
图 1.
FLPs催化的2, 3-二取代2H-1, 4-苯并噁嗪氢化反应
Figure 1.
Hydrogenation of 2, 3-disubstituted 2H-1, 4-benzoxazines by FLPs

不饱和有机化合物的催化氢化反应以氢气作为反应物, 具有原子经济性高、绿色清洁等特点, 是一类高效的反应, 因而一直备受科学家们的关注[1~8].长期以来, 各国的科研工作者致力于开发不同种类的催化氢化反应体系, 而过渡金属催化剂在该领域一直占主导地位, 并且已经取得了很多辉煌的成就[9~20], 但是这类催化剂一般是钌、铑、铱、钯等贵金属, 价格昂贵, 在工业应用中存在金属残留等问题; 此外这类催化剂还可能被反应体系毒化导致失活, 使反应成本增加, 这些因素制约了以过渡金属为基础的催化剂在催化氢化不饱和化合物领域的发展.因而, 发展新型的氢化反应催化剂具有非常重要的意义. 2006年, Stephan课题组[21]首次发现“受阻路易斯酸碱对”(Frustrated Lewis Pairs, 简称为FLPs) Mes2P-(p-C6F4)-B(C6F5)2 (Mes代表 2, 4, 6- Me3C6H2)可以在25 ℃下使氢气发生异裂, 并且其还原形式(Mes2HP-(p-C6F4)-BH(C6F5)2)可以在100 ℃时释放氢气, 首次实现了非金属催化剂对氢气的可逆活化, 这一研究为长期以来由金属主导的催化氢化领域开辟了全新的途径, 引起各国科研工作者极大的研究兴趣[22~25].截止目前, 以FLPs作为催化剂已成功实现了亚胺、烯胺、酮、烯烃和炔烃等不饱和化合物的氢化[22, 26~35], 但是相对于快速发展的FLPs催化反应而言, 这类反应的机理研究相对滞后, 这在一定程度上限制了这一领域的发展.
在生物医药领域, 3, 4-二氢-2H-1, 4-苯并噁嗪是一类重要的活性化合物, 比如在隐脉白坚木定(obscurinervi- dine), 尼布林宁(niblinine)和左氧氟沙星(levofloxacin)等中[36~39].直接的氢化2H-1, 4-苯并噁嗪是化学合成3, 4-二氢-2H-1, 4-苯并噁嗪最简单和直接的方法.因此我们课题组在之前的研究中利用2.5%的全氟苯基硼(B(C6F5)3简写为BCF)与底物原位形成FLPs顺利实现了3-取代2H-1, 4-苯并噁嗪的氢化反应[40].底物拓展中我们发现当3号位被苯基或者取代的苯基甚至是噻吩取代时, 都能取得较好的转化; 然而, 当底物为2, 3-二取代2H-1, 4-苯并噁嗪时, 在相同的反应条件下, 底物2号位和3号位被不同类型的取代基取代后会导致反应活性的巨大差异.具体来说, 当底物3号位为苯基取代, 2号位为苯基(1o)或者甲基(1p)取代时能得到接近完全转化的顺式产物; 然而当3号位变为甲基取代后(2号位为甲基取代, 1q), 氢化反应完全不能发生, 如图 1所示.很明显, 在FLPs催化的2, 3-二取代2H-1, 4-苯并噁嗪氢化反应中, 3号位取代基会对氢化反应产生巨大的影响.那么, 2, 3-二取代2H-1, 4-苯并噁嗪中3号位取代基是怎样影响氢化反应的, 我们对氢化反应机理进行了进一步的研究.
在本工作中, 我们使用密度泛函理论, 在PCM/ M06-2X/6-311++G(d, p)理论水平上对FLPs催化的2, 3-二取代2H-1, 4-苯并噁嗪氢化反应机理进行了研究.我们以化合物1o、1p和1q为模型化合物建立了氢化反应势能面, 通过比较(1)受阻路易斯酸碱对以及经典路易斯酸碱加合物的稳定性; (2)氢气裂解; (3)氢负离子(H-)转移等过程解释了2, 3-二取代-2H-1, 4-苯并噁嗪氢化反应中3号位取代基对反应活性的影响.我们发现:在氢化反应中, 化合物1o和1p与B(C6F5)3形成的FLPs以及路易斯酸碱加合物的稳定性相当, 在催化体系中路易斯酸碱加合物容易转变为具有催化活性的FLPs; 并且其后的氢气裂解以及氢负离子(H-)转移等过程也很容易发生, 因而氢化反应在室温条件下就很容易发生.然而, 化合物1q与B(C6F5)3容易形成更加稳定的路易斯酸碱加合物, 其不具备催化活性, 不能裂解氢气; 并且因为其与FLPs稳定性相差较大, 在我们的催化体系中并不容易转化为具有催化活性的FLPs化合物, 因而在实验中并没有观察到氢气加成产物.进一步的自然键轨道(Natural Bond Orbital, 简称为NBO)以及Mulliken电荷分析化合物1o, 1p和1q中N4位点所带电荷和直接的计算化合物1o, 1p和1q对质子的亲合能表明: 3号位苯基及甲基取代对N4位的电子效应几乎可以忽略, 从而推测造成化合物1q与B(C6F5)3形成的路易斯酸碱加合物和FLPs稳定性差别较大的原因是化合物1q中3号位甲基位阻太小.随后, 通过计算3号位不同位阻基团取代后形成的FLPs和加合物的稳定性进一步确认位阻效应在氢化反应中起主要作用.这些结果不仅对我们之前的实验结果给出了清晰合理的解释, 而且还可能会为设计新的FLPs催化反应体系提供思路.
在建立2, 3-二取代2H-1, 4-苯并噁嗪被FLPs催化的氢化反应势能面揭示反应机理之前, 我们首先对该反应的反应物与产物的化学计量数进行了研究.根据我们之前的实验研究, 室温条件下, 在甲苯溶液中充入20 bar的氢气, 当B(C6F5)3(5%)存在时, 2, 3-二取代2H-1, 4-苯并噁嗪就可以被H2还原为顺式的3, 4-二氢-2H-1, 4-苯并噁嗪[40], 如图 1可以所示.在本文中, 我们选择反应活性具有较大差别的三种化合物: 1o, 1p和1q作为模型化合物, 甲苯作为溶剂.具体结构如图 1所示, 优化得到的结构见下文.
为了揭示FLPs催化的2, 3-二取代2H-1, 4-苯并噁嗪氢化反应机理我们选用密度泛函M06-2X方法[41]在6-311G(d, p)基组水平上对该反应势能面中各物种的构型进行优化.之所以选用M06-2X泛函首先是因为其在处理有弱相互作用的体系时能取得较好的结果[42, 43]; 其次, 这一方法已经被应用于建立FLPs催化的氢化反应体系势能面, 并且取得了与实验事实相符的结果[44~48]; 为了得到更精确的能量, 我们使用带有弥散的基组6-311++G(d, p)对势能面中包含的各化合物进行了单点能校正; 最后考虑到该反应在甲苯溶液中反应效果最佳, 我们在计算构型以及能量校正过程中也充分考虑了甲苯的溶剂化效应, 使用经典的极化连续介质模型(PCM)进行模拟.为了验证所使用泛函的合理性, 我们在相同的基组水平上使用WB97XD泛函对化合物1o和1q的势能面进行了计算, 得到的势能面与M06-2X泛函的结果类似(图S1).因此本文我们只讨论M06-2X密度泛函在6-311++G(d, p)基组水平上计算的结果, 其中溶剂化效应用PCM模型模拟.
根据文献报道, 在FLPs催化的不饱和亚胺氢化反应中, B(C6F5)3首先会和路易斯碱形成FLPs, 其可以活化氢气形成带有H+和H-的正负离子对化合物, 该化合物中带H+的部分首先将H+转移到不饱和底物, 然后B(C6F5)3再将H-转移到底物中C+中心, 从而使整个氢化反应完成[44, 48], 因此我们也对这些过程进行了研究.
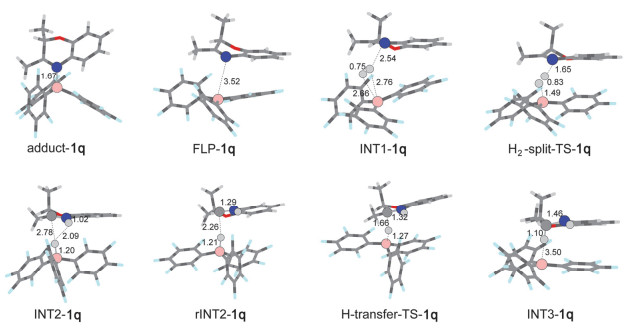
根据我们之前的研究, 在FLPs催化的2, 3-二取代2H-1, 4-苯并噁嗪氢化反应中, B(C6F5)3作为路易斯酸而2, 3-二取代2H-1, 4-苯并噁嗪作为路易斯碱首先形成FLPs[40], 这种底物充当路易斯碱的体系在之前的研究中已有报道[49].分析化合物1o、1p和1q的结构发现:化合物中的氮和氧位点都可以充当路易斯碱与B(C6F5)3形成FLPs, 但是由于氢化反应发生在C3=N4双键上, 因此我们只研究了B(C6F5)3与N4位点形成FLPs后活化氢气并最终还原化合物1o、1p和1q的过程. FLPs催化的化合物1o氢化反应势能面如图 2所示, 势能面中涉及的反应物、中间体、过渡态和产物的优化结构如图 3和图 4所示.势能面中以B(C6F5)3、氢气以及不饱和底物的能量和作为零点参照.
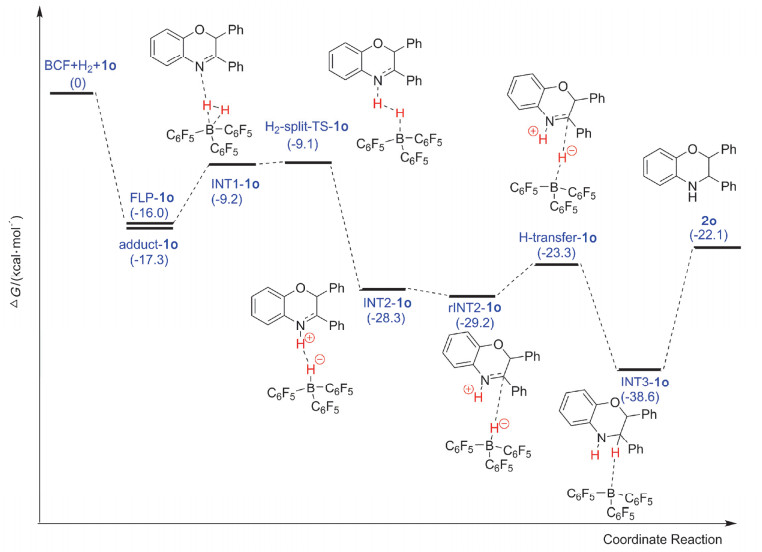
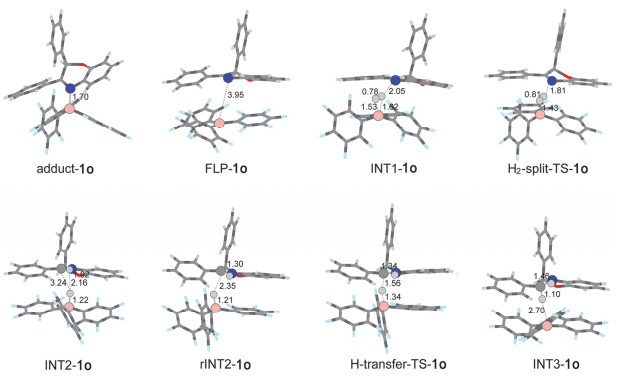
一般认为FLPs催化的氢化反应从FLPs活化氢气开始.因此, 我们首先优化得到了B(C6F5)3与化合物1o中N4位点形成的FLPs化合物(FLP-1o)构型.在FLP-1o中硼-氮键(B—N)的键长为3.95 Å, 和文献报道的B—N FLPs键长一致[44], 并且B—N键的键长明显比B(C6F5)3和化合物1o形成的路易斯酸碱加合物(adduct-1o, 1.70 Å)要长; 同时, FLP-1o中位于B(C6F5)3中心的BC3保持近平面构型, 这表明在FLP-1o中B—N键形成(dative bond).这可以从计算得到的路易斯酸碱加合物(adduct-1o)结构得到验证, 从图 4我们可以清楚地看到adduct-1o中位于B(C6F5)3中心的BC3呈现明显的角度.相对于B(C6F5)3+化合物1o, 形成FLP-1o后放热16.0 kcal/mol, 而形成adduct-1o后也会释放17.3 kcal/mol的能量, 只比FLP-1o低1.3 kcal/mol, 如图 2所示.不难看出B(C6F5)3和化合物1o形成的FLP-1o与adduct-1o在稳定性上相差不大, 这意味着在该体系中FLP-1o与adduct-1o都会存在; 但是在FLPs催化的化合物1o氢化反应中, 只有FLP-1o是具有催化活性的物质, 会对氢化反应产生促进作用, 因此我们推测在反应过程中adduct-1o可能通过分子内重排反应转化为FLP-1o.
随后H2分子被逐步异裂, 首先H2分子会和FLP-1o中的B中心通过配位相互作用形成INT1-1o; 然后INT1-1o会通过过渡态H2-split-TS-1o形成N4被质子化的化合物1o和氢硼酸负离子(hydridoborate anion)两性离子对中间体INT2-1o.在INT1-1o中氢气分子与B(C6F5)3中的B原子以及化合物1o中N4的距离分别为1.53, 1.62和2.05 Å; 氢气分子中两个氢原子之间的距离(0.78 Å)与自由的氢气分子(0.74 Å)相比稍微变长, 这表明在初始形成的INT1-1o中, 氢气分子已经被FLP-1o活化.在此基础上, 随后的氢气裂解过程很容易发生, 只需要经过0.1 kcal/mol的势垒就能发生, 并且伴随有19.1 kcal/mol的能量释放.在过渡态H2-split-TS-1o中, 氢气分子中两个氢原子之间的距离进一步增加到0.81 Å, 部分的B—H键(1.43 Å)和N—H键(1.81 Å)已经形成.在形成的两性离子对中间体INT2-1o中, B—H键(1.22 Å)和N—H键(1.02 Å)完全形成, 氢气被裂解后形成的H+和H-之间的距离为2.16 Å, 这表明在INT2-1o中H2分子已被完全裂解.整个氢气裂解过程需要克服的能垒为6.9 kcal/mol.
在进行氢负离子转移之前, 两性离子对中间体INT2-1o会通过分子内重排反应形成更利于氢负离子转移的异构体rINT2-1o, 该过程伴随有轻微的放热(0.9 kcal/mol).在rINT2-1o中, B—H键会指向化合物1o中阳离子中心, 即HN—C+键中的C+, B—H键中的H-与HN—C+键中的C+之间的距离从INT2-1o中的3.24 Å缩短到rINT2-1o中的2.35 Å.随后, B—H键中的H-就可以经过过渡态H-transfer-TS-1o转移到HN—C+键中的C+上形成中间体INT3-1o, 这一步需要克服5.9 kcal/mol的能垒并伴随有15.3 kcal/mol能量的释放.在过渡态H-transfer-TS-1o中, B—H键被拉长至1.34 Å (在rINT2-1o中B—H键长为1.21 Å), 同时N=C键中的C原子和B—H中H-的距离从rINT2-1o中的2.35 Å缩短到1.56 Å, N=C键长也从1.30 Å轻微拉长到1.34 Å.经过过渡态H-transfer-TS-1o后, B—H中的H-完全转移到化合物1o中N=C键的C上形成中间体INT3-1o.在中间体INT3-1o中, B和H-之间的距离增大到2.70 Å, 相互作用已经很弱, 而C与H之间的距离缩短到1.10 Å形成C—H键, 同时N=C键再次被拉长至1.46 Å, 以单键形式存在.
最后, 因为在中间体INT3-1o中B(C6F5)3与氢气加成之后的化合物1o之间的相互作用已经很弱, 所以中间体INT3-1o会迅速解离形成产物2o, 这个过程相对于INT3-1o需要吸热16.5 kcal/mol.最终形成的产物优化结构如图 3所示, 从图中可以看出氢气是从C=N键的同一面进攻并最终生成顺式产物, 和我们报道的晶体数据完全一致[40], 这表明我们建立的势能面可以描述该反应过程.从势能面图上还可以看出:在298 K, FLPs催化的化合物1o氢化反应放热22.1 kcal/mol, 整个氢化反应很容易发生, 这和我们的实验结果完全一致.
随后, 我们也对化合物1p被FLPs催化的氢化反应势能面进行了计算, 如图S2所示.化合物1p和1o的反应过程基本类似.化合物1p和B(C6F5)3混合后也会首先形成FLP-1p和adduct-1p的混合物, 之后氢气会与FLP-1p中的B原子中心通过配位相互作用形成中间体INT1-1p, INT1-1p经过过渡态H2-split-TS-1p (势垒: 0.3 kcal/mol)将H2裂解形成中间体INT2-1p, 整个H2裂解过程的能垒为6.2 kcal/mol. INT2-1p随后会经过分子内的重排反应形成更容易发生H-转移的中间体rINT2-1p.之后rINT2-1p经过过渡态H-transfer-TS-1p生成INT3-1p, 氢负离子转移过程需要克服的势垒为4.9 kcal/mol.最后INT3-1p会迅速解离为最终的氢化产物2p, 氢化反应放热23.4 kcal/mol.势能面中涉及的反应物、中间体、过渡态以及产物的优化结构如图S3和图S4所示.
化合物1q被FLPs催化的氢化反应势能面如图 5所示.与化合物1o和1p类似, 当B(C6F5)3和化合物1q混合形成FLPs后都会放热, 但是对于化合物1q放热只有11.1 kcal/mol, 比化合物1o和1p分别小4.9和5.0 kcal/mol; 这表明化合物1q与B(C6F5)3形成的FLP-1q没有FLP-1o和FLP-1p稳定.更令我们惊讶的是, 当B(C6F5)3和化合物1q形成经典路易斯酸碱加合物后放热高达22.0 kcal/mol, 这意味着当B(C6F5)3和化合物1q混合后更容易形成经典的路易斯酸碱加合物adduct-1q; 并且adduct-1q比adduct-1o和adduct-1p更稳定.由于FLP-1q与adduct-1q稳定性相差较大(10.9 kcal/mol), 导致FLP-1q和adduct-1q之间并不存在一个平衡, 体系中主要以adduct-1q形式存在, 其在室温下并不能通过分子内的重排转化为FLP-1q.然而在氢化反应中, 有催化活性的是FLP-1q, 这极有可能是导致化合物1q在我们的催化体系中不能被氢化的主要原因.
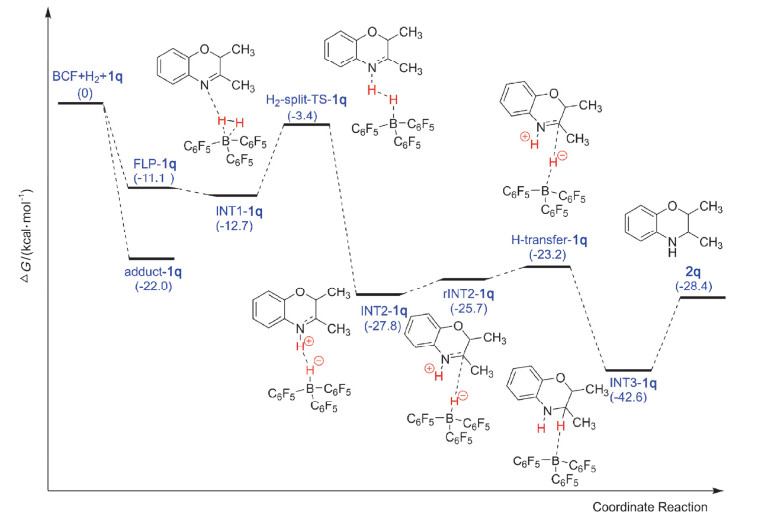
接着, 我们继续对FLP-1q催化的化合物1q氢化反应势能面进行了计算.出乎我们的意料, H2分子并没有像化合物1o和1p中一样与FLP-1q中的B中心通过配位相互作用结合. INT1-1q的结构如图 6所示.在INT1-1q中, H—H键虽然也被轻微拉长(0.75 Å), 表明FLP-1q和H2的相互作用很弱, 但是B…H及N…H的距离明显比INT1-1o和INT1-1p中更大, 分别为2.66, 2.76, 2.54 Å, 造成这种差异的可能原因是FLP-1q活化氢气的方式与FLP-1o和FLP-1p不同.在化合物1q中, 由于N4附近的空间位阻较小, 使得FLP-1q可以与氢气分子充分接触而一步协同裂解氢气, 而在化合物1o和1p中, 由于N4附近有苯环的存在, 其大的空间位阻会阻碍氢气分子接近FLPs中的B中心, 所以一般会分步裂解氢气.这种空间位阻效应改变FLPs活化氢气路径的推测在之前的研究中已被报道[50].随后INT1-1q需要越过9.3 kcal/mol的势垒将氢气裂解形成中间体INT2-1q, 该过程放热15.1 kcal/mol.在化合物1q中氢气裂解需要越过的能垒与化合物1o和1p近似.之后, INT2-1q也会经过分子内重排将B—H键指向化合物1q中HN—C+的C+中心形成rINT2-1q, 但是和化合物1o和1p中(放热, 分别为0.9 kcal/mol和1.1 kcal/mol)不同的是, 化合物1q中的INT2-1q重排需要吸热2.1 kcal/mol.形成的rINT2-1q只需越过2.5 kcal/mol的势垒就能将B—H键中的H-转移到化合物1q中HN—C+的C+中心形成中间体INT3-1q, 在化合物1q中氢负离子转移过程放热高达19.4 kcal/mol, 最后INT3-1q会迅速解离形成最终产物2q(图 7), 整个反应放热28.4 kcal/mol.从势能面图上可以清楚地看出:如果体系中一旦形成了FLP-1q, 氢化反应还是很容易发生的.
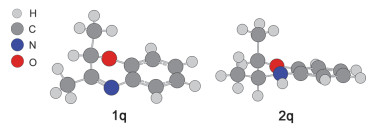
根据我们的计算结果:在298 K时, 对于化合物1o和1p, 其与B(C6F5)3会形成FLPs与经典路易斯酸碱加合物的混合物, 氢气裂解以及H-转移过程都是容易发生的, 氢化反应可以顺利进行, 这和我们之前的实验结果一致.但是对于化合物1q, 虽然氢气裂解以及H-转移过程容易发生, 但是其与B(C6F5)3混合后会形成更加稳定的路易斯酸碱加合物adduct-1q, 而其并不具备催化活性, 因而不能裂解氢气; 此外, 由于adduct-1q和FLP-1q之间能量相差较大, 导致adduct-1q在该反应体系中不能通过分子内重排反应生成具有催化活性的FLP-1q.因此, 我们在实验中并没有检测到化合物1q被氢化后的最终产物2q.那么是什么因素导致化合物1o和1p与化合物1q之间反应活性的巨大差别, 即:是什么因素导致2, 3-二取代2H-1, 4-苯并噁嗪与B(C6F5)3形成的路易斯酸碱加合物和FLPs之间的稳定性不同?
根据我们之前的报道, 当2, 3-二取代2H-1, 4-苯并噁嗪中的3号位被苯环或者其衍生物以及噻吩取代时, 氢化反应能顺利进行, 但是将2, 3-二取代2H-1, 4-苯并噁嗪中的3号位用甲基取代后, 反应完全不发生.很明显, 3号位取代基的变化导致了氢化反应活性的巨大差别.一般的, 取代基会通过电子效应以及位阻效应对反应产生影响.因为在FLPs催化的2, 3-二取代2H-1, 4-苯并噁嗪氢化反应中, 化合物1o, 1p及1q中的N4位点作为路易斯碱中心与B(C6F5)3形成FLPs, N4位点碱性(负电荷)的不同将会导致其与B(C6F5)3形成加合物的能力不同.为了考察3号位取代基对N4位点供电子能力的影响, 我们对化合物1o, 1p及1q进行了自然键轨道(NBO)计算, 分析了C3和N4位点所带电荷.如表 1所示, 在化合物1o, 1p及1q中, C3位点所带电荷为0.292 |e|, 0.290 |e|, 0.319 |e|, 而N4位点所带电荷分别为-0.473 |e|, -0.477 |e|, -0.489 |e|.可以看出3号位不同取代基对C3位点电荷分布的影响相对于N4稍大, 但是整体变化并不明显, 特别是N4位点的变化更小.而Mulliken电荷分析N4位点(-0.371 |e|, -0.391 |e|, -0.354 |e|)也显示了类似的结果.
 下载:
导出CSV
下载:
导出CSV
| NBO charge | Mulliken charge | ||||
| C3 | N4 | C3 | N4 | ||
| 1o | 0.292 | -0.473 | 0.146 | -0.371 | |
| 1p | 0.290 | -0.477 | 0.194 | -0.391 | |
| 1q | 0.319 | -0.489 | 0.110 | -0.354 | |
| -CH2CH3 | 0.324 | -0.492 | 0.120 | -0.349 | |
| -CH(CH3)2 | 0.326 | -0.491 | 0.167 | -0.348 | |
| -C(CH3)3 | 0.326 | -0.498 | 0.227 | -0.360 | |
此外, 我们也直接计算了化合物1o, 1p和1q对H+的亲和能力以评价N4位点碱性的强弱, 我们以H+为能量零点, 分别计算了化合物1o, 1p和1q的中性形式以及N4位点被质子化后阳离子形式的自由能, 得到了质子亲和能(PA).如表 2所示, 化合物1o, 1p和1q对于H+的亲和能分别为254.6, 255.0和255.1 kcal/mol, 几乎相同, 表明这3种化合物中N4位的碱性基本相同, 这和我们通过NBO计算及Mulliken电荷分析得到的N4位点电荷分布结果一致.因此可以推测在FLPs催化的2, 3-二取代2H-1, 4-苯并噁嗪氢化反应中3号位取代基的位阻效应对反应起主要作用, 而电子效应几乎可以忽略.
下载:
导出CSV
| Neutral form(Hartree) | Protonated form(Hartree) | PA (kcal/mol) | |
| 1o | -900.9 | -901.4 | 254.6 |
| 1p | -709.2 | -709.6 | 255.0 |
| 1q | -517.5 | -517.9 | 255.1 |
为了验证我们的猜想, 我们进行了进一步的计算研究.我们将2-甲基-3-取代2H-1, 4-苯并噁嗪中的3号位分别用乙基、异丙基以及叔丁基取代(取代基位阻逐渐增大), 评价3号位取代基位阻效应对FLPs及路易斯酸碱加合物稳定性的影响. NBO计算及Mulliken电荷分析结果表明: 3位乙基、异丙基以及叔丁基取代对N4位点供电子能力影响不大(表 1), 从而排除电子效应对反应的影响.从表 3中可以清楚地看到随着3号位取代基位阻逐渐增大, B(C6F5)3与2, 3-二取代2H-1, 4-苯并噁嗪形成的加合物与FLPs之间的能量差越来越小, 当位阻足够大时, 形成的FLPs甚至会比配位化合物更稳定, 这意味着增大3号位取代基位阻可以促进B(C6F5)3与2, 3-二取代2H-1, 4-苯并噁嗪形成的路易斯酸碱加合物与FLPs平衡的形成或者形成更稳定的FLPs.
下载:
导出CSV
| Gibbs free energy, G | ΔG (Gadduct-GFLP) | ||
| adduct | FLP | ||
| 1q | -23.5 | -12.7 | -10.8 |
| -CH2CH3 | -23.1 | -12.8 | -10.3 |
| -CH(CH3)2 | -14.3 | -15.1 | 0.8 |
| -C(CH3)3 | -7.5 | -12.3 | 5.1 |
| 1p | -19.7 | -18.6 | -1.1 |
此外, 我们计算得到的化合物1o, 1p和1q的质子亲和能也能反映N4位点被质子化后形成的阳离子化合物的稳定性, 其在一定程度上会影响H2-split-TS的稳定性继而影响氢气裂解的势垒.根据文献报道, 路易斯碱对H+的亲和能(PA)与路易斯酸对H-的亲和能(HA)的差值越大越有利于氢气裂解[51].在本文中, 因为体系中的路易斯酸都是B(C6F5)3, 其HA应该相同, 因此可以直接通过PA的大小判断化合物1o, 1p和1q对氢气裂解能力的大小.根据上文得到的PA, 化合物1o, 1p和1q近似相同, 表明其裂解氢气的能力近似相同, 这和我们从势能面中得到的氢气裂解势垒近似相同的结论一致.这些结果进一步表明化合物1q与B(C6F5)3容易形成更稳定的路易斯酸碱加合物是导致其没有反应活性的主要原因.
本文中, 我们通过选择反应活性具有较大差别的模型化合物2, 3-二苯基-2H-1, 4-苯并噁嗪(1o)、2-甲基-3-苯基-2H-1, 4-苯并噁嗪(1p)和2, 3-二甲基-2H-1, 4-苯并噁嗪(1q)对FLPs催化的2, 3-二取代2H-1, 4-苯并噁嗪氢化反应机理进行了研究.我们发现: (1)当3号位为苯基取代时(化合物1o和1p), 其与B(C6F5)3混合后会形成路易斯酸碱加合物与FLPs的混合物, 这两种物质稳定性相差较小容易相互转化; 而当3号位为甲基取代时(化合物1q), 其与B(C6F5)3混合后会形成更加稳定的路易斯酸碱加合物, 其比FLPs稳定10.9 kcal/mol, 这可能是导致化合物1o和1p与1q氢化反应活性有巨大差别的主要原因; (2)随后的势能面计算发现, 无论是没有反应活性的化合物1q还是接近完全转化的化合物1o和1p, 当其与B(C6F5)3形成FLPs之后, 氢气裂解以及H-转移过程都很容易发生, 从而确认了化合物1q与化合物1o和1p氢化反应活性剧烈变化的主要原因就是化合物1q与B(C6F5)3混合后会形成更加稳定的路易斯酸碱加合物, 导致体系中缺少具有催化活性的FLPs物种; (3)进一步的NBO计算, Mulliken电荷分析以及质子亲和能计算表明: 3号位取代基的电子效应对氢化反应的影响可以忽略, 从而推测影响氢化反应活性的因素主要来自于位阻效应; 接着, 通过改变2-甲基-3-取代2H-1, 4-苯并噁嗪中3号位取代基的位阻, 发现随着3号位取代基位阻增大, 底物与B(C6F5)3形成的路易斯酸碱加合物与FLPs稳定性趋于相同, 并且当位阻足够大时, 形成的FLPs甚至比路易斯加合物更稳定; 从而确认了在FLPs催化的2, 3-二取代2H-1, 4-苯并噁嗪氢化反应中, 3号位取代基的位阻效应影响路易斯酸碱加合物与FLPs的稳定性, 并最终影响氢化反应活性.我们的理论研究很好地解释了之前实验研究的结果, 并有望为以后设计新的FLPs催化反应体系提供理论基础.
本文中涉及的所有化合物, 包括反应物、中间体、过渡态以及最终形成的产物构型均在M06-2X/6-311G (d, p)理论水平上获得.通过振动频率分析确认反应物、中间体以及产物没有虚频, 而过渡态有且仅有一个虚频.在此基础上, 我们依据反应途径内禀坐标理论(IRC)对优化得到的过渡态进行了极小能量途径计算, 以正确关联反应物和产物.计算中甲苯的溶剂化效应采用经典的极化连续介质模型(PCM)模拟.为了获得更加精确的能量, 我们对所有物种使用带有弥散的基组6-311++G(d, p)进行了单点能校正, 势能面中所示的能量都经过零点能校正.所有计算都通过Gaussian 09程序包进行[52].
Hey, D. A.; Reich, R. M.; Baratta, W.; Kuhn, F. E. Coord. Chem. Rev. 2018, 374, 114. doi: 10.1016/j.ccr.2018.06.005
Lux, S.; Baldauf-Sommerbauer, G.; Siebenhofer, M. ChemSusChem 2018, 11, 3357. doi: 10.1002/cssc.v11.19
Liu, W. P.; Sahoo, B.; Junge, K.; Beller, M. Acc. Chem. Res. 2018, 51, 1858. doi: 10.1021/acs.accounts.8b00262
Ye, R. P.; Lin, L.; Li, Q. H.; Zhou, Z. F.; Wang, T. T.; Russell, C. K.; Adidharma, H.; Xu, Z. H.; Yao, Y. G.; Fan, M. H. Catal. Sci. Technol. 2018, 8, 3428. doi: 10.1039/C8CY00608C
Song, J. J.; Huang, Z. F.; Pan, L.; Li, K.; Zhang, X. W.; Wang, L.; Zou, J. J. Appl. Catal. B-Environ. 2018, 227, 386. doi: 10.1016/j.apcatb.2018.01.052
Rayhan, U.; Kowser, Z.; Islam, M. N.; Redshaw, C.; Yamato, T. Top. Catal. 2018, 61, 560. doi: 10.1007/s11244-018-0994-2
Orlandi, M.; Brenna, D.; Harms, R.; Jost, S.; Benaglia, M. Org. Process Res. Dev. 2018, 22, 430. doi: 10.1021/acs.oprd.6b00205
Filonenko, G. A.; van Putten, R.; Hensen, E. J. M.; Pidko, E. A. Chem. Soc. Rev. 2018, 47, 1459. doi: 10.1039/C7CS00334J
Meemken, F.; Baiker, A. Chem. Rev. 2017, 117, 11522. doi: 10.1021/acs.chemrev.7b00272
Schauermann, S. J. Phys. Chem. Lett. 2018, 9, 5555. doi: 10.1021/acs.jpclett.8b01782
Meemken, F.; Rodriguez-Garcia, L. J. Phys. Chem. Lett. 2018, 9, 996. doi: 10.1021/acs.jpclett.7b03360
谢建华, 周其林, 化学学报, 2012, 70, 1427. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract341445.shtmlXie, J. H.; Zhou, Q. L. Acta Chim. Sinica 2012, 70, 1427. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract341445.shtml
Yamaguchi, R.; Ikeda, C.; Takahashi, Y.; Fujita, K.-i. J. Am. Chem. Soc. 2009, 131, 8410. doi: 10.1021/ja9022623
Monfette, S.; Turner, Z. R.; Semproni, S. P.; Chirik, P. J. J. Am. Chem. Soc. 2012, 134, 4561. doi: 10.1021/ja300503k
胡书博, 陈木旺, 翟小勇, 周永贵, 化学学报, 2018, 76, 103. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346425.shtmlHu, S. B.; Chen, M. W.; Zhai, X. Y.; Zhou, Y. G. Acta Chim. Sinica 2018, 76, 103. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346425.shtml
张琪, 刘奥, 于海珠, 傅尧, 化学学报, 2018, 76, 113. doi: 10.3866/PKU.WHXB201707101Zhang, Q.; Liu, A.; Yu, H. Z.; Fu, Y. Acta Chim. Sinica 2018, 76, 113. doi: 10.3866/PKU.WHXB201707101
刘旭, 韩召斌, 王正, 丁奎岭, 化学学报, 2014, 72, 849. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract344492.shtmlLiu, X.; Han, Z. B.; Wang, Z.; Ding, K. L. Acta Chim. Sinica 2014, 72, 849. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract344492.shtml
Jiang, W.; Zhao, Q.; Tang, W. Chin. J. Chem. 2018, 36, 153. doi: 10.1002/cjoc.201700645
Xia, J. Z.; Nie, Y.; Yang, G. Q.; Liu, Y. G.; Gridnev, I. D.; Zhang, W. B. Chin. J. Chem. 2018, 36, 612. doi: 10.1002/cjoc.v36.7
张亦伟, 陈艺林, 方霄龙, 袁友珠, 朱红平, 有机化学, 2017, 37, 2275.Zhang, Y. W.; Chen, Y. L.; Fang, X. L.; Yuan, Y. Z.; Zhu, H. P. Chin. J. Org. Chem. 2017, 37, 2275.
Welch, G. C.; Juan, R. R. S.; Masuda, J. D.; Stephan, D. W. Science 2006, 314, 1124. doi: 10.1126/science.1134230
Stephan, D. W. Acc. Chem. Res. 2015, 48, 306. doi: 10.1021/ar500375j
刘勇兵, 杜海峰, 化学学报, 2014, 72, 771. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract344459.shtmlLiu, Y. B.; Du, H. F. Acta Chim. Sinica 2014, 72, 771. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract344459.shtml
Meng, W.; Feng, X. Q.; Du, H. F. Acc. Chem. Res. 2018, 51, 191. doi: 10.1021/acs.accounts.7b00530
王辉, 郑亿, 潘振涛, 傅鸿樑, 凌飞, 钟为慧, 有机化学, 2017, 37, 301.Wang, H.; Zheng, Y.; Pan, Z. T.; Fu, H. L.; Ling, F.; Zhong, W. H. Chin. J. Org. Chem. 2017, 37, 301.
Mömming, C. M.; Frömel, S.; Kehr, G.; Fröhlich, R.; Grimme, S.; Erker, G. J. Am. Chem. Soc. 2009, 131, 12280. doi: 10.1021/ja903511s
Mahdi, T.; Heiden, Z. M.; Grimme, S.; Stephan, D. W. J. Am. Chem. Soc. 2012, 134, 4088. doi: 10.1021/ja300228a
Zhang, Z.; Du, H. Angew. Chem. Int. Ed. 2015, 54, 623.
Liu, Y. B.; Du, H. F. J. Am. Chem. Soc. 2013, 135, 6810. doi: 10.1021/ja4025808
Liu, Y. B.; Du, H. F. J. Am. Chem. Soc. 2013, 135, 12968. doi: 10.1021/ja406761j
Wei, S. M.; Du, H. F. J. Am. Chem. Soc. 2014, 136, 12261. doi: 10.1021/ja507536n
Ren, X. Y.; Du, H. F. J. Am. Chem. Soc. 2016, 138, 810. doi: 10.1021/jacs.5b13104
Fasano, V.; Curless, L. D.; Radcliffe, J. E.; Ingleson, M. J. Angew. Chem.-Int. Ed. 2017, 56, 9202. doi: 10.1002/anie.201705100
Mahdi, T.; Stephan, D. W. J. Am. Chem. Soc. 2014, 136, 15809. doi: 10.1021/ja508829x
Scott, D. J.; Fuchter, M. J.; Ashley, A. E. J. Am. Chem. Soc. 2014, 136, 15813. doi: 10.1021/ja5088979
Brown, K. S.; Djerassi, C. J. Am. Chem. Soc. 1964, 86, 2451. doi: 10.1021/ja01066a031
McAllister, S. D.; Rizvi, G.; Anavi-Goffer, S.; Hurst, D. P.; Barnett-Norris, J.; Lynch, D. L.; Reggio, P. H.; Abood, M. E. J. Med. Chem. 2003, 46, 5139. doi: 10.1021/jm0302647
Wang, A. H.; Prouty, C. P.; Pelton, P. D.; Yong, M.; Demarest, K. T.; Murray, W. V.; Kuo, G. H. Bioorg. Med. Chem. Lett. 2010, 20, 1432. doi: 10.1016/j.bmcl.2009.12.096
Shim, J. Y.; Collantes, E. R.; Welsh, W. J.; Subramaniam, B.; Howlett, A. C.; Eissenstat, M. A.; Ward, S. J. J. Med. Chem. 1998, 41, 4521. doi: 10.1021/jm980305c
Wei, S. M.; Feng, X. Q.; Du, H. F. Org. Biomol. Chem. 2016, 14, 8026. doi: 10.1039/C6OB01556E
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215. doi: 10.1007/s00214-007-0310-x
王英辉, 节家龙, 赵红梅, 白羽, 秦佩萱, 宋迪, 化学学报, 2018, 76, 475. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346556.shtmlWang, Y. H.; Jie, J. L.; Zhao, H. M.; Bai, Y.; Qin, P. X.; Song, D. Acta Chim. Sinica 2018, 76, 475. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346556.shtml
Huang, F.; Jiang, J. L.; Wen, M. W.; Wang, Z. X. J. Theor. Comput. Chem. 2014, 13, 1350074. doi: 10.1142/S0219633613500740
Zhao, J. Y.; Wang, G. Q.; Li, S. H. Dalton Trans. 2015, 44, 9200. doi: 10.1039/C5DT00978B
Rokob, T. A.; Hamza, A.; Papai, I. J. Am. Chem. Soc. 2009, 131, 10701. doi: 10.1021/ja903878z
Antinolo, A.; Carrillo-Hermosilla, F.; Fernandez-Galan, R.; Martinez-Ferrer, J.; Alonso-Moreno, C.; Bravo, I.; Moreno-Blazquez, S.; Salgado, M.; Villasenor, E.; Albaladejo, J. Dalton Trans. 2016, 45, 10717. doi: 10.1039/C6DT01237J
Zhao, L.; Li, H.; Lu, G.; Huang, F.; Zhang, C.; Wang, Z.-X. Dalton Trans. 2011, 40, 1929. doi: 10.1039/c0dt01297a
Rokob, T. A.; Hamza, A.; Stirling, A.; Pápai, I. J. Am. Chem. Soc. 2009, 131, 2029. doi: 10.1021/ja809125r
Das, S.; Pati, S. K. Chem.-Eur. J. 2017, 23, 1078. doi: 10.1002/chem.201602774
Lu, Z. P.; Cheng, Z. H.; Chen, Z. X.; Weng, L. H.; Li, Z. H.; Wang, H. D. Angew. Chem.-Int. Ed. 2011, 50, 12227. doi: 10.1002/anie.v50.51
Gao, S. L.; Wu, W.; Mo, Y. R. Int. J. Quantum Chem. 2011, 111, 3761.
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Ha-segawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannen-berg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 09, Revision A. 01, Gaussian, Inc, Wallingford, CT, 2009.

图 1 FLPs催化的2, 3-二取代2H-1, 4-苯并噁嗪氢化反应
Figure 1 Hydrogenation of 2, 3-disubstituted 2H-1, 4-benzoxazines by FLPs

图 2 在M06-2X/6-311++G(d, p)理论水平上计算得到的FLPs催化化合物1o氢化反应势能面图, 溶剂化效应用PCM模型模拟
Figure 2 Potential energy profile obtained at M06-2X/6-311++G(d, p) level for the hydrogenation of 2, 3-diphenyl-2H-1, 4-benzoxazines by FLPs; the solvent effect is simulated with PCM

图 3 在M06-2X/6-311G(d, p)理论水平上优化得到的氢气(H2)、全氟苯基硼(BCF)及化合物1o和2o的结构, 溶剂效应使用PCM模型模拟
Figure 3 Optimized structures at M06-2X/6-311G(d, p) level for hydrogen (H2), B(C6F5)3 (BCF), compound 1o and 2o; The solvent effect is simulated with PCM

图 4 在M06-2X/6-311G(d, p)理论水平上优化得到的FLPs催化化合物1o氢化反应势能面中所包含的反应物、中间体、过渡态以及产物的构型, 溶剂化效应用PCM模型模拟, 其中浅灰色、深灰色、蓝色以及粉色球分别代表氢原子、碳原子、氮原子以及硼原子
Figure 4 Optimized structures obtained at M06-2X/6-311G(d, p) level for the hydrogenation of 1o by FLPs; the solvent effect is simulated with PCM. Hydrogen, carbon, nitrogen and boron atoms are denoted with lightgray (globule), dark gray (big ball), blue and pink balls, respectively

图 5 在M06-2X/6-311++G(d, p)水平下计算得到的FLPs催化化合物1q氢化反应势能面图, 溶剂化效应用PCM模型模拟
Figure 5 Potential energy profile obtained at the M06-2X/6-311++G(d, p) level for the hydrogenation of 1q by FLPs; the solvent effect is simulated with PCM

图 6 M06-2X/6-311G(d, p)理论水平上优化得到的FLPs催化化合物1q氢化反应势能面中所包含的反应物、中间体、过渡态以及产物的构型, 溶剂化效应使用PCM模型模拟, 其中浅灰色小球、深灰色大球、蓝色以及粉色球分别代表氢原子、碳原子、氮原子以及硼原子
Figure 6 Optimized structures obtained at M06-2X/6-311G(d, p) level for the hydrogenation of 1q by FLPs; the solvent effect is simulated with PCM. Hydrogen, carbon, nitrogen and boron atoms are denoted with light gray (globule), dark gray (big ball), blue and pink balls, respectively

图 7 在M06-2X/6311G(d, p)理论水平上优化得到的化合物1q和2q的结构, 溶剂化效应用PCM模型模拟
Figure 7 Optimized structures at M06-2X/6-311G(d, p) level for complex 1q and 2q; The solvent effect is simulated with PCM
表 1 化合物1o, 1p, 1q以及3号位乙基、异丙基和叔丁基取代的2-甲基-3-取代2H-1, 4-苯并噁嗪中C3和N4位点的电荷分布(|e|)
Table 1. Charge distribution (|e|) of C3 and N4 site of compound 1o, 1p and 1q as well as 2-methyl-3-substituted 2H-1, 4-benzoxazine
| NBO charge | Mulliken charge | ||||
| C3 | N4 | C3 | N4 | ||
| 1o | 0.292 | -0.473 | 0.146 | -0.371 | |
| 1p | 0.290 | -0.477 | 0.194 | -0.391 | |
| 1q | 0.319 | -0.489 | 0.110 | -0.354 | |
| -CH2CH3 | 0.324 | -0.492 | 0.120 | -0.349 | |
| -CH(CH3)2 | 0.326 | -0.491 | 0.167 | -0.348 | |
| -C(CH3)3 | 0.326 | -0.498 | 0.227 | -0.360 | |
 下载: 导出CSV
下载: 导出CSV
表 2 M06-2X/6-311++G(d, p)计算水平下得到的化合物1o, 1p和1q中性和质子化形式的能量以及对H+的亲和能, 其中以H+作为能量零点
Table 2. Computed energy of compound 1o, 1p and 1q in both neutral and protonated form as well as the proton affinities (PA) for 1o, 1p and 1q, respectively, at M06-2X/6-311++G(d, p) level with the energy for proton being taken as zero
| Neutral form(Hartree) | Protonated form(Hartree) | PA (kcal/mol) | |
| 1o | -900.9 | -901.4 | 254.6 |
| 1p | -709.2 | -709.6 | 255.0 |
| 1q | -517.5 | -517.9 | 255.1 |
下载: 导出CSV
表 3 2-甲基-3-取代2H-1, 4-苯并噁嗪3号位不同取代基取代时的吉布斯自由能以及与B(C6F5)3形成的路易斯酸碱加合物和受阻路易斯酸碱对化合物吉布斯自由能及差值, 其中以B(C6F5)3与各化合物的能量和作为能量零点, 单位为kcal/mol
Table 3. The Gibbs free energies (kcal/mol) of 2-methyl-3-substituted 2H-1, 4-benzoxazine, The free energy is relative to that of the B(C6F5)3+2-methyl-3-substituted 2H-1, 4-benzoxazine
| Gibbs free energy, G | ΔG (Gadduct-GFLP) | ||
| adduct | FLP | ||
| 1q | -23.5 | -12.7 | -10.8 |
| -CH2CH3 | -23.1 | -12.8 | -10.3 |
| -CH(CH3)2 | -14.3 | -15.1 | 0.8 |
| -C(CH3)3 | -7.5 | -12.3 | 5.1 |
| 1p | -19.7 | -18.6 | -1.1 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们