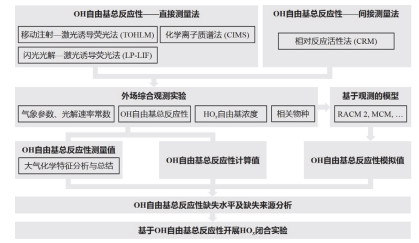
图 1.
研究框架图
Figure 1.
The research framework of this review

羟基自由基(OH)是大气中最重要的氧化剂, OH自由基与人类或生物排放到大气中的还原性气体发生反应, 可决定大气中绝大部分痕量气体(包括甲烷、一氧化碳、氮氧化物、二氧化硫、挥发性有机化合物等)的化学寿命, 并生成O3等二次污染物造成大气污染.因此, 系统研究OH自由基及其化学反应机制对于理解大气氧化性和大气污染形成过程等具有关键作用[1].
早期人们对OH自由基的研究主要侧重于其浓度水平和变化特征等方面, 并主要集中在以CO和CH4为主要污染物的清洁地区, 该类地区OH自由基浓度的模拟结果与测量结果能较好地吻合[1].随着测量技术的发展, OH自由基浓度已实现较为精准的测量[2], 然而在运用大气光化学数值模型对局部区域的OH自由基浓度进行模拟时, 在部分污染地区频繁出现OH自由基浓度实测值与大气光化学模型计算值不能完全吻合的问题.近年来, 大量外场测量研究尽可能多地测量能与OH自由基发生反应的大气组分, 但OH自由基浓度模拟值和测量值比对结果依然存在问题[3, 4], 说明光化学模型中所依赖的OH自由基化学机制依然不够完整.
现阶段的研究表明, OH自由基来源主要包括两部分, 即OH自由基的初级来源和OH自由基的循环再生, 其中初级来源过程包括臭氧光解、气态亚硝酸光解、臭氧烯烃反应等, 循环再生过程指HO2自由基与NO或O3反应再生OH自由基的循环过程, 此外OH自由基来源还包括其他未知部分[5].与OH自由基的来源过程相比, 其去除过程同样复杂.通常所认为的OH自由基反应物为CO、NOx、CH4、SO2以及VOCs等, 但VOCs种类繁多、数量巨大[6], 受限于测量仪器与技术, 很多活性物种无法测定, 又或存在未知的痕量组分, 导致OH自由基的去除难以准确定量.因此, OH自由基总生成速率或总去除速率的准确定量成为HOx自由基化学研究的核心问题之一.
基于以上背景, 发展直接测量OH自由基总生成速率或OH自由基总去除速率的技术有助于加深对OH自由基收支的认识. 1999年, Kovacs和Brune[7]首次提出OH自由基总反应性kOH的概念. OH自由基总反应性kOH的定义为大气中所有能与OH自由基反应的活性物质的浓度及其与OH自由基反应的反应速率常数乘积的加和, 是大气中各活性物质与OH自由基发生反应能力的综合量度, 数值上等于OH自由基寿命的倒数τ-1, 单位为s-1, 如式(1)所示:
|
$ \begin{array}{*{20}{l}} {{k_{{\rm{OH}}}} = {k_{{\rm{O}}{{\rm{H}}^ + }{\rm{N}}{{\rm{O}}_x}}}\left[ {{\rm{N}}{{\rm{O}}_x}} \right] + {k_{{\rm{O}}{{\rm{H}}^ + }{\rm{CO}}}}[{\rm{CO}}] + {k_{{\rm{O}}{{\rm{H}}^ + }{\rm{S}}{{\rm{O}}_2}}}\left[ {{\rm{S}}{{\rm{O}}_2}} \right] + }\\ {{k_{{\rm{O}}{{\rm{H}}^ + }{{\rm{O}}_3}}}\left[ {{{\rm{O}}_3}} \right] + {k_{{\rm{O}}{{\rm{H}}^ + }{\rm{vo}}{{\rm{c}}_i}}}\left[ {{\rm{VO}}{{\rm{C}}_i}} \right] + \ldots = \Sigma {k_{{\rm{O}}{{\rm{H}}^ + }{{\rm{X}}_i}}}\left[ {{{\rm{X}}_i}} \right]} \end{array} $ |
(1) |
其中, [Xi]表示可与OH自由基反应的活性物质的浓度, kOH+Xi表示各活性物质与OH自由基反应的反应速率常数, kOH+Xi[Xi]表示某种活性物质[Xi]的活性.
测量OH自由基总反应性对于深入了解以HOx自由基化学为核心的大气光化学过程而言具有十分重要的意义.首先, 直接测量得到的OH自由基总反应性与OH自由基浓度相乘, 可直接得到OH自由基总去除速率, 用于表征大气氧化能力, 如式(2)所示:
|
$ \begin{array}{l} {D_{{\rm{OH}}}} = {k_{{\rm{OH}} + {\rm{N}}{{\rm{O}}_x}}}\left[ {{\rm{N}}{{\rm{O}}_x}} \right][{\rm{OH}}] + {k_{{\rm{OH}} + {\rm{CO}}}}[{\rm{CO}}][{\rm{OH}}] + \\ {k_{{\rm{O}}{{\rm{H}}^ + }{\rm{S}}{{\rm{O}}_2}}}\left[ {{\rm{S}}{{\rm{O}}_2}} \right][{\rm{OH}}] + {k_{{\rm{O}}{{\rm{H}}^ + }{{\rm{O}}_3}}}\left[ {{{\rm{O}}_3}} \right][{\rm{OH}}] + \\ {k_{{\rm{O}}{{\rm{H}}^ + }{\rm{VO}}{{\rm{C}}_i}}}\left[ {{\rm{VO}}{{\rm{C}}_i}} \right][{\rm{OH}}] + \ldots = \Sigma {k_{{\rm{OH}} + {{\rm{X}}_i}}}\left[ {{{\rm{X}}_i}} \right][{\rm{OH}}]\\ = {k_{{\rm{OH}}}}[{\rm{OH}}] \end{array} $ |
(2) |
其次, OH自由基总反应性的测量可在一定程度上检验VOCs测量的完整度, VOCs活性可表示如下:
|
$ {k_{{\rm{VOCs}}}} \approx {k_{{\rm{OH}}}} - \Sigma {k_{{\rm{O}}{{\rm{H}}^ + }{{\rm{Y}}_i}}}\left[ {{{\rm{Y}}_i}} \right] $ |
(3) |
其中, [Yi]表示可与OH自由基反应的无机活性物质(如CO、NOx、SO2等)的浓度, kOH+Yi表示各无机活性物质与OH自由基反应的反应速率常数, kOH+Yi[Yi]表示某种无机活性物质Yi的活性.通过直接测量OH自由基总反应性, 可间接得到VOCs总活性测量值, 与通过加和各VOCs物种活性所得到的VOCs总活性计算值进行比对, 可判断VOCs测量的完整度[8].第三, 根据光稳态假设下OH自由基总生成速率与总去除速率的相等关系, 由OH自由基总去除速率间接定量OH自由基总生成速率, 进而对OH自由基的来源进行定量分析, 以进一步开展OH自由基化学的来源研究.总的来说, OH自由基总反应性的测量与研究具有十分重要的大气化学意义.
本文的研究内容如图 1所示, 整体研究框架可分为四个层次的总结, 即OH自由基总反应性测量仪器、大型综合观测实验中OH自由基总反应性大气化学行为的分析, 活性缺失及缺失来源, 以及OH自由基总反应性在HOx闭合实验中的应用.
目前应用较为广泛的OH自由基总反应性测量技术可分为两大类, 第一类方法为直接测量法, 即通过测量人为产生的OH自由基与环境空气中活性物种反应, 基于OH自由基浓度随反应时间的衰减直接获得OH自由基总反应性.现有的直接测量方法分为三种, 即移动注射-激光诱导荧光法(TOHLM, Total OH Loss-rate Measurement)、闪光光解-激光诱导荧光法(LP-LIF, Laser flash Photolysis-Laser Induced Fluorescence)和化学离子质谱法(CIMS, Chemical Ionisation Mass Spectrometry); 第二类方法为间接测量法, 即通过对比环境空气中活性物质和参比物种与OH自由基反应的相对竞争关系间接得到OH自由基总反应性, 通常所用的方法为相对反应活性法(CRM, Comparative Reactivity Method).
移动注射-激光诱导荧光法由美国宾夕法尼亚州立大学Kovacs和Brune开发, 并分别于1999年和2006年被首次应用于地面站点观测和航测[7, 9], 目前主要在美国宾夕法尼亚州立大学、美国印第安纳大学和英国利兹大学等单位有所应用[10]; Sadanaga等[11]提出了闪光光解-激光诱导荧光法, 并于2003年在日本进行了第一次外场观测实验, 目前主要在日本首都大学东京、英国利兹大学、法国里尔大学和德国于利希研究中心等单位有所应用[10].随后德国马克斯普朗克化学研究所Sinha和Williams等[12]开发了相对反应活性法, 并于2005年在德国美因茨站点进行了第一次外场观测, 目前主要在德国马普所、法国国立杜埃高等矿业学院、法国气候科学与环境实验室、印度科学与教育研究所、芬兰气象学院、北京大学、英国莱斯特大学和美国加州大学欧文分校等单位有所应用[10]. 2009年德国气象局DWD在运用CIMS方法连续测量OH自由基浓度的同时开始测量OH自由基总反应性, 而目前基于CIMS测量OH自由基总反应活性的方法主要在德国MOHp站点有所应用, 并于2009年至2017年间进行了长期观测实验[10, 13].
OH自由基总反应性的直接测量法分为三种, 即移动注射-激光诱导荧光法(TOHLM)、闪光光解-激光诱导荧光法(LP-LIF)和化学离子质谱法(CIMS).从基本原理来看, 三种直接测量方法均由人为发生的高浓度OH自由基和实际大气反应时随时间的衰减曲线来得到OH自由基总反应性.从本质上讲, 上述三种方法的系统结构具有一致性, 如图 2所示, 三种方法均包含了OH自由基发生装置, 反应流动管以及OH自由基检测系统, 但在具体设计上有一定区别.
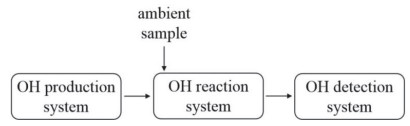
从OH自由基人为发生原理来讲, TOHLM和CIMS方法的OH自由基发生原理均为184.9 nm紫外光下的水蒸气光解, 即
|
$ {{\rm{H}}_2}{\rm{O}} + hv \to {\rm{H}} + {\rm{OH}} $ |
(R1) |
LP-LIF方法OH自由基发生原理为266 nm高能量激光脉冲下的臭氧光解生成O(1D), O(1D)和水汽反应生成OH自由基, 即
|
$ {{\rm{O}}_3} + hv \to {{\rm{O}}^1}({\rm{D}}) + {{\rm{O}}_2} $ |
(R2) |
|
$ {\rm{O}}\left( {^1{\rm{D}}} \right) + {{\rm{H}}_2}{\rm{O}} \to 2{\rm{OH}} $ |
(R3) |
从流动管的设计来看, TOHLM方法基于经典的排气流体技术, 通过移动OH自由基发生源注射器的位置来控制OH自由基在流动管中的停留时间(即和实际大气的反应时间), 从而得到不同反应时间下的OH自由基浓度, 再通过拟合OH浓度随反应时间的衰减来定量OH自由基总反应性[7, 14, 15]. LP-LIF方法的流动管是与激光光解泵-探针配合使用的[11, 16], 激光工作的一个脉冲过程中, 会先在流动管内实时产生一定量的OH自由基, 随后OH自由基的浓度将会随着在流动管中与活性物质反应而衰减, 再通过拟合OH自由基浓度衰减来定量OH自由基反应活性, 该方法的OH发生源和反应流动管是同一模块. TOHLM和LP-LIF方法中OH自由基检测系统均为激光诱导荧光系统LIF, LIF系统在之前研究中已有详细介绍[17~19]. CIMS方法是在反应流动管的前后两个不同位置分别测量OH自由基的浓度, 定量不同停留时间下的自由基浓度, 进而根据其浓度随时间的衰减得到OH自由基总反应性.该方法的关键在于采用化学离子质谱作为OH自由基检测系统, 其原理是通入SO2与OH自由基反应生成H2SO4, 通过定量H2SO4来获得OH自由基浓度, 反应式如下所示[13, 20]:
|
$ {{\rm{S}}{{\rm{O}}_2} + {\rm{OH}} + {\rm{M}} \to {\rm{HS}}{{\rm{O}}_3} + {\rm{M}}} $ |
(R4) |
|
$ {{\rm{HS}}{{\rm{O}}_3} + {{\rm{O}}_2} \to {\rm{S}}{{\rm{O}}_3} + {\rm{H}}{{\rm{O}}_2}} $ |
(R5) |
|
$ {{\rm{S}}{{\rm{O}}_3} + {{\rm{H}}_2}{\rm{O}} + {\rm{M}} \to {{\rm{H}}_2}{\rm{S}}{{\rm{O}}_4} + {\rm{M}}} $ |
(R6) |
生成的H2SO4利用负离子NO3-的电荷转移, 随后被离子化形成HSO4-, 由质谱法检测得到H2SO4浓度, 进而间接得到OH自由基浓度.
上述三种方法均通过拟合OH自由基浓度的指数衰减来得到OH自由基衰减速率, 即为OH自由基总反应性, 衰减曲线拟合式如下:
|
$ {k_{{\rm{OH}}}} = - \Delta \ln \left( {{S_{{\rm{OH}}}}} \right)/\Delta t $ |
(4) |
其中SOH为OH自由基浓度信号, Δt为积分时间.
三种方法的优缺点也各有异同.第一, 三者的共同优点在于检测结果准确性均较高, 缺点在于仪器维护成本较高、难度较大; 第二, TOHLM和LP-LIF方法相比于CIMS方法来讲, 前两者测量OH自由基浓度进而得到OH自由基总反应性的方法比化学离子质谱法更加直接; 第三, TOHLM和CIMS方法中OH自由基在水蒸气光解下的产率较高, 但在产生OH自由基的同时会产生等量的HO2自由基, 在NO浓度较高时会因HOx循环对测量结果产生干扰, 反应如下:
|
$ {{\rm{H}} + {{\rm{O}}_2} \to {\rm{H}}{{\rm{O}}_2}} $ |
(R7) |
|
$ {{\rm{H}}{{\rm{O}}_2} + {\rm{NO}} \to {\rm{OH}} + {\rm{N}}{{\rm{O}}_2}} $ |
(R8) |
LP-LIF方法因臭氧光解产生OH自由基, 故不存在此干扰问题, 但臭氧光解法的OH自由基产率较低; 第四, TOHLM方法因需手动移动注射器位置以得到衰减曲线, 故相比于LP-LIF方法来讲, 其时间分辨率较低, 且操作复杂; 第五, CIMS方法测量范围较窄, 一般适用于40 s-1以下的观测实验, 对于更高活性的测量应通过手动标定来进行实现[10].
相对反应活性法(CRM)基本原理为将空气中不存在的某种已知浓度的参比物质(通常为吡咯, Pyrrole)通入反应管与人为产生的高浓度OH自由基发生反应, 在零空气和环境空气中分别测得其浓度衰减, 检测装置包括气相色谱质谱Gas-Chromatograpy Mass-Spectrometry (GC-MS)、质子转移四极质谱Proton-Transfer-Reaction Mass-Spectrometry (PTR-QMS)、质子转移飞行时间质谱Proton-Transfer-Reaction Time-of-Flight-Mass-Spectro- metry (PTR-ToFMS)或气相色谱光电离检测Gas- Chromatographic Photo-Ionization-Detector (GC-PID)等[12, 21, 22]. OH自由基人为发生原理为184.9 nm紫外光下的水蒸气光解, 如反应式(R1)所示.通过比较零空气和环境空气中参比物质的浓度确定参比物质分别与环境空气中活性物质和OH自由基反应的竞争关系, 从而间接测量得到OH自由基总反应性.
该方法整个测量过程是在三种不同模式下进行的, 模式1为“零OH自由基模式”, 即仅通入参比物质; 模式2为“零空气模式”, 即通入参比物质和一定量的OH自由基; 模式3为“工作模式”, 即同时通入参比物质、OH自由基和环境空气.根据三种不同模式下参比物质浓度C1、C2和C3的关系即可实现对OH自由基总反应性的测量, 计算公式为:
|
$ {k_{{\rm{OH}}}} = \left( {{C_3} - {C_2}} \right)/\left( {{C_1} - {C_3}} \right) \cdot {C_1} \cdot {k_{{\rm{X}} + {\rm{OH}}}} $ |
(5) |
该方法的优点在于无需对OH自由基的浓度变化进行测量, 同时因化学分析仪器的商业化应用使得该方法的操作维护较为简便[23].然而, 该方法也具有一定的缺点:第一, 在高NO环境下的干扰严重, 其原理如反应式(R7)、(R8)所示.由于本法无法直接测量HO2自由基或者OH自由基浓度, 因此较难直接通过定量计算的方法进行修正.目前实验者通过稀释和NO干扰实验的方法进行校正, 但结论的可靠性仍有待印证, 因此不适于高NOx, 尤其是高NO条件下的测量; 第二, 作为参比物质的吡咯在紫外光条件下存在一定的光解, 可能会对计算过程造成一定影响.应保证模式1~3全过程均是在有吡咯光解的条件下进行, 以尽量排除吡咯光解对于定量结果造成的不利影响; 第三, 测量结果受相对湿度变化的影响, 通常使用零空气来确定吡咯基线, 但因零空气和环境空气间相对湿度存在差别, 会导致真实大气环境条件下所扣除的基线信号与零空气下的基线信号相比有所偏差, 从而对OH自由基总反应性的测量产生干扰.不过此干扰可以通过使用Nafion管对零空气进行湿度补偿以减小相对湿度对测量结果的干扰; 第四, 该方法的前提假设是该系统中参比物质与OH自由基的反应为准一级反应, 为满足此要求, 需参比物质浓度远高于OH自由基浓度, 而在此条件下, 会导致C1和C2十分接近, 因此误差会大大加大.实际过程中往往选取较为合适的Pyrrole/OH比, 一般在1.7:1~5:1之间, 而此条件并不严格满足计算的假设, 这也从实验结果中得到了一定的验证.对此, 研究者通常通过引入较为简单的一元化学反应模型对实验过程进行模拟, 通过模拟结果和测量结果的比较来确定较为合适的校正比, 从而将计算得到的OH自由基总反应性进行适当校正, 但此模型的实用范围和准确性还有待进一步考证[12].
OH自由基总反应性测量方法的测量原理及时间分辨率、检测限、不确定度和优缺点等进行总结, 如表 1所示.自OH自由基测量仪器开发以来, OH自由基总反应性测量仪器的大型比对实验主要有三次, 包括同一方法内部的比对, 以及不同方法间的比对.同一方法间的比对为2013年夏季Zannoni等[24]于地中海盆地进行的两台CRM仪器内部比对实验; 不同方法间的成功比对有两次, 分别为2012年秋季Hansen等[25]于法国里尔大学进行的CRM和LP-LIF比对实验, 以及2015年、2016年Fuchs等[10]于德国于利希研究所SAPHIR烟雾箱中开展的TOHLM、LP-LIF、CIMS和CRM方法的大型比对实验.
 下载:
导出CSV
下载:
导出CSV
| Method | Direct measurement method | Indirect measurement method | ||
| Specific methods | Total OH Loss-rate Measurement (TOHLM) | Laser Photolysis-Laser Induced Fluorescence (LP-LIF) | Chemical Ionisation Mass Spectrometry (CIMS) | Comparative Reactivity Method (CRM) |
| OH source | the photolysis of H2O (g) | the photolysis of O3 | the photolysis of H2O (g) | the photolysis of H2O (g) |
| Detectors | LIF | LIF | CIMS | PTR-MS/GC-PID |
| Time resolution/s | 50~200 | 30~180 | 60~300 | 180~300 |
| Detection limit/s-1 | 1 | 1 | 1 | 3~5 |
| Uncertainties | 10%~15% | 10%~15% | 3%~7% | 15%~30% |
| Advantages | (1) high OH radical yield, (2) direct and high accuracy | (1) the little interference, (2) direct and high accuracy | (1) high accuracy | (1) high OH radical yield, (2) well commercialized detector |
| Disadvantages | (1) the interference at NO condition, (2) high cost and complicated operation | (1) low OH radical yield, (2) high cost and complicated operation | (1) narrow measurement range, (2) high cost and complicated operation | (1) the interference at NO condition |
| Reference | 7 | 11 | 13 | 12 |
同一方法间的第一次比对实验为2015年7月于地中海盆地进行的两台CRM仪器内部比对实验.在此次比对观测实验中, 两台CRM的OH自由基总反应性测量结果具有较好的一致性(线性最小二乘拟合的斜率为1, R2为0.75).
2012年10月CRM和LP-LIF方法间的比对为第一次成功开展的OH自由基总反应性测量方法比对实验, 此次比对实验之前, 研究者利用CRM法对OH自由基总反应性进行测量大多在低NOx条件下进行, 而此次比对实验是在对CRM法具有较大挑战的高NOx条件下进行, 因此该比对实验最重要的意义之一在于证明了CRM法在高NOx条件下经过校正可实现OH自由基总反应性的准确测量.比对结果表明, CRM和LP-LIF方法对OH自由基总反应性测量值在不确定性范围之内一致性较好, 但两种方法对OH自由基总反应性的测量均存在一定程度的低估, LP-LIF法由于归零问题对OH自由基总反应性的测量存在低估, 该低估不随OH自由基总反应性的变化而变化, 准确定量零空气的反应活性对于准确测量OH自由基总反应性是至关重要的, 尤其是在低OH自由基总反应性的条件下; CRM法由于VOCs在采样器内的光解导致对OH自由基总反应性的测量存在低估, 低估程度随OH自由基总反应性的变化而变化, 提高OH自由基总反应性测量准确性的方法有以下三种, 第一, 通过改变汞灯位置减少采样器内部的光解过程; 第二, 采用零空气发生器动态调节相对湿度以模拟环境空气湿度, 从而减少零空气和环境空气的相对湿度差来减少湿度校正; 第三, 通过使用不产生HO2的OH源来减少HO2+NO校正[25].
2015年10月和2016年4月, OH自由基测量不同方法间的大型比对实验在德国于利希研究所SAPHIR烟雾箱中开展, 比对仪器包括TOHLM、LP-LIF、CIMS和CRM方法等.比对结果表明, LIF和CIMS方法的检测限和时间分辨率均优于CRM法; 对于复杂化学条件来说, LP-LIF法的精度高于TOHLM和CIMS法, 而CRM法精度较差, 数据较离散; CIMS方法作为一种新兴的测量方法, 适用于低活性和低NOx环境下的测量.总的来看, LP-LIF法在测量精度、检测限、时间分辨率、环境适用性等方面均优于其他方法, 为不同的大气环境下进行OH自由基总反应性准确测量的最佳方法[10].
随着测量技术的成功开发, OH自由基总反应性相关的外场综合观测实验在多种类型的站点相继展开. 表 2对全球不同站点OH自由基总反应性外场观测情况进行了汇总整理, 包括测量站点、测量时间、测量方法、OH自由基总反应性水平以及活性缺失等情况.近二十年来, OH自由基总反应性外场测量已在城市、郊区、森林等多种类型站点展开, 不同大气环境下的OH自由基总反应性呈现出不同的大气化学特征.总体来说, 研究者对于清洁海洋环境或高NOx环境下的OH自由基总反应性认识较为清晰, 但对于低NOx高VOCs环境(如森林地区)和受大陆性气团影响严重的大气环境下的OH自由基总反应性的认识还存在不足[26].
下载:
导出CSV
| Campaign | Site | Time | Condition | Instrument | kOH/s-1 | kOH missing | REF |
| PROPHET 2000 | Michigan, USA | summer, 2000 | forest | TOHLM | 1~12a | 33% | 27 |
| PMTACS-NY | New York, USA | summer, 2002 | forest | TOHLM | 5.6b | agreed well | 28 |
| OP3 | Borneo, Malaysian | spring, 2008 | forest | TOHLM | 29.1b | 38% | 26 |
| GABRIEL | Brownsberg, Suriname | autumn, 2005 | forest | CRM | 53b | 65% | 12 |
| BFORM | Hyytiala, Finland | summer, 2008 | forest | CRM | 9b | 50.6% | 29 |
| HUMPPA-COPEC 2010 | Hyytiala, Finland | summer, 2010 | forest | CRM | 3~76a | (1) 58% (normal) (2) 89% (stressed) | 22 |
| BEACHON-SRM08 | Colorado, USA | summer, 2008 | forest | LP-LIF | 6.7b | 29.5% | 30 |
| CABINEX | Michigan, USA | summer, 2009 | forest | TOHLM | 3~33a | (1) 55.8% (6 m) (2) 51.7% (21 m) (3) 57.3% (31 m) | 15 |
| CANOPEE | Haute Provence, France | spring, 2014 | forest | CRM | 24b | (1) agreed well (day) (2) 50% (night) | 24 |
| SOS | Nashville, USA | summer, 1999 | urban | TOHLM | 11b | 29% | 31 |
| — | Helsinki, Finland | winter, 2016 | semi-urban | CRM | 7.6c | 47% | 32 |
| PMTACS-NY2001 | New York, America | summer, 2001 | urban | TOHLM | 19b | within 10% | 33 |
| MCMA 2003 | Mexico City, Mexico | spring, 2003 | urban | TOHLM | 20b | 30% | 34 |
| TEXAQS2000 | Houston, USA | summer, 2000 | urban | TOHLM | 7~12a | agreed well | 35 |
| TRAMP2006 | Houston, USA | summer, 2006 | urban | TOHLM | 9~22a | agreed well | 35 |
| — | Tokyo, Japan | 2003~2004 | suburban | LP-LIF | 10~100a | 30% | 11 |
| — | Tokyo, Japan | summer, 2007 | urban | LP-LIF | 10~55a | 29% | 36 |
| — | Tokyo, Japan | spring, 2009 | suburban | LP-LIF | 10~35a | 20% | 37 |
| — | Tokyo, Japan | summer, 2007 | urban | LP-LIF | 33.4b | 26.5% | 38 |
| — | Tokyo, Japan | autumn, 2009 | urban | LP-LIF | 32.3b | 34.7% | 38 |
| — | Mainz, Germany | summer, 2005 | urban | CRM | 10.4b | — | 12 |
| MEGAPOLI | Paris, France | winter, 2010 | urban | CRM | 10~130a | 54% | 39 |
| — | Lille, France | autumn, 2012 | urban | CRM, LP-LIF | 70b | agreed well | 25 |
| — | Wangdu, China | summer, 2014 | urban | LP-LIF | 10~20a | agreed well | 40 |
| — | Pennsylvania, USA | spring, 2002 | rural | TOHLM | 6.1b | — | 41 |
| TORCH-2 | Weybourne, England | spring, 2004 | rural | TOHLM | 4.9b | 39% | 14 |
| CAREBeijing 2006 | Yufa, China | summer, 2006 | rural | LP-LIF | 10~30a | 67% | 2 |
| PRIDE-PRD | Backgarden, China | summer, 2006 | rural | LP-LIF | 10~120a | 50% | 16 |
| DOMINO | EI Arenosillo | winter, 2008 | rural | CRM | 3.5~84a | — | 42 |
| HOxComp | Jülich, German | summer, 2005 | rural | LP-LIF | 8.8b | 40% | 43 |
| a The range of the measured reactivity during the whole campaign. b The mean of the measured reactivity during the whole campaign. c The median of the measured reactivity during the whole campaign. | |||||||
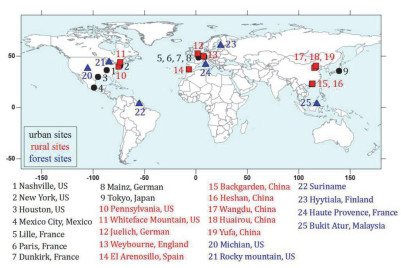
从观测站点上来说, OH自由基总反应性外场测量的大部分站点分布于北美洲、欧洲和东亚地区, 东亚地区的观测主要集中于中国京津冀和珠三角地区, 如图 3所示.
OH自由基总反应性大气化学特征的观测主要在两种大气环境下开展, 即人为源排放影响的区域和生物源排放影响的区域.对不同大气环境下的OH自由基总反应性大气化学特征进行总结, 一方面有利于全面了解世界范围内OH自由基总反应性特征, 另一方面可为研究者测量参数的选择提供一定的参考, 即可根据其研究区域的实际大气环境状况选择合适的测量参数.
典型城市地区和部分郊区一般受人为源排放影响较大, 当该类地区受到交通源和工业源排放的高浓度NOx和VOCs影响, 或受到大陆性气团影响时, 其OH自由基总反应性通常会呈现出相似的化学特征.
从活性水平来看, 该类地区OH自由基总反应性变化范围较大, 从低于10 s-1(如德国美因茨, 2005年夏季)至200 s-1不等(如墨西哥城, 2003年春季)[12, 34], 且活性水平通常与观测点位基本特征及观测季节等因素密切相关.
从日变化来看, 该类地区的OH自由基总反应性通常会呈现早晚高峰现象[31, 33~35, 39], 第一个峰值大约于清晨出现, 在上午晚些时候略有下降, 到傍晚时分出现第二个高峰, 主要原因有三个:第一, 早晚受局地污染排放(如机动车排放)的活性, 即NOx和VOCs浓度升高故而活性增强; 第二, 早晚光照条件较弱, 氧化剂浓度较低, 化学反应对这些活性气体的去除较慢; 第三, 晨间逆温现象较为普遍, 造成污染物在近地面的积累, 表现为OH反应活性水平在早高峰明显高于晚高峰.在墨西哥城的观测中发现了比其他城市更高的早高峰现象, 活性值达到了120 s-1, 主要与当地极强的排放与特殊的城市地形有关, 其中早高峰期间总碳氢化合物的浓度大约为其他城市的5倍以上[34].
从活性组成来看, 该类地区的OH自由基总反应性组成可根据所在区域的污染特征分为两大类, 第一类为以交通源排放为主导或受交通源排放类气团传输影响较大的污染地区, 第二类为以工业源排放为主导或受工业源排放类气团传输影响较大的污染地区.在第一类污染地区, OH自由基总反应性组成主要为NOx.如法国巴黎、日本东京、美国纽约等地[11, 33, 39], NOx贡献超过OH总活性的50%, 其次是NMHCs (non-methane volatile organic compounds)、CO和OVOCs (oxygenated volatile organic compounds)等.在第二类污染地区, OH自由基总反应性组成主要为NMHC及其氧化物种.如墨西哥城和美国休斯敦等地[34, 35], NMHCs大约贡献了OH总反应活性的50%, 其次为NOx、OVOCs和CO, 其中墨西哥城的高NMHCs主要受生物质燃烧的影响, 而美国休斯敦的工业排放非常强, 且芳香烃物种所占比重较大[44].当站点受到大陆性气团影响时, 因长距离输送的影响气团为老化气团, 以醛酮类为主的二次有机氧化物种OVOCs对OH总反应性有较大贡献.综上, 受人为源排放影响较大地区的OH自由基总反应性主要来源于NOx和NMHCs及其氧化物种, 但具体情况与观测站点所在地区的排放结构和强度等均有直接关系.以人为源排放为主的典型站点中OH自由基总反应性的组成情况如图 4上半部分所示.
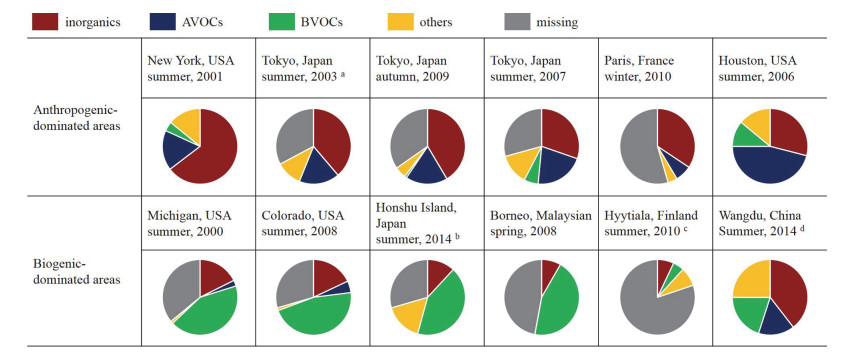
a Calculated contributions from 15:00-17:00 JST on August 20, 2003, b calculated contributions towards the noontime OH reactivity, c calculated contributions above canopy, d calculated contributions after 20 June, 2014
研究表明, 全球天然源VOCs (BVOCs)排放量(约1150 TgC/year)比人为源VOCs (AVOCs)排放量(约160 TgC/year)高一个数量级[45]. BVOCs的天然排放主要来自于森林地区和部分偏远郊区, 包括异戊二烯、单萜烯、倍半萜烯等[6, 46, 47]. BVOCs可被氧化为二次物种, 在低NO环境下(尤其是北方森林)易形成过氧化物(日间被OH自由基氧化)、醛和有机酸(日间和夜间被O3氧化)等[48], 可对环境造成进一步影响.
此类地区的第一次OH自由基总反应性外场观测由Di Carlo等[27]在PROPHET项目中于北方密歇根森林进行, 之后研究者在热带[12, 26]、温带[15, 24, 30]和寒带森林[22, 29]等多个站点进行了观测, 同时对不同高度上的OH自由基总反应性进行了观测及比对分析[15, 49].研究表明, 森林地区OH总反应性与测量高度有关[50], 截至目前森林站点中OH自由基总反应性外场观测大多在树冠之上进行, 因为多数观测中排放的VOCs及其氧化产物、远距离传输而来的痕量气体及太阳辐射均位于树冠之上, 故该大气环境被认为是森林中化学活性最强的区域[15].然而, 树冠层之上和之下均能观测到VOCs氧化产物, 表明树冠层以下也可为OH自由基活性缺失提供有用信息[49].
BVOCs的化学成分和排放速率高度依赖于树种[51], 目前在该类地区的OH自由基总反应性外场观测通常分为两种环境, 第一种环境为异戊二烯主导的BVOCs排放地区[12, 14, 26, 27], 主要集中于热带森林地区.异戊二烯排放的主控因子为温度和光照, 两者通过影响异戊二烯合成酶的活性进而影响其排放速率, 且对植物排放异戊二烯排放速率的影响均存在一个饱和值.随环境温度或光照的升高, 植物对异戊二烯的排放速率增大, 但升至一定温度或光照时, 又会使排放速率减小[52, 53]; 第二种环境为单萜烯主导的BVOCs排放地区[22, 29], 主要集中于温带和寒带森林地区, 单萜烯排放的主控因子为温度, 温度主要通过控制单萜合成酶的活性影响其排放速率, 研究表明单萜烯排放速率与温度变化呈较好的指数关系但同样存在饱和值.与异戊二烯相比, 大多植物单萜烯的合成与排放受光照影响不大, 主要原因是叶片内单萜合成酶的活性受光照影响较小[54].
从活性组成和水平来看, 热带森林地区的OH自由基总反应性比温带、寒带森林地区高.热带森林在午后可出现50 s-1的高值[12, 26], 而温带、寒带森林地区反应性均值通常在10 s-1以下[29, 30].主要原因是热带地区的BVOC排放量高于温、寒带地区, 尤其是异戊二烯; 同时OH和异戊二烯的反应速率常数高于OH和单萜烯类物质的反应速率常数[55], 从而导致热带森林地区的OH自由基总反应性水平一般高于温、寒带.从活性组成来看, 热带森林地区的OH自由基总反应性组成以异戊二烯占主导, 甚至可占据总活性的最大比重, 如美国密歇根森林站点(比重为49%)、美国白脸山站点(比重为14%)、马来西亚热带雨林站点(比重为30%)及苏里南热带森林站点等[12, 26~28].而在低异戊二烯排放、高单萜烯类排放的寒带森林地区, 活性的最大组成部分为单萜烯活性, 如芬兰站点[22, 29], 以及地中海森林地区等[24].以生物源排放为主的典型站点中OH自由基总反应性的组成情况如图 4下半部分所示.
从日变化来看, 受生物源排放影响较大地区的OH自由基总反应性日变化呈现出以下三个特征.第一, 总体呈现日间高、夜间低的变化趋势, 从早上开始增加至午后达到高值, 随后下降至夜晚达到低值, 即与光照和温度呈现正相关关系, 主要原因为森林地区BVOCs排放与温度及光照相关[12, 26].第二, 热带森林地区的活性日变化比温带、寒带森林地区明显, 主要原因为热带森林地区的活性主导物种为异戊二烯, 其排放受光照影响较大, 温带、寒带森林地区的活性主导物种为单萜烯类, 单萜烯类受温度影响较大, 热带地区的日间光照变化幅度大于温带、寒带地区的日间温度变化幅度; 此外, 北方森林虽日间排放大于夜间, 但夜间边界层高度会低于日间边界层高度, 排放与边界层高度变化的补偿效应使得活性日变化不明显导致其日变化较小.第三, 平均偏差和标准偏差所反映的OH自由基总反应性的波动在日间较夜间更大, 表明日间和夜间的化学机制是不同的[26].如Edwards等[26]在东南亚热带森林的观测中发现活性值日间波动大于夜间, 主要由于观测站点夜间通常会位于边界层以上, 导致稀释度增加, OH自由基总反应性及其变化幅度减小; 而日间由于边界层上升使得站点处于边界层以下, 易受到森林排放BVOCs以及更多变化性强的气团的影响.
总体来看, 在受BVOCs排放影响较大的地区, 尤其是物种丰富、生物排放量大的热带森林地区, 异戊二烯和单萜烯为BVOCs的主要成分.该类地区BVOCs排放量大, 且BVOCs的氧化过程较为复杂, 因此其本身对于大气氧化过程具有重要影响, 其氧化产物在对流层化学中也非常重要, 尤其是异戊二烯的氧化机制, 研究者对其存在不同的理解与看法.
OH自由基总反应性缺失是指观测实验中OH自由基总反应性的测量值kmea与计算值kcal或模拟值kmod之间的差值.测量活性值kmea指外场观测中所直接测得的OH自由基总反应性值; 计算活性值kcal指外场观测中将各活性物种浓度测量值及其与OH自由基反应的反应速率常数的乘积, 即根据式(R1)进行加和得到的活性值.在近二十年的OH自由基总反应性外场观测实验中, 不同类型站点的OH自由基总反应性测量中虽表现出不同的大气化学特征, 但普遍出现了活性缺失现象.对活性缺失现象进行研究有助于深入了解以HOx自由基化学为核心的大气光化学反应过程.限于外场观测中各项观测能力, 很多OH自由基反应活性物种并不能用仪器直接测得, 因此通过模型引入部分未被实际测量得到的活性物种, 模拟所得到的活性值称为模拟活性值kmod.
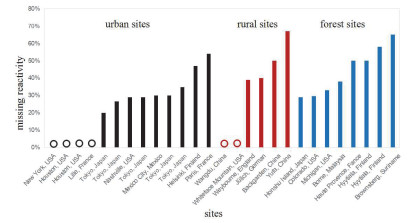
图 5总结了城市、郊区和森林不同类型站点观测的活性缺失情况, 由统计结果可以看出, 活性缺失最普遍的区域为受天然源排放最为严重的森林地区, 活性缺失大约在30%~70%之间[12, 22, 26, 27, 29, 30].城市地区和郊区在部分观测实验中活性缺失现象较为严重[11, 31, 34, 36~38], 最高缺失活性可达到70%左右[39], 但在部分条件下城市和郊区也存在活性不缺失的情况[25, 32, 33, 35, 39].
在此背景下, 研究OH自由基总反应性缺失的来源对于OH自由基总反应性研究来讲具有重要意义.通常所认为OH自由基无机活性物种的测量准确性较高, 且活性物种与OH自由基的反应速率常数较为明确, 所以活性缺失一般归因于有机物种, 即主要来源于未测或未知的一次有机物种、有机物的氧化产物或两者的共同作用.对OH自由基总反应性缺失来源进行总结, 可为研究区域内活性缺失来源等提供一定的参考, 即研究者可根据其观测站点的点位特征等选择合适的测量物种, 亦可对拟模拟的未测物种选择具有一定的侧重.
研究表明, 有机物一次排放物种为OH自由基总反应性缺失的重要来源, 有机物一次排放物种的人为源主要为化工生产、溶剂使用和汽车尾气等, 天然源主要为植物排放.在受人为源影响比较严重的地区, 未测或未知的AVOCs为活性缺失一次来源的重要组成.在1999年美国纳什维尔观测中, 机动车排放的短寿命VOCs为活性缺失的重要来源[31]; 此外, 在2004年春季在英国海岸站点的观测中, 发现活性缺失大部分来源于高分子芳香类化合物[56].
对于受生物源排放影响较大的地区来讲, 未测或未知的BVOCs为活性缺失一次来源的重要组成.国内外对于全球BVOCs的研究主要集中于异戊二烯和单萜烯[57], 两者在外场观测中一般可被准确测量, 但其他萜烯类物质的测量依然不够完全[58], 因此可能为OH自由基总反应性缺失的重要组成部分.如Di Carlo等[27]研究表明在美国密歇根森林站点的活性缺失可能主要来源于与温度相关性较高的萜烯类BVOCs; 此外, Sinha等[29]在芬兰森林的观测中推测OH自由基总反应性缺失主要来源于未测得的生物源VOCs排放.
在受人为源影响比较严重的地区, 即受工业排放或者典型大陆性气团影响严重的地区, 活性缺失一般来源于AVOCs的氧化态产物. OVOCs作为光化学烟雾过程中的重要中间产物, 为该类地区最重要的活性缺失组成之一, 对OH自由基总反应性影响较大的OVOCs一般为甲醛、乙醛、乙二醛和异戊二烯氧化产物(甲基乙烯基酮、甲丙烯醛)等.因物种组成的复杂性和测量仪器的限制, 仅在少数OVOCs物种在外场观测中被测量到[11, 58], 在部分研究中, 还可以通过模型模拟的OVOCs来进行活性缺失分析[16, 59].目前观测实验中OVOCs的测量和模拟还比较缺乏, 但现阶段的研究认为OVOCs可能是活性缺失的重要组成部分, 甚至在一些条件下能弥补所有的活性缺失[16].此外, 大陆性气团所携带的高氧化态产物对活性缺失也具有一定贡献, 如2010年冬季法国巴黎观测期间, 受大陆气团影响, 高达74%的活性缺失表现出与长寿命化合物和光化学生成的气溶胶相似的变化, 表明该站点的大陆性气团可能携带高氧化态产物[39].
在受天然源影响较为严重的地区, BVOCs氧化产物对活性缺失可能具有很大贡献[22, 26, 60, 61]. OH自由基与异戊二烯之间的光氧化反应所产生的二次物种被认为是HOx化学过程中的关键物种之一[30], 因异戊二烯氧化产物活性可高达异戊二烯活性的150%, 故未测得的异戊二烯氧化产物可在一定程度上有助于解释活性缺失[60, 61].因此对于该类地区所出现的活性缺失现象, 应对BVOCs氧化过程开展全面研究, 以确定二次物种是否足以解释观测中出现的活性缺失, 亦或存在其他活性气体对活性缺失有所贡献.
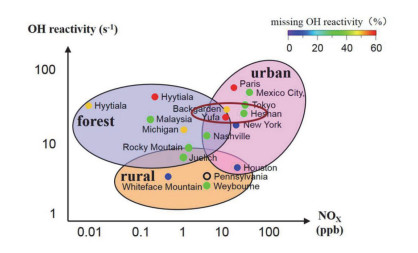
图 6总结了现有的部分城市、郊区和森林不同类型站点上OH自由基总反应性、NOx浓度和活性缺失的相对大小情况.城市站点和部分郊区站点分布于高活性、高NOx区域, 森林站点一般分布于高活性、低NOx区域, 大部分郊区站点分布于低活性、低NOx区域, 表明了三种类型站点的活性情况与排放源排放状况具有紧密联系.其中中国北京榆垡站点和广东后花园虽为郊区站点, 但其OH自由基总反应性和NOx浓度的相对大小分布于偏向城市站点的区域内, 且活性缺失均在50%以上.说明中国郊区的污染状况与世界范围内其他国家郊区站点的污染状况具有显著区别.
开展HOx化学机制研究的收支闭合实验最为常见的形式为对HOx浓度的测量值和光化学模式的模拟值进行比对.对于OH自由基而言, 因其大气化学寿命较短, 故在通常考虑的时间尺度下处于光化学稳态, 即其生成速率和去除速率相等[2]. OH自由基总反应性为进行OH自由基收支分析的重要参数.基于该参数, 结合OH、HO2和NO等浓度数据, OH自由基收支闭合分析即可基于观测实验开展, 而无需依赖于模型.因为OH自由基总反应性kOH结合OH自由基浓度[OH]可直接定量OH自由基总去除速率DOH, 即DOH=kOH*[OH], 光稳态条件下的OH自由基总生成速率POH与总去除速率DOH相等, 进而得到OH自由基总生成速率.因此, 可根据式(6)判断目前已知的OH自由基来源机制是否正确.
|
$ {k_{{\rm{OH}}}} \times [{\rm{OH}}] = P({\rm{OH}}) + {k_8}\left[ {{\rm{H}}{{\rm{O}}_2}} \right][{\rm{NO}}] $ |
(6) |
目前, 已有多个研究基于OH自由基总反应性的观测实验进行了OH自由基来源分析, 包括墨西哥墨西哥城, 美国纽约、纳什维尔, 英国伦敦以及中国珠三角后花园、北京榆垡、河北望都、北京怀柔等地.在1999年美国纳什维尔、2001年英国纽约和2003年墨西哥城等地的观测中, 均出现了早高峰期间OH自由基总生成速率高于总去除速率的现象, 纳什维尔和纽约观测均认为此差异最可能的解释为没有正确修正NO对移动注射-激光诱导荧光系统测量方法的干扰, 从而导致了OH自由基总反应性的低估; 墨西哥观测中研究者对此不平衡现象的解释是, HO2和NO的部分反应产物可能不是OH和NO2, 而是造成了HOx的直接去除, 或者伴随着某些循环反应使得反应产物不经过OH直接迅速变为HO2, 因此导致计算得到的OH自由基总生成速率比实际情况偏大[33, 34, 62].而在纳什维尔和纽约观测中, 均发现夜间OH自由基总去除速率高于总生成速率, 表明OH自由基存在未知来源, 该未知源可能来自于未知反应通道, 如HO2、O3和HCHO等化合物参与的未知反应机制[62]; 而基于纽约观测认为此差异可能为观测中未测量到的生物排放的高活性不饱和萜烯的臭氧分解产生OH自由基, 或者人为源VOCs与O3或NO3的反应生成的OH自由基[33].
自2006年以来, 中国地区关于OH自由基总反应性的大型外场观测实验逐渐在广东后花园(2006年)、北京榆垡(2006年)、河北望都(2014年)、北京怀柔(2016年)等地逐渐展开.近十年中国地区的研究结果表明, OH自由基未知来源可能有HO2或RO2自由基向OH自由基转化的未知机制、萜类化合物的臭氧分解、PAN等自由基储物通过夜间边界层的向下输送、异戊二烯氧化机制等[5, 18, 19, 58, 59].在研究过程中, 可通过在模型中对新机理的添加, 如异戊二烯氧化机制、温室气体七氟醚降解产生的七氟醚自由基与O2反应再生OH的微观机理等[19, 63], 以完善大气HOx自由基光化学过程.
自20世纪90年代研究者对OH自由基总反应性进行首次测量以来, 作为以OH自由基为核心的大气光化学过程研究的关键参数, 其测量技术在进行不断发展与完善.目前OH自由基总反应性主要有三种测量技术, 即直接测量法(移动注射-激光诱导荧光法和闪光光解-激光诱导荧光法)、半直接测量法(化学离子质谱法)和间接测量法(相对反应活性法), 相比于闪光光解-激光诱导荧光法来讲, 另外三种方法易受NO干扰, 一般适用于低NOx, 尤其是低NO环境下的测量; 此外, 移动注射-激光诱导荧光法和相对反应活性法分辨率较低, 化学离子质谱法测量范围相对较窄.因此, 闪光光解-激光诱导荧光法凭借其高时空分辨率、较低的检测限、较低的不确定性及较小的干扰等优势逐渐成为了国际上OH自由基总反应性的主要测量方法.
近二十年以来, OH自由基总反应性相关的外场综合观测实验在城市、郊区及森林等多种类型的站点相继展开.相比于森林和郊区站点来讲, 城市站点的OH自由基总反应性水平更高, 日变化因受机动车排放影响而通常呈现典型的双峰现象, NOx和NMHC一般在城市站点的OH自由基总反应性组成中占较大比例; 森林站点的OH自由基总反应性的主要组成部分为BVOCs, 日变化因BVOCs排放与温度和光照密切相关而呈现日间高、夜间低的变化趋势; 郊区站点因不同站点所受人为源排放和生物源排放程度不同而不同.此外, 活性缺失为外场综合观测实验中的一个普遍现象, 缺失部分一般来源于未测或未知的一次排放VOCs, 以及未测或未知的光氧化OVOCs等.
总的来说, 关于OH自由基总反应性测量技术和外场观测中所出现的研究难题可以总结为如下五个方面:第一, 闪光光解-激光诱导荧光法在高NO条件下的数据拟合问题有待优化, 而移动注射-激光诱导荧光法和相对反应活性法在高NO条件下的干扰问题有待解决; 第二, 不同模型对于OH自由基总反应性模拟结果的比对较为缺乏, 增加模型间的比对分析将有助于理解具体化学机理在大气光化学循环中的作用; 第三, 全球范围内不同类型站点OH自由基总反应性水平、活性组成、活性缺失大小及缺失来源等问题, 大多停留在对数值进行报道及初步分析猜测阶段, 而对背后真正的化学机理缺乏深入分析; 第四, 目前全球范围内的各个观测之间仍缺乏普遍性和共通性, 需要对OH自由基总反应性缺失提炼参数化公式; 第五, OH自由基总反应性长期自动观测站的建立将成为该领域的一个待努力方向.
Ehhalt, D. H. Phys. Chem. Chem. Phys. 1999, 1, 5401. doi: 10.1039/a905097c
陆克定, 张远航, 化学进展, 2010, 22, 500. http://manu56.magtech.com.cn/progchem/CN/abstract/abstract10259.shtmlLu, K.-D.; Zhang, Y.-H. Prog. Chem. 2010, 22, 500(in Chinese). http://manu56.magtech.com.cn/progchem/CN/abstract/abstract10259.shtml
Hofzumahaus, A.; Aschmutat, U.; Hessling, M.; Holland, F.; Ehhalt, D. H. Geophys. Res. Lett. 1996, 23, 2541. doi: 10.1029/96GL02205
Heard, D. E.; Pilling, M. J. Chem. Rev. 2003, 103, 5163. doi: 10.1021/cr020522s
Hofzumahaus, A.; Rohrer, F.; Lu, K.; Bohn, B.; Brauers, T.; Chang, C.-C.; Fuchs, H.; Holland, F.; Kita, K.; Kondo, Y.; Li, X.; Lou, S.; Shao, M.; Zeng, L.; Wahner, A.; Zhang, Y. Science 2009, 324, 1702. doi: 10.1126/science.1164566
Goldstein, A. H.; Galbally, I. E. Environ. Sci. Technol. 2007, 41, 1514. doi: 10.1021/es072476p
Kovacs, T. A.; Brune, W. H. J. Atmos. Chem. 2001, 39, 105. doi: 10.1023/A:1010614113786
Yang, Y.; Shao, M.; Wang, X.; Noelscher, A. C.; Kessel, S.; Guenther, A.; Williams, J. Atmos. Environ. 2016, 134, 147. doi: 10.1016/j.atmosenv.2016.03.010
Mao, J.; Ren, X.; Brune, W. H.; Olson, J. R.; Crawford, J. H.; Fried, A.; Huey, L. G.; Cohen, R. C.; Heikes, B.; Singh, H. B.; Blake, D. R.; Sachse, G. W.; Diskin, G. S.; Hall, S. R.; Shetter, R. E. Atmos. Chem. Phys. 2009, 9, 163. doi: 10.5194/acp-9-163-2009
Fuchs, H.; Novelli, A.; Rolletter, M.; Hofzumahaus, A.; Pfannerstill, E. Y.; Kessel, S.; Edtbauer, A.; Williams, J.; Michoud, V.; Dusanter, S.; Locoge, N.; Zannoni, N.; Gros, V.; Truong, F.; Sarda-Esteve, R.; Cryer, D. R.; Brumby, C. A.; Whalley, L. K.; Stone, D.; Seakins, P. W.; Heard, D. E.; Schoemaecker, C.; Blocquet, M.; Coudert, S.; Batut, S.; Fittschen, C.; Thames, A. B.; Brune, W. H.; Ernest, C.; Harder, H.; Muller, J. B. A.; Elste, T.; Kubistin, D.; Andres, S.; Bohn, B.; Hohaus, T.; Holland, F.; Li, X.; Rohrer, F.; Kiendler-Scharr, A.; Tillmann, R.; Wegener, R.; Yu, Z.; Zou, Q.; Wahner, A. Atmos. Meas. Tech. 2017, 10, 4023. doi: 10.5194/amt-10-4023-2017
Sadanaga, Y.; Yoshino, A.; Watanabe, K.; Yoshioka, A.; Wakazono, Y.; Kanaya, Y.; Kajii, Y. Rev. Sci. Instrum. 2004, 75, 2648. doi: 10.1063/1.1775311
Sinha, V.; Williams, J.; Crowley, J. N.; Lelieveld, J. Atmos. Chem. Phys. 2008, 8, 2213. doi: 10.5194/acp-8-2213-2008
Muller, J. B. A.; Elste, T.; Plass-Duelmer, C.; Stange, G.; Holla, R.; Claude, A.; Englert, J.; Gilge, S.; Kubistin, D. Atmos. Meas. Tech. 2018, 11, 4413. doi: 10.5194/amt-11-4413-2018
Ingham, T.; Goddard, A.; Whalley, L. K.; Furneaux, K. L.; Edwards, P. M.; Seal, C. P.; Self, D. E.; Johnson, G. P.; Read, K. A.; Lee, J. D.; Heard, D. E. Atmos. Meas. Tech. 2009, 2, 465. doi: 10.5194/amt-2-465-2009
Hansen, R. F.; Griffith, S. M.; Dusanter, S.; Rickly, P. S.; Stevens, P. S.; Bertman, S. B.; Carroll, M. A.; Erickson, M. H.; Flynn, J. H.; Grossberg, N.; Jobson, B. T.; Lefer, B. L.; Wallace, H. W. Atmos. Chem. Phys. 2014, 14, 2923. doi: 10.5194/acp-14-2923-2014
Lou, S.; Holland, F.; Rohrer, F.; Lu, K.; Bohn, B.; Brauers, T.; Chang, C. C.; Fuchs, H.; Haeseler, R.; Kita, K.; Kondo, Y.; Li, X.; Shao, M.; Zeng, L.; Wahner, A.; Zhang, Y.; Wang, W.; Hofzu-mahaus, A. Atmos. Chem. Phys. 2010, 10, 11243. doi: 10.5194/acp-10-11243-2010
Holland, F.; Hessling, M.; Hofzumahaus, A. J. Atmos. Sci. 1995, 52, 3393. doi: 10.1175/1520-0469(1995)052<3393:ISMOTO>2.0.CO;2
Lu, K. D.; Rohrer, F.; Holland, F.; Fuchs, H.; Bohn, B.; Brauers, T.; Chang, C. C.; Haeseler, R.; Hu, M.; Kita, K.; Kondo, Y.; Li, X.; Lou, S. R.; Nehr, S.; Shao, M.; Zeng, L. M.; Wahner, A.; Zhang, Y. H.; Hofzumahaus, A. Atmos. Chem. Phys. 2012, 12, 1541. doi: 10.5194/acp-12-1541-2012
Tan, Z.; Rohrer, F.; Lu, K.; Ma, X.; Bohn, B.; Broch, S.; Dong, H.; Fuchs, H.; Gkatzelis, G. I.; Hofzumahaus, A.; Holland, F.; Li, X.; Liu, Y.; Liu, Y.; Novelli, A.; Shao, M.; Wang, H.; Wu, Y.; Zeng, L.; Hu, M.; Kiendler-Scharr, A.; Wahner, A.; Zhang, Y. Atmos. Chem. Phys. 2018, 18, 12391. doi: 10.5194/acp-18-12391-2018
Berresheim, H.; Elste, T.; Plass-Dulmer, C.; Eisele, F. L.; Tanner, D. J. Int. J. Mass Spectrom. 2000, 202, 91. doi: 10.1016/S1387-3806(00)00233-5
Kumar, V.; Sinha, V. Int. J. Mass Spectrom. 2014, 374, 55. doi: 10.1016/j.ijms.2014.10.012
Noelscher, A. C.; Williams, J.; Sinha, V.; Custer, T.; Song, W.; Johnson, A. M.; Axinte, R.; Bozem, H.; Fischer, H.; Pouvesle, N.; Phillips, G.; Crowley, J. N.; Rantala, P.; Rinne, J.; Kulmala, M.; Gonzales, D.; Valverde-Canossa, J.; Vogel, A.; Hoffmann, T.; Ouwersloot, H. G.; De Arellano, J. V.-G.; Lelieveld, J. Atmos. Chem. Phys. 2012, 12, 8257. doi: 10.5194/acp-12-8257-2012
Williams, J.; Brune, W. Atmos. Environ. 2015, 106, 371. doi: 10.1016/j.atmosenv.2015.02.017
Zannoni, N.; Gros, V.; Lanza, M.; Sarda, R.; Bonsang, B.; Ka-logridis, C.; Preunkert, S.; Legrand, M.; Jambert, C.; Boissard, C.; Lathiere, J. Atmos. Chem. Phys. 2016, 16, 1619. doi: 10.5194/acp-16-1619-2016
Hansen, R. F.; Blocquet, M.; Schoemaecker, C.; Leonardis, T.; Locoge, N.; Fittschen, C.; Hanoune, B.; Stevens, P. S.; Sinha, V.; Dusanter, S. Atmos. Meas. Tech. 2015, 8, 4243. doi: 10.5194/amt-8-4243-2015
Edwards, P. M.; Evans, M. J.; Furneaux, K. L.; Hopkins, J.; Ingham, T.; Jones, C.; Lee, J. D.; Lewis, A. C.; Moller, S. J.; Stone, D.; Whalley, L. K.; Heard, D. E. Atmos. Chem. Phys. 2013, 13, 9497. doi: 10.5194/acp-13-9497-2013
Di Carlo, P.; Brune, W. H.; Martinez, M.; Harder, H.; Lesher, R.; Ren, X. R.; Thornberry, T.; Carroll, M. A.; Young, V.; Shepson, P. B.; Riemer, D.; Apel, E.; Campbell, C. Science. 2004, 304, 722. doi: 10.1126/science.1094392
Ren, X.; Brune, W. H.; Oliger, A.; Metcalf, A. R.; Simpas, J. B.; Shirley, T.; Schwab, J. J.; Bai, C.; Roychowdhury, U.; Li, Y.; Cai, C.; Demerjian, K. L.; He, Y.; Zhou, X.; Gao, H.; Hou, J. J. Geophys. Res.-Atmos. 2006, 111, D10S03.
Sinha, V.; Williams, J.; Lelieveld, J.; Ruuskanen, T. M.; Kajos, M. K.; Patokoski, J.; Hellen, H.; Hakola, H.; Mogensen, D.; Boy, M.; Rinne, J.; Kulmala, M. Environ. Sci. Technol. 2010, 44, 6614. doi: 10.1021/es101780b
Nakashima, Y.; Kato, S.; Greenberg, J.; Harley, P.; Karl, T.; Turnipseed, A.; Apel, E.; Guenther, A.; Smith, J.; Kajii, Y. Atmos. Environ. 2014, 85, 1. doi: 10.1016/j.atmosenv.2013.11.042
Kovacs, T. A.; Brune, W. H.; Harder, H.; Martinez, M.; Simpas, J. B.; Frost, G. J.; Williams, E.; Jobson, T.; Stroud, C.; Young, V.; Fried, A.; Wert, B. J. Environ. Monit. 2003, 5, 68. doi: 10.1039/b204339d
Praplan, A. P.; Pfannerstill, E. Y.; Williams, J.; Hellen, H. Atmos. Environ. 2017, 169, 150. doi: 10.1016/j.atmosenv.2017.09.013
Ren, X. R.; Harder, H.; Martinez, M.; Lesher, R. L.; Oliger, A.; Shirley, T.; Adams, J.; Simpas, J. B.; Brune, W. H. Atmos. Environ. 2003, 37, 3627. doi: 10.1016/S1352-2310(03)00460-6
Shirley, T. R.; Brune, W. H.; Ren, X.; Mao, J.; Lesher, R.; Cardenas, B.; Volkamer, R.; Molina, L. T.; Molina, M. J.; Lamb, B.; Velasco, E.; Jobson, T.; Alexander, M. Atmos. Chem. Phys. 2006, 6, 2753. doi: 10.5194/acp-6-2753-2006
Mao, J.; Ren, X.; Chen, S.; Brune, W. H.; Chen, Z.; Martinez, M.; Harder, H.; Lefer, B.; Rappenglueck, B.; Flynn, J.; Leuchner, M. Atmos. Environ. 2010, 44, 4107. doi: 10.1016/j.atmosenv.2009.01.013
Chatani, S.; Shimo, N.; Matsunaga, S.; Kajii, Y.; Kato, S.; Nakashima, Y.; Miyazaki, K.; Ishii, K.; Ueno, H. Atmos. Chem. Phys. 2009, 9, 8975. doi: 10.5194/acp-9-8975-2009
Kato, S.; Sato, T.; Kajii, Y. Atmos. Environ. 2011, 45, 5531. doi: 10.1016/j.atmosenv.2011.05.074
Yoshino, A.; Nakashima, Y.; Miyazaki, K.; Kato, S.; Suthawaree, J.; Shimo, N.; Matsunaga, S.; Chatani, S.; Apel, E.; Greenberg, J.; Guenther, A.; Ueno, H.; Sasaki, H.; Hoshi, J.-Y.; Yokota, H.; Ishii, K.; Kajii, Y. Atmos. Environ. 2012, 49, 51. doi: 10.1016/j.atmosenv.2011.12.029
Dolgorouky, C.; Gros, V.; Sarda-Esteve, R.; Sinha, V.; Williams, J.; Marchand, N.; Sauvage, S.; Poulain, L.; Sciare, J.; Bonsang, B. Atmos. Chem. Phys. 2012, 12, 9593. doi: 10.5194/acp-12-9593-2012
Fuchs, H.; Tan, Z.; Lu, K.; Bohn, B.; Broch, S.; Brown, S. S.; Dong, H.; Gomm, S.; Haeseler, R.; He, L.; Hofzumahaus, A.; Holland, F.; Li, X.; Liu, Y.; Lu, S.; Min, K.-E.; Rohrer, F.; Shao, M.; Wang, B.; Wang, M.; Wu, Y.; Zeng, L.; Zhang, Y.; Wahner, A.; Zhang, Y. Atmos. Chem. Phys. 2017, 17, 645. doi: 10.5194/acp-17-645-2017
Ren, X. R.; Brune, W. H.; Cantrell, C. A.; Edwards, G. D.; Shirley, T.; Metcalf, A. R.; Lesher, R. L. J. Atmos. Chem. 2005, 52, 231. doi: 10.1007/s10874-005-3651-7
Sinha, V.; Williams, J.; Diesch, J. M.; Drewnick, F.; Martinez, M.; Harder, H.; Regelin, E.; Kubistin, D.; Bozem, H.; Hosaynali-Beygi, Z.; Fischer, H.; Andres-Hernandez, M. D.; Kartal, D.; Adame, J. A.; Lelieveld, J. Atmos. Chem. Phys. 2012, 12, 7269. doi: 10.5194/acp-12-7269-2012
Elshorbany, Y. F.; Kleffmann, J.; Hofzumahaus, A.; Kurtenbach, R.; Wiesen, P.; Brauers, T.; Bohn, B.; Dorn, H. P.; Fuchs, H.; Holland, F.; Rohrer, F.; Tillmann, R.; Wegener, R.; Wahner, A.; Kanaya, Y.; Yoshino, A.; Nishida, S.; Kajii, Y.; Martinez, M.; Kubistin, D.; Harder, H.; Lelieveld, J.; Elste, T.; Plass-Duelmer, C.; Stange, G.; Berresheim, H.; Schurath, U. J. Geophys. Res.-Atmos. 2012, 117, D03307.
Leuchner, M.; Rappenglueck, B. Atmos. Environ. 2010, 44, 4056. doi: 10.1016/j.atmosenv.2009.02.029
Haque, M. M.; Kawamura, K.; Kim, Y. Atmos. Environ.2016, 130, 95. doi: 10.1016/j.atmosenv.2015.09.075
Guenther, A.; Hewitt, C. N.; Erickson, D.; Fall, R.; Geron, C.; Graedel, T.; Harley, P.; Klinger, L.; Lerdau, M.; Mckay, W. A.; Pierce, T.; Scholes, B.; Steinbrecher, R.; Tallamraju, R.; Taylor, J.; Zimmerman, P. J. Geophys. Res.-Atmos. 1995, 100, 8873. doi: 10.1029/94JD02950
Fehsenfeld, F.; Calvert, J.; Fall, R.; Goldan, P.; Guenther, A.; Hewitt, C.; Lamb, B.; Liu, S.; Trainer, M.; Westberg, H.; Zimmerman, P. Global Biogeochem. Cycles. 1992, 6, 389. doi: 10.1029/92GB02125
Atkinson, R.; Arey, J. Atmos. Environ.2003, 37, S197. doi: 10.1016/S1352-2310(03)00391-1
Holzinger, R.; Lee, A.; Paw, K. T.; Goldstein, A. H. Atmos. Chem. Phys. 2005, 5, 67. doi: 10.5194/acpd-4-5345-2004
Mogensen, D.; Smolander, S.; Sogachev, A.; Zhou, L.; Sinha, V.; Guenther, A.; Williams, J.; Nieminen, T.; Kajos, M. K.; Rinne, J.; Kulmala, M.; Boy, M. Atmos. Chem. Phys. 2011, 11, 9709. doi: 10.5194/acp-11-9709-2011
Laothawornkitkul, J.; Taylor, J. E.; Paul, N. D.; Hewitt, C. N. New Phytol. 2009, 184, 276. doi: 10.1111/nph.2009.184.issue-1
杨丹菁, 白郁华, 李金龙, 潘南明, 俞开衡, 唐丽, 彭立新, 苏行, 中国环境科学, 2001, 21, 422. doi: 10.3321/j.issn:1000-6923.2001.05.009Yang. D.-J.; Bai, Y.-H.; Li, J.-L.; Pan, N.-M.; Yu, K.-H.; Tang, L.; Peng, L.-X.; Su, H. China Environ. Sci. 2001, 21, 422(in Chinese). doi: 10.3321/j.issn:1000-6923.2001.05.009
彭立新, 唐孝炎, 白郁华, 李金龙, 中国环境科学, 2000, 20, 132. doi: 10.3321/j.issn:1000-6923.2000.02.009Peng, L.-X.; Tang, X.-Y.; Bai, Y.-H.; Li, J.-L. China Environ. Sci. 2000, 20, 132(in Chinese). doi: 10.3321/j.issn:1000-6923.2000.02.009
徐天莹, 硕士论文, 甘肃农业大学, 2018 http://cdmd.cnki.com.cn/Article/CDMD-10733-1018711496.htmXu, T.-Y. Master Dissertation, Gansu Agricultural University, 2018(in Chinese). http://cdmd.cnki.com.cn/Article/CDMD-10733-1018711496.htm
Lee, J. D.; Young, J. C.; Read, K. A.; Hamilton, J. F.; Hopkins, J. R.; Lewis, A. C.; Bandy, B. J.; Davey, J.; Edwards, P.; Ingham, T.; Self, D. E.; Smith, S. C.; Pilling, M. J.; Heard, D. E. J. Atmos. Chem. 2009, 64, 53. doi: 10.1007/s10874-010-9171-0
白建辉, Guenther Alex; Turnipseed Andrew, 环境科学学报, 2012, 32, 2236. http://www.cqvip.com/QK/91840X/201209/43111208.htmlBai, J.-H; Guenther, A.; Turnipseed, A. J. Environ. Sci.-China 2012, 32, 2236(in Chinese). http://www.cqvip.com/QK/91840X/201209/43111208.html
Fuchs, H.; Tan, Z.; Lu, K.; Bohn, B.; Broch, S.; Brown, S. S.; Dong, H.; Gomm, S.; Haeseler, R.; He, L.; Hofzumahaus, A.; Holland, F.; Li, X.; Liu, Y.; Lu, S.; Min, K.-E.; Rohrer, F.; Shao, M.; Wang, B.; Wang, M.; Wu, Y.; Zeng, L.; Zhang, Y.; Wahner, A.; Zhang, Y. Atmos. Chem. Phys. 2017, 17, 645. doi: 10.5194/acp-17-645-2017
Lu, K. D.; Hofzumahaus, A.; Holland, F.; Bohn, B.; Brauers, T.; Fuchs, H.; Hu, M.; Haeseler, R.; Kita, K.; Kondo, Y.; Li, X.; Lou, S. R.; Oebel, A.; Shao, M.; Zeng, L. M.; Wahner, A.; Zhu, T.; Zhang, Y. H.; Rohrer, F. Atmos. Chem. Phys. 2013, 13, 1057. doi: 10.5194/acp-13-1057-2013
Karl, T.; Guenther, A.; Turnipseed, A.; Tyndall, G.; Artaxo, P.; Martin, S. Atmos. Chem. Phys. 2009, 9, 7753. doi: 10.5194/acp-9-7753-2009
Kim, S.; Guenther, A.; Karl, T.; Greenberg, J. Atmos. Chem. Phys. 2011, 11, 8613. doi: 10.5194/acp-11-8613-2011
Martinez, M.; Harder, H.; Kovacs, T. A.; Simpas, J. B.; Bassis, J.; Lesher, R.; Brune, W. H.; Frost, G. J.; Williams, E. J.; Stroud, C. A.; Jobson, B. T.; Roberts, J. M.; Hall, S. R.; Shetter, R. E.; Wert, B.; Fried, A.; Alicke, B.; Stutz, J.; Young, V. L.; White, A. B.; Zamora, R. J. J. Geophys. Res.-Atmos. 2003, 108, 4617. doi: 10.1029/2003JD003551
武卫荣, 员晓敏, 侯华, 王宝山, 化学学报, 2018, 76, 793. doi: 10.7503/cjcu20170561Wu, W.-R.; Yuan, X.-M.; Hou, H.; Wang, B.-S. Acta Chim. Sinica 2018, 76, 793(in Chinese). doi: 10.7503/cjcu20170561

图 2 OH自由基总反应性直接测量系统的模块
Figure 2 The modules of OH reactivity direct measurement system

图 3 OH自由基总反应性测量不同类型站点分布
Figure 3 The distribution of different types of sites for OH reactivity measurements

图 4 全球典型站点OH自由基总反应性组成(此图中所有物种活性均为计算值,且活性缺失均为OH自由基总反应性测量值与计算值之差)
Figure 4 OH reactivity and calculated contributions in some typical sites all over the world (Here the contributions are calculated results, and the missing reactivity is the difference between the measured and calculated reactivity)
a Calculated contributions from 15:00-17:00 JST on August 20, 2003, b calculated contributions towards the noontime OH reactivity, c calculated contributions above canopy, d calculated contributions after 20 June, 2014

图 5 不同类型站点活性缺失汇总(此图中活性缺失均为OH自由基总反应性测量值与计算值之差; 黑色代表城市站点, 红色代表郊区站点, 蓝色代表森林站点, 空心圆代表不存在活性缺失的情况)
Figure 5 The missing OH reactivity of different types of site (Here the missing reactivity is the difference between the measured and calculated reactivity; open circle represents no missing reactivity)

图 6 不同类型站点OH自由基总反应性、NOx浓度和活性缺失的相对大小关系
Figure 6 Relative relationship of OH reactivity, NOx concentration and the missing reactivity at different types of sites
表 1 OH自由基总反应性测量方法汇总
Table 1. The summary of OH reactivity measurement methods
| Method | Direct measurement method | Indirect measurement method | ||
| Specific methods | Total OH Loss-rate Measurement (TOHLM) | Laser Photolysis-Laser Induced Fluorescence (LP-LIF) | Chemical Ionisation Mass Spectrometry (CIMS) | Comparative Reactivity Method (CRM) |
| OH source | the photolysis of H2O (g) | the photolysis of O3 | the photolysis of H2O (g) | the photolysis of H2O (g) |
| Detectors | LIF | LIF | CIMS | PTR-MS/GC-PID |
| Time resolution/s | 50~200 | 30~180 | 60~300 | 180~300 |
| Detection limit/s-1 | 1 | 1 | 1 | 3~5 |
| Uncertainties | 10%~15% | 10%~15% | 3%~7% | 15%~30% |
| Advantages | (1) high OH radical yield, (2) direct and high accuracy | (1) the little interference, (2) direct and high accuracy | (1) high accuracy | (1) high OH radical yield, (2) well commercialized detector |
| Disadvantages | (1) the interference at NO condition, (2) high cost and complicated operation | (1) low OH radical yield, (2) high cost and complicated operation | (1) narrow measurement range, (2) high cost and complicated operation | (1) the interference at NO condition |
| Reference | 7 | 11 | 13 | 12 |
 下载: 导出CSV
下载: 导出CSV
表 2 全球不同站点OH自由基总反应性测量情况汇总
Table 2. OH reactivity measurements in different sites all over the world
| Campaign | Site | Time | Condition | Instrument | kOH/s-1 | kOH missing | REF |
| PROPHET 2000 | Michigan, USA | summer, 2000 | forest | TOHLM | 1~12a | 33% | 27 |
| PMTACS-NY | New York, USA | summer, 2002 | forest | TOHLM | 5.6b | agreed well | 28 |
| OP3 | Borneo, Malaysian | spring, 2008 | forest | TOHLM | 29.1b | 38% | 26 |
| GABRIEL | Brownsberg, Suriname | autumn, 2005 | forest | CRM | 53b | 65% | 12 |
| BFORM | Hyytiala, Finland | summer, 2008 | forest | CRM | 9b | 50.6% | 29 |
| HUMPPA-COPEC 2010 | Hyytiala, Finland | summer, 2010 | forest | CRM | 3~76a | (1) 58% (normal) (2) 89% (stressed) | 22 |
| BEACHON-SRM08 | Colorado, USA | summer, 2008 | forest | LP-LIF | 6.7b | 29.5% | 30 |
| CABINEX | Michigan, USA | summer, 2009 | forest | TOHLM | 3~33a | (1) 55.8% (6 m) (2) 51.7% (21 m) (3) 57.3% (31 m) | 15 |
| CANOPEE | Haute Provence, France | spring, 2014 | forest | CRM | 24b | (1) agreed well (day) (2) 50% (night) | 24 |
| SOS | Nashville, USA | summer, 1999 | urban | TOHLM | 11b | 29% | 31 |
| — | Helsinki, Finland | winter, 2016 | semi-urban | CRM | 7.6c | 47% | 32 |
| PMTACS-NY2001 | New York, America | summer, 2001 | urban | TOHLM | 19b | within 10% | 33 |
| MCMA 2003 | Mexico City, Mexico | spring, 2003 | urban | TOHLM | 20b | 30% | 34 |
| TEXAQS2000 | Houston, USA | summer, 2000 | urban | TOHLM | 7~12a | agreed well | 35 |
| TRAMP2006 | Houston, USA | summer, 2006 | urban | TOHLM | 9~22a | agreed well | 35 |
| — | Tokyo, Japan | 2003~2004 | suburban | LP-LIF | 10~100a | 30% | 11 |
| — | Tokyo, Japan | summer, 2007 | urban | LP-LIF | 10~55a | 29% | 36 |
| — | Tokyo, Japan | spring, 2009 | suburban | LP-LIF | 10~35a | 20% | 37 |
| — | Tokyo, Japan | summer, 2007 | urban | LP-LIF | 33.4b | 26.5% | 38 |
| — | Tokyo, Japan | autumn, 2009 | urban | LP-LIF | 32.3b | 34.7% | 38 |
| — | Mainz, Germany | summer, 2005 | urban | CRM | 10.4b | — | 12 |
| MEGAPOLI | Paris, France | winter, 2010 | urban | CRM | 10~130a | 54% | 39 |
| — | Lille, France | autumn, 2012 | urban | CRM, LP-LIF | 70b | agreed well | 25 |
| — | Wangdu, China | summer, 2014 | urban | LP-LIF | 10~20a | agreed well | 40 |
| — | Pennsylvania, USA | spring, 2002 | rural | TOHLM | 6.1b | — | 41 |
| TORCH-2 | Weybourne, England | spring, 2004 | rural | TOHLM | 4.9b | 39% | 14 |
| CAREBeijing 2006 | Yufa, China | summer, 2006 | rural | LP-LIF | 10~30a | 67% | 2 |
| PRIDE-PRD | Backgarden, China | summer, 2006 | rural | LP-LIF | 10~120a | 50% | 16 |
| DOMINO | EI Arenosillo | winter, 2008 | rural | CRM | 3.5~84a | — | 42 |
| HOxComp | Jülich, German | summer, 2005 | rural | LP-LIF | 8.8b | 40% | 43 |
| a The range of the measured reactivity during the whole campaign. b The mean of the measured reactivity during the whole campaign. c The median of the measured reactivity during the whole campaign. | |||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
