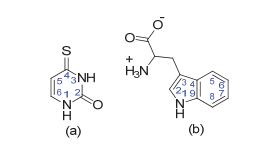
图 1.
反应物分子式(a) 4-硫代尿嘧啶(4-TU)和(b)色氨酸(TrpH)
Figure 1.
Molecular structures of (a) 4-thiouracil (4-TU) and (b) tryptophan

蛋白质(protein)和核糖核酸(ribonucleic acid, RNA)在细胞内许多重要的生命活动中发挥着至关重要的作用.核糖核酸与蛋白质相互作用(RNA-protein interactions)是生物学中一个普遍存在的现象, 是细胞在生命过程中进行信号传导及代谢活动的基础[1].这种相互作用通常是以形成复合物(ribonucleic protein complex RNPs)的方式来行使其功能, 并且广泛存在于RNA剪接、翻译、蛋白质合成以及转录后调控等很多关键的生理过程.因此, 了解和研究核糖核酸与蛋白质相互作用机理有助于我们深入地理解生命过程, 进而通过调控生物大分子之间的相互作用来影响和控制生理过程[2, 3].
通过实验手段确定蛋白质结合了哪种RNA以及结合位点是研究二者相互作用的重要内容.在20世纪60年代, 人们发现紫外光照射会导致蛋白质和核糖核酸交联, 基于这一发现, 紫外交联免疫共沉淀技术(UV-crosslinking and immunoprecipitation, CLIP)应运而生[4]. CLIP技术是研究RNA和蛋白质相互作用的重要技术, 利用的是相互靠近的蛋白质与RNA(<10 Å )在256 nm的紫外光照射下会发生共价交联的特性, 2003年《科学》杂志报道了Ule等[2]采用CLIP技术研究小鼠脑内Nova蛋白与RNA相互作用对调节自身免疫性神经系统疾病中编码抑制性突触的多个组分进行剪接的过程.但因为普通的核糖核酸碱基与蛋白质发生紫外交联的效率很低(<5%), CLIP技术的应用受到了限制.为提高交联效率, Hafner等[5]利用光活化核苷酸衍生物4-硫代尿苷的光致交联效率远远高于普通的核苷酸(尿苷、腺苷及鸟苷等)的特性, 开发了PAR-CLIP (photoactivatable-ribonucleotide-enhanced CLIP)技术. 4-硫代尿苷经365 nm的紫外光照射会与邻近蛋白质中的氨基酸发生共价交联.与普通的254 nm紫外光照射天然核糖核酸尿嘧啶相比较, PAR-CLIP可以将交联效率提高100~1000倍, 并且可以有效地降低紫外光对其他生物组织的辐射损伤[6]. PAR-CLIP技术的紫外交联结果表明蛋白质中的氨基酸会与单链RNA中的光活化核苷酸4-硫代尿苷交联, Urlaub等[7]利用高分辨率质谱确认交联位点为4-硫代尿苷的C(4)位, 交联同时会脱去H2S.
然而, 目前的研究还不能解释光激发4-硫代尿苷与氨基酸共价交联的反应机理.对于常规的核糖核酸碱基, 有研究推测认为激发态的碱基可以和数埃距离内的氨基酸通过自由基机制或者电子转移机制引发共价交联反应[8]. 20种天然氨基酸都有可能同RNA发生潜在的交联反应, 其中蛋白质中的芳香族氨基酸以及RNA中的尿嘧啶更容易发生交联反应[5].在本工作中, 我们选取可有效发生共价交联的尿嘧啶衍生物4-硫代尿嘧啶(4-TU)[9, 10]和芳香氨基酸色氨酸(TrpH)作为模型体系, 通过纳秒时间分辨的紫外-可见激光闪光光解技术, 捕捉到4-TU三重态(550 nm)与色氨酸发生电子转移后的瞬态自由基物种4-TU自由基阴离子(370 nm, 4-TU•-)和色氨酸自由基阳离子(570 nm, TrpH•+), 以及色氨酸自由基阳离子发生质子转移后生成的瞬态物种色氨酸中性自由基(510 nm, Trp•).光谱上这些关键的瞬态中间体的特征吸收表明在4-TU三重态上发生的光致电子转移是引发4-TU与色氨酸光致共价交联过程中关键的第一步反应.通过测量色氨酸猝灭4-TU三重态的衰减动力学, 得到三重态4-TU与色氨酸发生电子转移反应的准一级反应速率常数为2.88×109 L•mol―1•s-1, 这是典型的由扩散控制的电子转移速率常数, 并且pH值对电子转移反应的速率常数无影响.我们进一步用经典的Rehm-Weller经验公式计算了光致电子转移过程的驱动力, 即吉布斯自由能的变化值为-0.15 eV, 表明4-TU三重态与色氨酸间的光致电子转移反应是一个热力学上允许的有利的过程.
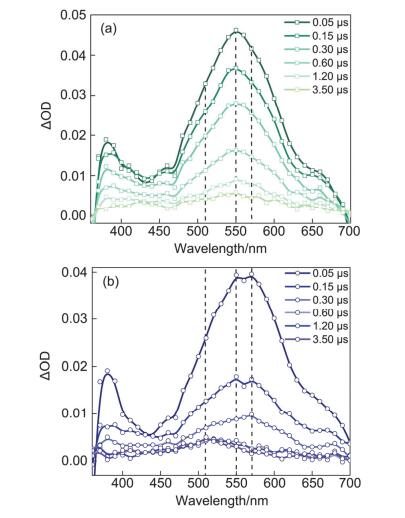
纳秒时间分辨瞬态吸收光谱手段可用于实时探测分子的激发态和瞬态中间体的衰减或生成动力学.实验首先在氮气条件下采集了355 nm激光激发的4-TU (pH=7的磷酸钠缓冲溶液)的瞬态光谱(见图 2a), 380 nm和550 nm处的正峰为其最低激发三重态T1的特征吸收, 通过拟合550 nm的衰减动力学得到自由的4-TU三重态寿命约为320 ns, 这些结果与已有的文献报道[11~13]完全吻合.当4-TU缓冲溶液中加入色氨酸后, 瞬态吸收光谱有了变化.如图 2b所示, 初始时刻4-TU三重态在550 nm处的吸收峰强度有所减弱, 表明色氨酸猝灭了4-TU三重态, 但是, 三重态在380 nm的吸收峰非但没有被猝灭, 相反, 380 nm相对于550 nm处的吸收峰强度有所增加, 并且在570 nm处出现了新的吸收峰、叠加在4-TU三重态吸收光谱之上.瞬态吸收光谱上的这些变化源于光激发4-TU与色氨酸发生反应后新生成的瞬态物种, 我们推测新的瞬态物种是色氨酸将电子转移给4-TU三重态生成4-TU自由基阴离子(4-TU•-)和色氨酸自由基阳离子(TrpH•+).根据已有文献报道[12], 在给电子试剂N, N-二乙基苯胺(DEA)存在条件下, 4-TU三重态得到一个电子后生成瞬态产物自由基阴离子(4-TU•-)的吸收峰位于380 nm.因此4-TU与色氨酸反应的瞬态光谱上增强的380 nm的吸收应该也归属为4-TU•-.同时, 有研究表明[14]色氨酸的氧化电势较低可作为优秀的电子给体, 并且文献[15]中通过激光辐解色氨酸与钌的络合物[Ru(bpy)3]2+的方式产生色氨酸自由基阳离子, TrpH•+在瞬态紫外吸收光谱上的特征峰即为570 nm, 因此, 图 2b中570 nm处[16]新的吸收峰来源于电子转移反应中生成的产物TrpH•+.瞬态光谱上观测到的两个物种的特征吸收峰证实了光激发4-TU与色氨酸发生了电子转移反应.随着瞬态光谱上380 nm和570 nm处吸收峰的衰减, 510 nm处有新物种生成并且寿命很长(~300 μs), 归属为电子转移产物TrpH•+脱掉吲哚环N(1)(图 1b)位的质子形成的色氨酸中性自由基(Trp•)[17, 18].
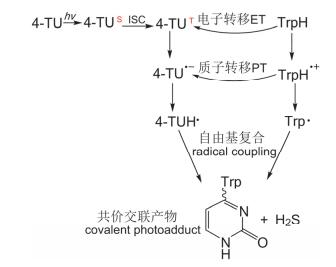
瞬态光谱上380 nm与570 nm, 510 nm处自由基的吸收信号证实了在355 nm激光激发4-TU后, 首先发生的是4-TU三重态与色氨酸间的光致电子转移反应, 即4-TU被紫外光激发到单重态后, 经系间窜越到三重态, 4-TU三重态得到色氨酸的一个电子生成4-TU•-, 同时色氨酸变为TrpH•+, TrpH•+随后会脱质子变为中性自由基Trp•, 而4-TU•-会得到一个质子生成中性自由基4-TUH•, 两个中性自由基会进一步复合, 根据文献中高分辨率质谱的实验结果[19], 可得知色氨酸与4-TU的C(4)号位形成共价键, 生成最终的交联产物(如图 3).
对于4-TU三重态与色氨酸间的光致电子转移反应:
|
$ 4-\mathrm{TU}^{*}+\mathrm{TrpH} \rightarrow \operatorname{Tr} \mathrm{pH}^{·+}+4-\mathrm{TU}^{·-} $ |
(1) |
为了得到反应驱动力的大小, 可采用公式(2)所示的Rehm-Weller经验公式[20], 通过实验数据计算得到电子转移过程的吉布斯自由能变化值:
|
$ \Delta G=E\left(\mathrm{D}^{+} / \mathrm{D}\right)-E\left(\mathrm{A} / \mathrm{A}^{-}\right)-E\left(\mathrm{A}^{*}\right)-C $ |
(2) |
公式(2)中ΔG代表电子转移反应的吉布斯自由能, 即光致电子转移的驱动力; E(D+/D)代表色氨酸的氧化电势, 采用的数据是文献中给出的1.21 eV[14]; E(A/A-)代表 4-TU的还原电势, 采用的是文献报道在pH=7.1的磷酸缓冲溶液中测得的数据-1.52 eV[21, 22]; E(A*)代表 4-TU的三重态的能量, 可以通过磷光发射峰位置来确定, 采用的数据是文献报道的520 nm[10]的磷光发射峰, 对应的能量是2.38 eV; C代表的是溶剂对离子的稳定化作用, 为溶剂影响的校正系数, 通常是一个较小的数值, 一般为0.5 eV(以上数据的单位均已统一).经过计算, 得到电子转移的吉布斯自由能变化值为-0.15 eV, 进一步证实了4-TU三重态与色氨酸间可发生有效的热力学上允许的光致电子转移反应.为了与色氨酸作比较, 我们计算了另一种芳香族氨基酸酪氨酸与4-TU发生光致电子转移反应的驱动力, 酪氨酸的还原电势为1.34 eV[23], 由Rehm-Weller经验公式计算出电子转移反应的驱动力为ΔG=-0.02 eV, 小于色氨酸与4-TU电子转移反应的吉布斯自由能变化值.理论估算值进一步说明在芳香氨基酸中色氨酸更易与激发态的4-TU发生光致电子转移反应, 从而有效地引发后续的共价交联反应.
为了得到电子转移反应速率常数, 我们进一步对不同浓度的色氨酸与4-TU三重态(550 nm)反应的动力学进行了测量(见图 4a).未加入色氨酸时(红色曲线), 4-TU三重态衰减最慢且可衰减到基线.随着色氨酸浓度的增加, 4-TU三重态衰减加快, 且未回复到基线, 这是因为电子转移产物TrpH•+(特征峰在570 nm)及其脱质子产物长寿命的Trp(特征峰在510 nm)在550 nm处也有吸收.对4-TU三重态在550 nm处的动力学衰减曲线进行拟合得到4-TU三重态的寿命.由于体系中产生的4-TU三重态浓度远远小于色氨酸的浓度, 4-TU三重态与色氨酸之间的双分子反应近似为准一级反应, 故可采用公式(3)的Stern-Volmer曲线求解准一级反应速率常数:
|
$ \tau^{-1}=\tau_{0}^{-1}+K q[\mathrm{Q}] $ |
(3) |
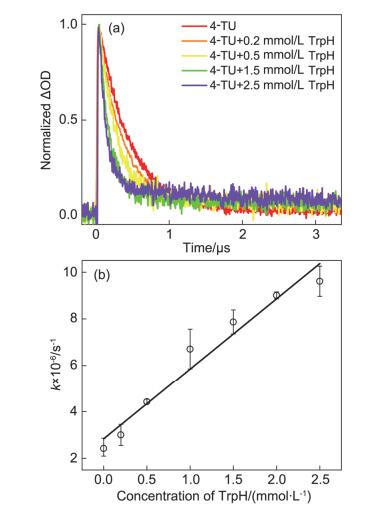
公式(3)中τ代表 4-TU在不同浓度的色氨酸溶液中拟合得到的三重态寿命; τ0代表 4-TU三重态固有的寿命; 斜率Kq代表 4-TU三重态与色氨酸发生电子转移反应的准一级反应速率常数; [Q]代表实验中的色氨酸浓度.如图 4b拟合得到的Stern-Volmer曲线的斜率Kq=2.88×109 L•mol-1•s-1, 即为二者发生电子转移的准一级反应速率常数, 这是典型的由扩散控制的电子转移反应的速率常数[24].这说明4-TU与色氨酸之间的光致电子转移反应极易发生, 并由此进一步引发后续的交联反应.
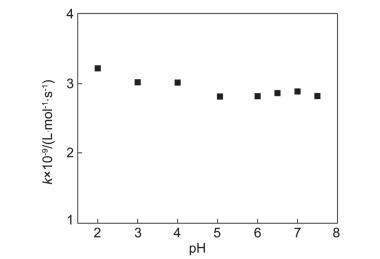
我们进一步研究pH值对4-TU三重态与色氨酸电子转移反应的影响.在pH<8的溶液中, 4-TU是中性的结构(图 1a)[25], 因此其激发三重态的性质不会随着pH值而产生变化.与色氨酸反应时, 4-TU三重态在550 nm处的衰减动力学过程不受pH值的影响.如图 5所示, 在pH=2~7.5范围内电子转移的准一级反应速率常数为2.78×109~3.20×109 L•mol-1•s-1, 仅有微小变化, 显示出光致电子转移的速率常数对pH值没有依赖性的特点[26~28].
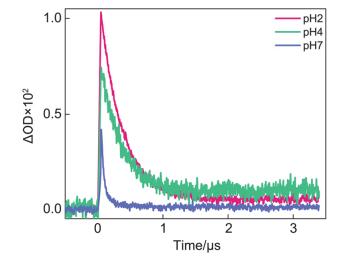
同时, 我们对电子转移产物TrpH•+在不同pH值条件下570 nm处的动力学进行了测量.由于4-TU三重态的摩尔消光系数(ε550 nm=4×103 L•mol-1•cm-1)[11]比色氨酸自由基阳离子(ε570 nm=3×103 L•mol-1•cm-1)[17]和中性色氨酸自由基(ε510 nm=2.3×103 L•mol-1•cm-1)[17]的摩尔消光系数大, 4-TU三重态的吸收光谱与色氨酸自由基阳离子的吸收光谱叠加且该反应的光致电子转移过程较快, 导致570 nm处的动力学不能观测到明显的上升过程即电子转移产物TrpH•+的生成过程.根据文献[13]得知TrpH•+的pKa为4.3, 因此pH值会影响TrpH•+的脱质子过程.如图 6所示, 随着pH值的降低, TrpH•+衰减过程变慢, 即脱质子生成Trp•的过程变慢, 这使得初始观测到的TrpH•+吸收强度会增加.进一步, 通过拟合570 nm的衰减动力学曲线得到pH值为7时的脱质子速率常数为7.3×106 s-1, pH值为2时的脱质子速率常数为3.0×106 s-1, 这与文献中报道的速率常数吻合[14].这个现象进一步证明电子转移产物TrpH•+后续发生了质子转移过程, 因此其衰减与pH值有关.实验表明, 4-TU与色氨酸的光致电子转移反应与pH值无关, 但后续的质子转移过程受pH值影响, 这些研究结果有助于进一步了解由光致电子转移反应引发的RNA与蛋白质光致共价交联的机理, 从而帮助理解RNA与蛋白质光致共价交联的本质并对发生交联的位点或交联的氨基酸种类进行调控.
本研究工作选取光活化核苷酸衍生物4-硫代尿嘧啶与芳香氨基酸色氨酸的反应作为研究RNA与蛋白质光致共价交联的模型体系.通过纳秒时间分辨的紫外-可见瞬态吸收光谱技术观测到4-硫代尿嘧啶三重态与色氨酸间光致电子转移反应的重要中间体: 4-硫代尿嘧啶自由基阴离子(370 nm, 4-TU•-), 色氨酸自由基阳离子(570 nm, TrpH•+)及TrpH•+脱质子生成的色氨酸中性自由基(510 nm, Trp•).通过测量和拟合色氨酸猝灭4-硫代尿嘧啶三重态的动力学, 得到二者发生电子转移的准一级反应速率常数为2.88×109 L•mol-1•s-1, 表现为扩散控制的动力学过程, 并且通过Rehm-Weller经验公式计算出该过程的吉布斯自由能变化为-0.15 eV, 是热力学上有利的过程.在pH=2~7.5范围内, 电子转移速率常数不受pH值影响, 同时, 我们也发现电子转移产物TrpH•+的后续的脱质子过程受pH值影响.研究表明电子转移反应是引发4-硫代尿嘧啶与色氨酸的光致共价交联的关键的第一步反应, 随后会发生质子转移及自由基复合的过程, 最终生成共价交联的加合物.这些结果说明光活化核苷酸衍生物可以在三重态上与蛋白质发生光致电子转移反应, 从而引发后续的质子转移及自由基复合过程而形成共价交联产物.本研究测量的动力学数据可为后续探索复杂的RNA结构与多肽及蛋白质体系光致共价交联的机理提供实验基础.
4-硫代尿嘧啶(4-TU, 98%)和色氨酸(TrpH, 99%)均购买于百灵威科技有限公司(J & K Scientific), 4-TU的浓度通过在327 nm处的稳态紫外吸收光谱测定(327 nm处的摩尔消光系数为19380 L•mol-1•cm-1), 实验中的4-TU浓度为100 μmol•L-1, 色氨酸浓度为0.2~2.5 mmol•L-1.高纯水及各种pH条件下的磷酸钠缓冲溶液作为溶剂使用.混合均匀的溶液样品在实验前及实验过程中均通入氮气饱和.实验中的温度为室温25 ℃.
通过配有Nd:YAG激光器的纳秒激光闪光光解仪Edinburgh LP920获得纳秒时间分辨的瞬态吸收光谱, 样品由355 nm的脉冲激光激发, 由450 W脉冲氙灯产生分析检测光, 采集300~700 nm的光谱进行分析, 获得的数据用LP920软件进行分析, 每次采集的实验数据都重复三次取平均值以确保所得数据的准确性[29].
Lin, R. J. RNA-Protein Interaction Protocols, Humana Press, Totowa, USA, 2008, p. 85.
Ule, J.; Jensen, K. B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R. B. Science 2003, 302, 1212. doi: 10.1126/science.1090095
Licatalosi, D. D.; Mele, A.; Fak, J. J.; Ule, J.; Kayikci, M.; Chi, S. W.; Clark, T. A.; Schweitzer, A. C.; Blume, J. E.; Wang, X.; Dar-nell, J. C.; Darnell, R. B. Nature 2008, 456, 464. doi: 10.1038/nature07488
Kishore, S.; Jaskiewicz, L.; Burger, L.; Hausser, J.; Khorshid, M.; Zavolan, M. Nat. Methods 2011, 8, 559. doi: 10.1038/nmeth.1608
Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M. Jr.; Jungkamp, A. C.; Munschauer, M.; Ulrich, A.; Wardle, G. S.; Dewell, S.; Zavolan, M.; Tuschl, T. Cell 2010, 141, 129. doi: 10.1016/j.cell.2010.03.009
Ascano, M.; Hafner, M.; Cekan, P.; Gerstberger, S.; Tuschl, T. WIRES RNA 2012, 3, 159. doi: 10.1002/wrna.v3.2
Kstharina, K.; Timo, S.; Benedikt, M. B.; Saadia, Q.; Kum, L. B.; Matthias, W. H.; Oliver, K.; Urlaub, H. Nat. Methods 2014, 11, 1064. doi: 10.1038/nmeth.3092
周德建, 叶克穷, 生命科学, 2014, 26, 207.Zhou, D. J.; Ye, K. Q. Chin. Bull. Life Sci. 2014, 26, 207(in Chinese).
Favre, A.; Saintome, C.; Fourrey, J. L.; Clivio, P.; Laugaa, P. J. Photochem. Photobiol. B-Biol. 1998, 42, 109. doi: 10.1016/S1011-1344(97)00116-4
Shalitin, N.; Feitelson, J. Biochemistry 1976, 15, 2092. doi: 10.1021/bi00655a010
Zou, X. R.; Dai, X. J.; Liu, K. H.; Zhao, H. M.; Song, D.; Su, H. M. J. Phys. Chem. B 2014, 118, 5864. doi: 10.1021/jp501658a
Milder, S. J.; Kliger, D. S. J. Am. Chem. Soc. 1985, 107, 7365. doi: 10.1021/ja00311a025
Khvorostov, A.; Lapinski, L.; Rostkowska, H.; Nowak, M. J. Photochem. Photobiol. 2005, 81, 1205. doi: 10.1562/2005-05-19-RA-534
Gagliardi, C. J.; Binstead, R. A.; Thorp, H. H.; Meyer, T. J. J. Am. Chem. Soc. 2011, 133, 19594. doi: 10.1021/ja207379n
Zhang, M. T.; Hammarström, L. J. Am. Chem. Soc. 2011, 133, 8806. doi: 10.1021/ja201536b
Miller, J. E.; Gradinaru, C.; Crane, B. R.; Di Bilio, A. J.; Wehbi, W. A.; Un, S.; Winkler, J. R.; Gray, H. B. J. Am. Chem. Soc. 2003, 125, 14220. doi: 10.1021/ja037203i
Lassmann, G.; Lendzian, F.; MacMillian, F.; Bittl, R.; Potsch, S.; Sahlin, M.; Sjoberg, B. M.; Graslund, A.; Lubitz, W. FASEB J. 1997, 11, A882.
Solar, S.; Getoff, N.; Surdhar, P. S.; Armstrong, D. A.; Singh, A. J. Phys. Chem. 1991, 95, 3639. doi: 10.1021/j100162a038
Katharina, K.; Petra, H.; He, H. H.; Xiao, L.; Markus, W.; Urlaub, H. Int. J. Mass Spectrom. 2011, 304, 184. doi: 10.1016/j.ijms.2010.10.009
Rehm, D.; Weller, A. Isr. J. Chem. 1970, 8, 259. doi: 10.1002/ijch.v8.2
Holzer, K. P.; Wrona, Z. Bioelectrochem. Bioenerg. 1983, 11, 3. doi: 10.1016/0302-4598(83)85096-X
Wrona, Z.; Czochralska, B.; Shugar, D. J. Electroanal. Chem. 1976, 68, 355. doi: 10.1016/S0022-0728(76)80275-6
Sjödin, M.; Styring, S.; Åkermark, B.; Sun, L. C.; Hammarström, L. J. Am. Chem. Soc. 2000, 122, 3932. doi: 10.1021/ja993044k
Tsentalovich, Y. P.; Lopez, J. J.; Hore, P. J.; Sagdeev, R. Z. Spectrochim. Acta, Part A 2002, 58, 2043. doi: 10.1016/S1386-1425(01)00652-7
Psoda, A.; Kazimierczuk, Z.; Shugar, D. J. Am. Chem. Soc. 1974, 96, 6832. doi: 10.1021/ja00829a003
Cardoso, D. R.; Franco, D. W.; Olsen, K.; Andersen, M. L.; Skibsted, L. H. J. Agric. Food Chem. 2004, 52, 6602. doi: 10.1021/jf0401165
Heelis, P. F.; Parsons, B. J.; Phillips, G. O. Biochim. Biophys. Acta 1979, 587, 455. doi: 10.1016/0304-4165(79)90449-5
Sjödin, M.; Styring, S.; Wolpher, H.; Xu, Y.; Sun, L.; Ham-marström, L. J. Am. Chem. Soc. 2005, 127, 3855. doi: 10.1021/ja044395o
吴丽丹, 节家龙, 刘坤辉, 苏红梅, 化学学报, 2014, 72, 1182. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract344673.shtmlWu, L. D.; Jie, J. L.; Liu, K. H.; Su, H. M. Acta Chim. Sinica 2014, 72, 1182(in Chinese). http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract344673.shtml

图 1 反应物分子式(a) 4-硫代尿嘧啶(4-TU)和(b)色氨酸(TrpH)
Figure 1 Molecular structures of (a) 4-thiouracil (4-TU) and (b) tryptophan

图 2 355 nm激光辐照(a) 4-TU和(b) 4-TU与TrpH体系的瞬态吸收光谱
Figure 2 Transient absorption spectra of (a) 4-TU and (b) 4-TU with TrpH after 355 nm laser excitation

图 3 4-TU与TrpH光致共价交联机理示意图
Figure 3 Proposed mechanism of photo-crosslinking between 4-TU and TrpH

图 4 (a) 4-TU三重态在550 nm处的衰减动力学曲线和(b)不同浓度的色氨酸与4-TU三重态反应的Stern-Volmer曲线
Figure 4 (a) Triplet decay dynamics of 4-TU at 550 nm and (b) Stern-Volmer plot for reactions of 4-TU triplet with different concentrations of TrpH

图 5 不同pH下色氨酸与4-TU三重态电子转移反应速率常数
Figure 5 The rate constants of electron transfer from TrpH to 4-TU triplet at different pHs

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:
