxidant (2.0 equiv.), 10 W Blue LED under N2 at room temperature for 16 h. b Yield of isolated product. c At 50 ℃, without light irradiation. d At room temperature, without light irradiation. e NR=no reaction. | |||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们