Received Date:
04 January 2019 Available Online:
15 April 2019
Fund Project:
Project supported by the Funds for Creative Research Group of NSFC (No. 21621005)
Abstract:
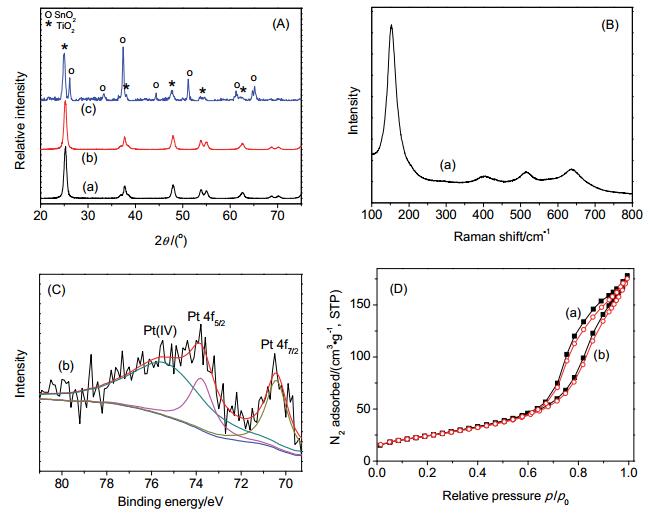
It is known that fluoride and phosphate in aqueous solution can accelerate the photocatalytic degradation of phenol over anatase or P25 TiO2. But the mechanism still remains under debate. In this work, an anion-free anatase TiO2 is prepared, followed by deposition with 0.52 wt% Pt (Pt/TiO2). Reaction was performed in aqueous solution at initial pH 5.2, where 99% of anions were in the form of F- or H2PO4-. On the addition of 0.1~30 mmol/L anions, the rate constants of phenol degradation (kobs) were all increased, confirming the positive effect of fluoride and phosphate, respectively. Interestingly, there was a linear relationship between the increase of kobs and the amounts of anion adsorption, the slope of which became larger in the order of fluoride>phosphate, and Pt/TiO2>TiO2. These observations indicate that the positive effect of anions originates from the adsorbed anions on solid, and that fluoride was more active than phosphate. A (photo)electrochemical measurement showed that fluoride and phosphate were negative and positive, respectively, to O2 reduction, but they were all beneficial to phenol oxidation. Furthermore, in the presence of fluoride and phosphate, the flat band potentials of TiO2 were shifted by -159 and 89 mV, respectively. The former favors orbital overlapping of phenol with TiO2 valence band, and the latter favors orbital overlapping of O2 with TiO2 conduction band, all of which promotes the interfacial charge transfers. Since inorganic anions are widely present, this result would benefit the mechanism study of a semiconductor photocatalyis and its application. As a reference, pure anatase was prepared from the hydrolysis of tetrabutyl titanate, followed by calcination in air at 400℃ for 2 h. The solid was then deposited with Pt, produced in situ from the photocatalytic reduction of H2PtCl6 in the presence of methanol. Solid was characterized with X-ray diffraction, N2 adsorption, Raman, and X-ray photoelectron spectroscopy. After Pt deposition, anatase phase remained unchanged, but the solid pores were blocked by a mixture of Pt and PtO2. Photoreactions were performed at room temperature under UV light at wavelengths equal to and longer than 320 nm. Organic compounds and inorganic anions were quantitatively analyzed with a high performance liquid and ionic chromatography, respectively. (Photo)electrochemical measurement was performed in a three-electrode compartment, where a Pt gauze was used as counter electrode, and a AgCl/Ag as reference electrode.
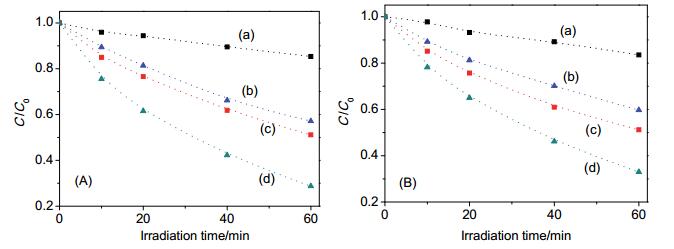
Figure 2.
Effect of fluoride (A) and phosphate (B) on photocatalytic degradation of phenol. Samples were (a) sAT, (b) Pt/sAT, (c) sAT+10 mmol/L anions, and (d) Pt/sAT+10 mmol/L anions
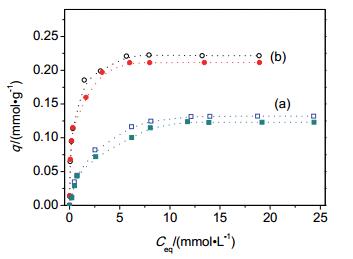
Figure 4.
Concentration and adsorption effect of fluoride (A, A') and phosphate (B, B'), where k0 and kx represent the rate constants of phenol degradation measured in absence and presence of anions, respectively. Samples were (a) sAT, and (b) Pt/sAT
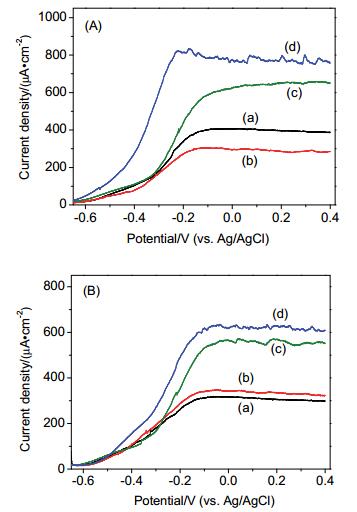
Figure 5.
Photoelectrochemical oxidation of water and phenol in the presence of fluoride (A) and phosphate (B). Experiment was performed under N2 in 0.5 mol/L NaClO4 at pH 5.2, (a) TiO2, (b) TiO2+5 mmol/L anions, (c) TiO2+0.43 mmol/L phenol, and (d) TiO2+0.43 mmol/L phenol +5 mmol/L anions
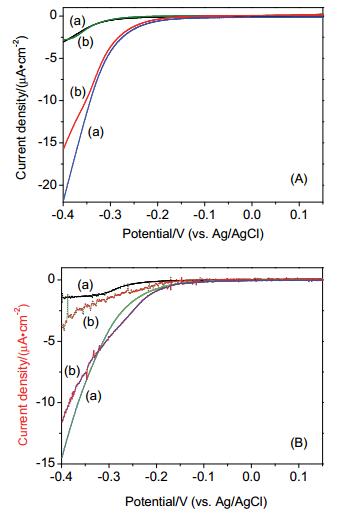
Figure 6.
Effect of fluoride (A) and phosphate (B) on electrochemical reduction of O2
Experiment was performed over a film electrode of (a) TiO2, and (b) TiO2+5 mmol/L anions, in 0.5 mol/L NaClO4 at pH 5.2 under N2 (dotted lines) or O2 (solid lines)
锐钛矿TiO2是n-型半导体, 其导带边电位(ECB)略接近Efb.在pH 0水溶液中, 锐钛矿的ECB和价带边电位(EVB)分别等于-0.12和3.08 V vs. NHE, 而O2和苯酚的单电子氧化还原电位分别为-0.05和1.43 V vs. NHE.因此, O2能被TiO2(ecb-)生成HO2‒∙, 而TiO2(hvb+)既能将苯酚氧化成苯酚阳离子自由基, 也能将水氧化成O2 (1.23 V vs. NHE).但是, 在pH 3.5或pH 5.2水溶液中, 苯酚却比H2O易被hvb+氧化.这可能是苯酚和H2O氧化分别是单电子和多电子过程的缘故.
Chen, K. T.; Lu, C. S.; Chang, T. H.; Lai, Y. Y.; Chang, T. H.; Wu, C. W.; Chen, C. C. J. Hazard. Mater. 2010, 174, 598. doi: 10.1016/j.jhazmat.2009.09.094
Park, H.; Choi, W. J. Phys. Chem. B2004, 108, 4086. doi: 10.1021/jp036735i
[16]
Yu, J. C.; Zhang, L. Z.; Zheng, Z.; Zhao, J. C. Chem. Mater. 2003, 15, 2280. doi: 10.1021/cm0340781
[17]
Zhao, D.; Chen, C. C.; Wang, Y. F.; Ji, H. W.; Ma, W. H.; Zang, L.; Zhao, J. C. J. Phys. Chem. C2008, 112, 5993. doi: 10.1021/jp712049c
[18]
Zhang, X.; Xiong, X. Q.; Xu, Y. M. RSC Adv. 2016, 6, 61830. doi: 10.1039/C6RA10291C
[19]
Xiong, X. Q.; Xu, Y. M. J. Phys. Chem. C2016, 120, 3906.
[20]
Xiong, X. Q.; Zhang, X.; Xu, Y. M. J. Phys. Chem. C2016, 120, 25689. doi: 10.1021/acs.jpcc.6b07951
[21]
Mathpal, M. C.; Tripathi, A. K.; Singh, M. K; Gairola, S. P.; Pandey, S. N.; Agarwal, A. Chem. Phys. Lett. 2013, 555, 182. doi: 10.1016/j.cplett.2012.10.082
Figure 2
Effect of fluoride (A) and phosphate (B) on photocatalytic degradation of phenol. Samples were (a) sAT, (b) Pt/sAT, (c) sAT+10 mmol/L anions, and (d) Pt/sAT+10 mmol/L anions
Figure 4
Concentration and adsorption effect of fluoride (A, A') and phosphate (B, B'), where k0 and kx represent the rate constants of phenol degradation measured in absence and presence of anions, respectively. Samples were (a) sAT, and (b) Pt/sAT
Figure 5
Photoelectrochemical oxidation of water and phenol in the presence of fluoride (A) and phosphate (B). Experiment was performed under N2 in 0.5 mol/L NaClO4 at pH 5.2, (a) TiO2, (b) TiO2+5 mmol/L anions, (c) TiO2+0.43 mmol/L phenol, and (d) TiO2+0.43 mmol/L phenol +5 mmol/L anions
Figure 6
Effect of fluoride (A) and phosphate (B) on electrochemical reduction of O2
Experiment was performed over a film electrode of (a) TiO2, and (b) TiO2+5 mmol/L anions, in 0.5 mol/L NaClO4 at pH 5.2 under N2 (dotted lines) or O2 (solid lines)

 下载:
下载:


 下载:
下载:







