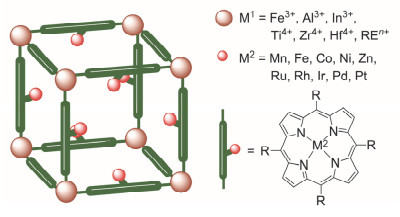
图 1.
卟啉金属-有机框架示意图
Figure 1.
Schematics of porphyrin metal-organic frameworks

近年来, 温室效应的加剧导致了许多全球性环境与气候问题的出现, 人们开始着手研究以二氧化碳(CO2)为主的温室气体的处理方法[1~10]. CO2捕捉和储存(CO2 capture and storage, CCS)是降低CO2含量的一种有效方法, 主要步骤包括:将在燃烧或其他工业过程中产生的CO2选择性移除、将其加压到超临界态、然后运输至“注射站”、最终在地下或水下永久储存[11]. CCS技术已取得飞速发展, 但仍然存在一些难题亟待解决.例如, 捕获过程中需要消耗大量能量, 成本居高不下; 安全隐患仍然存在, 储存时依靠地壳或水对CO2施加巨大的压力, 若发生泄漏, 埋存的CO2将重新回到大气中或对海洋和陆地的生态造成威胁[12].与CCS技术的最终目的不同, CO2捕获与转化(CO2 capture and conversion, C3)旨在将捕获得到的CO2转化为有用的产品[13, 14].尽管CO2在热力学上极其稳定, 发生反应需要消耗大量能量, 但其作为各类化学品和小分子燃料的原料, 具有无毒无害、化学性质稳定、廉价等优点, 因此具有成为可再生能源的巨大潜力.
金属-有机框架(Metal-Organic Frameworks, MOFs), 又称多孔配位聚合物(Porous Coordination Polymers, PCPs), 是一种由配体(或者金属有机配体)与金属节点组成的多孔配位聚合物[15]. MOFs具有长程有序的晶体结构, 以及合成简便、结构可设计调节、比表面积大等特点, 在客体分子的吸附、分离和催化等各个领域都有广泛应用[16].如图 1所示, 卟啉金属-有机框架(Porphy- rin Metal-Organic Frameworks, PMOFs)是一种基于卟啉或金属卟啉配体的多孔材料[17].卟啉配体具有良好的热、化学稳定性以及优异独特的光学性能.在卟啉环中心可以引入各种金属元素, 此外, 在卟啉环周围或环上可以引入具有位阻或电子效应的官能团[18].通过以上结构改造, PMOFs能够参与各类催化反应.从第一例基于卟啉配体的配位聚合物的成功合成[19]到去除客体分子后能保持框架稳定性的PMOFs的发现[20], 从对结构特性的单纯阐述到对PMOFs性能的深入研究, PMOFs已是当今的热门研究领域.
结合PMOFs的合成策略、结构特征和稳定性研究, 简要综述了近几年PMOFs在CO2捕获与转化上的应用, 主要包括含CO2混合气体的选择性吸附和分离、有机化学反应(主要为环加成反应)、光催化反应和电催化反应, 旨在归纳PMOFs在CO2捕获与转化中的优势和挑战, 并对PMOFs在这个学术研究领域的前景进行了展望.
构筑策略的发展与进步不仅推动了具有各类拓扑结构的PMOFs合成, 更是促进了PMOFs的特色催化应用研究.下文主要介绍拓扑导向、柱层和金属-有机笼三种构筑策略.通过这三种策略, 合成了一大批各具特色的PMOFs材料.
拓扑导向策略指在PMOFs的构筑过程中, 保持原有的拓扑结构, 对框架中的有机配体(包括金属有机配体)或金属簇进行调整, 有目的地进行功能化PMOFs的合成.通过这种策略: (1)可以对PMOFs的孔道大小进行调整, 例如改变卟啉配体中端基的长度; (2)可以对PMOFs材料的稳定性进行优化, 例如通过使用不同Lewis酸性的金属构筑金属簇, 从而调节金属簇与配体间配位键的键能大小; (3)可以对PMOFs的催化性能进行调控, 例如通过在卟啉环中心引入不同的金属离子.
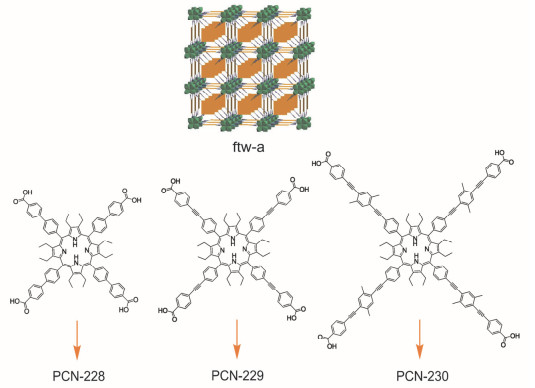
通过延长四羧酸卟啉的端基, Zhou等[21]合成了一系列具有ftw-a拓扑结构但孔道大小不同的PMOFs (PCN-228、PCN-229、PCN-230, PCN=Porous Coordination Framework). ftw-a网络是由具有12-连接Oh对称性的金属节点和4-连接D4h对称性的配体组装而成(图 2).孔洞大小随着羧酸卟啉配体端基的延长而增大, 三者的孔洞大小分别是2.5, 2.8和3.8 nm.孔道大小中等的PCN-229具有最高的BET比表面积与孔隙率.值得一提的是, 框架的稳定性并没有随着配体的延伸而下降, PCN-230表现出了极高的稳定性.
MOFs的化学稳定性与金属离子和配体间的配位键强度有很大的关联性, 主要是基于“软硬酸碱规则”.通过改变金属离子和配位阴离子的硬度, 可以调节PMOFs的稳定性.例如, 由硬酸高价金属离子和硬碱羧酸配体构筑的PMOFs在酸性环境下, 甚至强酸环境下, 都可以保持稳定, 但是在碱性环境下, 羧酸配体容易被取代, 框架坍塌. Zhou和Li等[22~24]发现使用软酸金属离子Ni2+盐和软碱含氮端基卟啉配体TPP [5, 10, 15, 20-四(吡唑-4-基)卟啉]、TPPP [5, 10, 15, 20-四(4-(1-氢-吡唑-4-基)苯基)卟啉]和TFPPPP [2, 3, 5, 6-四氟-4-(1-氢-吡唑-4-基)苯基)卟啉]构筑得到的PCN-601、PCN-602和PCN-624, 在强碱水溶液中保持了很高的稳定性, 拓宽了PMOFs催化剂的应用范围.
通过在卟啉环上引入不同种类的金属中心, 可以调控PMOFs的催化性能[25~31]. Zhou等[25]使用Zr4+盐和基于羧基端基的配体TCPP [[5, 10, 15, 20-四(4-羧基苯基)卟啉]合成了以过渡金属(Ni、Co、Fe)为中心的PCN-224.我们课题组近年来使用Zr4+盐/Hf4+盐和MTCPP (M=Ir[26~28]、Rh[29, 30]、Ru[31])配体合成了一系列具有与PCN-224相同的she拓扑结构但是以贵金属为中心的M-PMOF-1(图 3), 分别在有机催化, 包括CO2环加成反应和X—H (X=N, O, Si)插入反应, 与CO2光催化还原反应中具有优异的催化特性.
大部分具有柱层式结构的PMOFs由二维层状结构及连接两个层状结构的柱子(通常为联吡啶及其衍生物)组成.代表性的柱层式PMOFs大致分为两类, 一类是由Choe等[32]合成的PPFs系列(PPFs=Porphyrin Pad-dlewheel Frameworks), 一类是Hupp, Nguyen, Farha等[33]发展的用吡啶基卟啉作为柱子的系列RPMs (RPMs=Robust Porphyrin Materials).
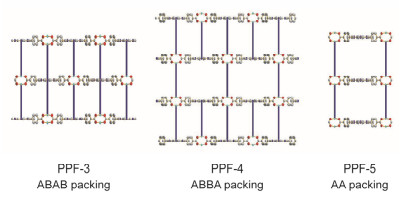
PPFs系列的PMOFs由金属卟啉配体、浆轮状M2(COO)4次级结构基元(Secondary Building Unit, SBU)和联吡啶柱组成. PPF-3、PPF-4和PPF-5是其中最具有代表性的结构[32].如图 4所示, 在反应过程中, 金属卟啉配体先与金属硝酸盐反应得到二维的片状结构, 4, 4'-联吡啶(BPY)再与片状结构相连, 最终得到PPF-3、PPF-4、PPF-5. BPY既可以与次级结构单元的金属也可以与卟啉配体中心的金属离子配位, 因此, 卟啉配体中心的金属离子的配位方式决定了框架的堆积方式, 包括ABAB(PPF-3)、ABBA(PPF-4)和AA(PPF-5).
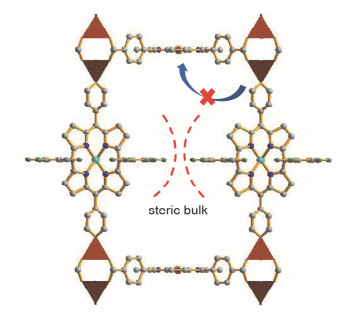
与PPFs使用联吡啶作为柱子不同, Hupp, Nguyen和Farha等[33]利用二吡啶基卟啉配体作为柱子合成了RPMs系列框架.当使用简单的二吡啶基卟啉配体来构筑RPMs时, 浆轮状M2(COO)4次级结构基元和卟啉环金属中心都会参与配位.而当使用具有较大位阻的二吡啶基卟啉配体作为连接柱子时, 可以避免吡啶端基与层状结构中的卟啉环金属中心发生配位作用(图 5).
基于金属卟啉配体的金属-有机笼(Metal-Organic Cage, MOC), 笼壁富含π电子与活性金属中心, 两者协同能与底物或目标客体分子发生良好的相互作用.基于上述观点, 将MOCs引入PMOFs中, 得到的框架具有可设计的孔道, 同时具有良好的气体吸附和催化性质.
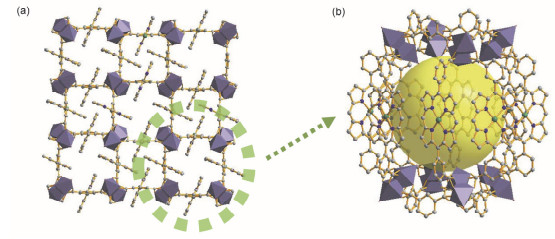
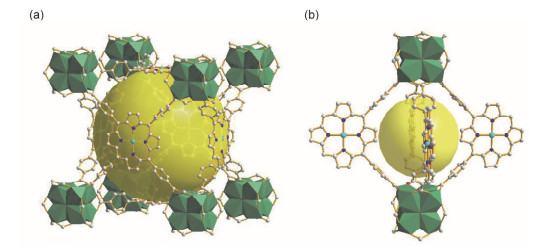
Ma等[34]于2011年合成的MMPF-1, 是第一例在框架中包含纳米级MOCs的PMOF(图 6).作者选择了特殊的四羧基卟啉配体BDCPP (5, 15-双(3, 5-二羧基苯基)卟啉)与Cu2(COO)4 SBU构筑框架. MMPF-1具有类lvt拓扑结构, 其框架可视作由无数个MOCs通过“ABAB”模式堆积而成.每一个MOC由8个Cu2(COO)4与16个BDCPP配体相互连接而成, 因此, 这样的MOC中具有高密度的活性金属位点.
具有高度对称性拓扑结构的PMOFs, 例如PCN- 221[35]和先前讨论的PCN-228, PCN-229和PCN-230, 在框架内部也存在MOCs. Zhou等[35]使用TCPP配体和ZrCl4的溶剂热反应合成了这一具有高度对称性ftw-a拓扑结构的框架PCN-221(图 7).框架中存在两种MOCs, 一种是正方体笼, 由8个位于顶点的Zr8簇与6个平面上的TCPP配体组成, 孔道直径约为2.0 nm; 另一种是八面体笼, 由2个Zr8簇与4个TCPP配体组成, 孔道直径约为1.1 nm.
利用上述拓扑导向、柱层、金属-有机笼等三种构筑策略合成了一大批各具特色的PMOFs材料, 促进了PMOFs领域的飞速发展.在三种构筑策略中, 由柱层或金属-有机笼策略构筑的PMOFs往往稳定性不高, 主要原因是这些PMOFs的簇中金属往往为软酸, 金属簇与有机配体之间的配位键容易被破坏.拓扑导向策略在金属簇及卟啉配体的种类上有更多的选择, 因此得到的PMOFs往往具有较高的稳定性.高稳定性是PMOFs材料在各类领域得到应用的基础.另一方面, 通过金属-有机笼策略构筑得到的PMOFs的孔道通常由多个金属卟啉配体包围而成, 活性金属位点密度较高, 因此拥有优异的气体吸附和催化性质.
本文选取了一些具有代表性的PMOFs, 根据次级结构基元(SBUs)进行系统分类, 包括基于低价态金属离子、桨轮状M2(COO)4、金属-氧无限长链和硬酸离子金属-氧簇四类, 叙述了各类PMOFs的结构特性和稳定性.
早期研究中, 用于构造PMOF的SBU主要是低价态的金属离子, 例如1991年Robson等[19]利用Cd2+构筑的PMOF.每一个Cd2+与两个顺式排列的
卟啉配体之间也可通过卟啉中心的金属离子与另一卟啉的端基配体的配位键进行连接. 1999年, Lin等[20]首次报道了除去客体分子后能稳定存在的SMTP-1 (SMTP=Supramolecular Materials in Taiwan porphyrin).框架中卟啉中心的Mn与相邻两个卟啉配体的端基吡啶配位, 通过这种方式六个卟啉配体围成了直径为20 Å的孔道.这样的六边形结构可在一个平面内无限延伸, 相邻的两个卟啉层平面间由π-π共轭作用进行堆积.
M2(COO)4桨轮状结构主要出现于柱层式PMOFs中, 基于MOCs的PMOFs中也有桨轮状SBU的出现.这样的浆轮状SBU通常在卟啉层中起着连接相邻卟啉配体的作用, 例如Choe等[32]合成的PPF系列.
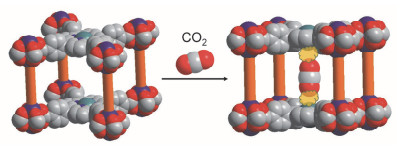
基于金属-氧无限长链SBU的MOFs往往在去除客体分子后仍能保持孔道的结构, 因此这类MOFs在实际应用中也具有很大的潜力. 2011年, Rosseinsky等[36]合成了一种以Al—O长链作为SBU的Al-PMOF(图 8).长链中每一个重复结构单元Al(OH)O4由1个Al原子、4个TCPP配体上羧基的O原子以及1个连接相邻两个Al原子的μ2-OH组成. Al-PMOF在水中拥有极好的稳定性.类似地, Stock等[37]利用与Al同族的Ga和In的氧化物长链作为SBU构筑了Ga-PMOF和In-PMOF.在氧化物长链中, Al、Ga、In三种金属都是六配位的. Jiang等[38]使用微溶的In(OH)3作为金属前体合成了基于In—O长链的USTC-8(M) (USTC=University of Science and Technology of China, M=In, Cu, Co, Ni).当金属中心为In时, 由于In3+较大的半径及轴向上与之配位的羟基的作用, In中心暴露于卟啉平面之外, 因此具有独特的催化性质.

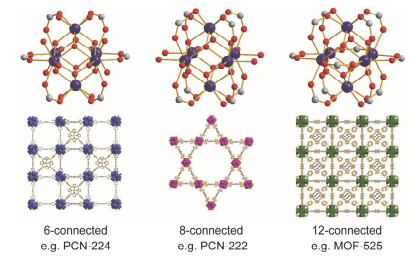
根据Pearson的软硬酸碱理论, 羧酸根离子(COO-)属于硬碱, 与软酸金属离子(如Zn2+、Cu2+、Cd2+)的配位能力较弱, 而与硬酸金属离子(例如Cr3+、Fe3+、Ti4+、Zr4+、Hf4+和稀土金属离子REn+)的配位能力很强[39].因此, 基于硬酸金属-氧簇的MOFs的稳定性大大提升[40].近年来, Yaghi和Morris[41], Zhou[21, 25, 35, 42~44], Ma[45], Farha、Hupp、Stoddart和Snurr[46], Feng和Bu[47], Su和Wu[48], Du和Fang[49], 和我们课题组[26~31]等先后合成了基于Zr/Hf-O簇的PMOFs. Zhou等通过调整反应条件如Zr4+/Hf4+和卟啉配体的比例以及苯甲酸等可以减慢自组装反应的添加剂的量, 合成得到了一系列基于Zr6簇但成键数目和拓扑结构不同的PMOFs(图 9).其中包括具有(4, 6)-连接的she拓扑结构的PCN-224[25], (4, 8)-连接的csq结构的PCN-222[42], (4, 8)-连接的sqc结构的PCN-225[43], (4, 12)-连接的shp-a结构的PCN-223[44], 以及(4, 12)-连接的ftw-a结构的PCN-228[21]等(括号内的两个数字分别代表卟啉配体的成键数以及Zr6簇的成键数).上述一系列基于Zr/Hf-O簇的PMOFs具有多孔结构(孔道直径高达3.7 nm)、高热稳定性(250~500 ℃)以及高化学稳定性(在不同pH的水溶液及各种有机溶剂中都能稳定存在).
除了Zr4+和Hf4+外, Fe3+和Ti4+等硬酸也能应用于PMOFs的构筑. Zhou等先后用上述两种硬酸构筑了PCN-600[50]和PCN-22[51]. Fe3+对人体的毒性较低, 在自然界中含量高, 以Fe3+构建的PCN-600在水中具有很好的稳定性.
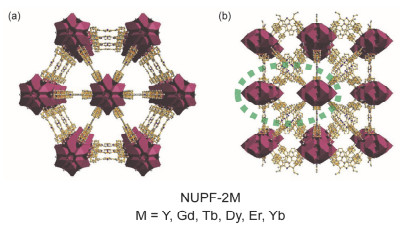
稀土金属离子REn+(n多为3或者4), 与羧酸根离子的O配位能力强, 结合稀土金属具有独特的光、电、磁性质, 通过稀土金属簇与卟啉配体的结合, 可以得到结构稳定、性能优异的PMOFs材料. 2016年, Du等[52]合成了具有shp-a拓扑结构的NUPF-2M (NUPF=Nanjing University Porphyrinic Framework, M=Y, Gd, Tb, Dy, Er, Yb; 图 10).以NUPF-2Y为例, 每个卟啉配体TCPP与四个Y9簇相连, 每个Y9簇与12个TCPP配体相连.框架中存在两种通道, 一种是直径为1.2 nm的三角形孔道, 另一种是直径为0.9 nm的菱形孔道. NUPF-2Y具有较大的BET比表面积(1948 m2•g-1)和极高的热稳定性(高于500 ℃时也能稳定存在).
基于浆轮状M2(COO)4的PMOFs一般是通过柱层策略或者金属-有机笼策略构筑得到.由于M2(COO)4中金属离子一般为低价态的Cu2+、Zn2+、Ni2+等, 与基于羧基端基的卟啉配体间形成的配位键容易被破坏, 这类PMOFs的稳定性相对较低.基于金属-氧无限长链和硬酸金属-氧簇的PMOFs通常由拓扑导向策略构筑得到, 它们通常具有较高的稳定性, 在气体的吸附与催化应用中具有明显优势.
过去二十多年, 结构稳定、孔隙率高、比表面积大的MOFs在CO2捕获与转化上的应用研究取得了很大的进展[53].相比之下, PMOFs在C3的应用仍处于起步阶段, 研究成果相对较少.本文选取了部分例子来讨论PMOFs在C3的应用, 包括有机反应(主要是环加成)、光催化还原和电催化还原反应, 阐述中穿插PMOFs作为催化剂的结构要求及所需表征手段.
MOFs中CO2的吸附位点可分为开放金属位点、Lewis碱位点和其他官能团位点.然而, 对CO2具有强吸附性的MOFs材料对其他小分子气体例如N2、CH4、CO、O2等的作用力也很强, 因此提高MOFs吸附CO2选择性仍然是一项具有挑战性的课题.
CO2是一种直线型分子, 碳原子和氧原子分别带部分正电荷和负电荷, 据此, Zhou等提出在MOFs的构筑过程中预先设计一对距离合适、带有正电荷的“锚”与两个氧原子相互作用, 能够提高MOFs对CO2的吸附能力.如图 11所示, 由两个Cu2(COO)4浆轮状SBU和四个二元羧酸配体组成, 通过配位不饱和的金属与氧原子配位的结构, 被命名为单分子肼(Single Molecular Trap, SMT)[54].
单分子肼策略被Zhang等[55]运用到UNLPF-2 (UNLPF=University of Nebraska-Lincoln Porous Frame- work)的构筑中.作者先在剑桥结构数据库(Cambridge Structure Database, CSD)中搜寻到两种同为pts拓扑结构的MOFs, 然后使用八元羧酸卟啉配体BCPPP (3, 5-二(4-羧基苯基)苯基卟啉)配体构筑了拓扑结构相同且两个配位不饱和的金属(M-M)距离减小的UNLPF-2.傅里叶变换红外光谱(FT-IR)、气相色谱(GC)等表征证明CO2分子被UNLPF-2捕获了.作者随后通过一系列控制实验表明, 框架中捕获的CO2分子是由N, N-二甲基甲酰胺(DMF)溶剂分子分解后被框架原位捕获的, 反应环境中的Lewis酸例如乙酸和Co(NO3)2能促进此分解反应的进行.
基于MOCs的PMOFs具有富含π电子与活性金属中心的多分子笼, 增强了与CO2的相互作用, 因此这样的框架具有良好的CO2吸附性质. Ma等[56]利用八羧基卟啉配体TDCPP (5, 10, 15, 20-四(3, 5-双羧基苯基)卟啉)与Zn(NO3)2合成了具有pcu拓扑结构的MMPF-4 (M=Zn).框架中每个菱形八面体MOC由6个Zn-TDCPP配体与8个Zn2(CO2)3构成, 内直径为21.5 Å, 窗口尺寸为7.8 Å×8.0 Å.此外, 框架中八面体MOCs通过“AA”堆积模式, 形成了另一种由8个Zn2(CO2)3围成的孔, 内直径为11.2 Å, 窗口尺寸为8.0 Å×8.0 Å.通过测量不同气体的吸附等温线, 以及应用理想吸附溶液理论(the Ideal Adsorption Solution Theory, IAST)计算框架对模拟燃烧后尾气(15% CO2和85%和N2)选择性吸附, 作者证明了构筑这样的多分子笼是增强CO2选择性吸附的有效策略.
CO2与环氧乙烷的环加成反应的产物环状碳酸酯是制药和化工行业中非常有价值的化合物, 可以作为高分子合成的前体, 亦可作为锂电池的电解液和非质子性溶剂[57].许多MOFs中存在大量配位不饱和的金属离子, 这样的Lewis酸位点对CO2的环加成反应有非常良好的催化作用.
2013年, Zhou等[25]合成了一种基于Zr-O簇的框架PCN-224.作者在TCPP配体的合成过程中预先在卟啉中心插入金属, 金属化得到的Co-TCPP再进行溶剂热反应合成得到PCN-224(Co).在100 ℃和2 MPa CO2的条件下, 在高压釜中加入环氧丙烷、四丁基氯化铵以及0.1 mol% PCN-224(Co)进行催化反应, 反应时间为4 h. PCN-224(Co)能进行三轮催化反应, 三轮反应的转化率分别为42%、33%和39%, 对应的转化频率(TOF)依次为115、104和129 h-1, 催化效果与均相钴卟啉催化剂相当.
具有π-π堆积结构的PMOFs由于拥有独特的孔道性质, 也能对环加成反应进行催化. Ma等[58]设计合成的MMPF-18由Zn(NO3)2•6H2O和BCPP (5, 15-二(4-羧基苯基)卟啉)配体的溶剂热反应合成得到.由于BCPP配体的π-π堆积作用较强, 结构中出现四重穿插的结构, 孔道面积大大减小, 促进了CO2/CH4选择性吸附.同时, 孔道中锌卟啉的密度增大, 使得MMPF-18也能在常温常压下催化环加成反应.作者还报道了该反应对底物具有尺寸选择性作用, 该作用是由底物自身特性和底物在狭窄孔道中的缓慢扩散共同导致的.
基于稀土金属RE6簇(RE=Eu, Y, Yb, Tb, Dy)的PCN-900也能应用到此反应中[59]. PCN-900具有tam拓扑结构.通过在RE6簇之间引入二元羧基配体CoBPYDC (BPYDC=2, 2'-联吡啶-4, 4'-二甲酸), 合成得到的PCN-900(Eu)-CoTCPP-CoBPYDC具有催化CO2环加成反应的多种活性位点.催化反应可以在50 ℃和常压的条件下进行, 反应可以循环三轮.
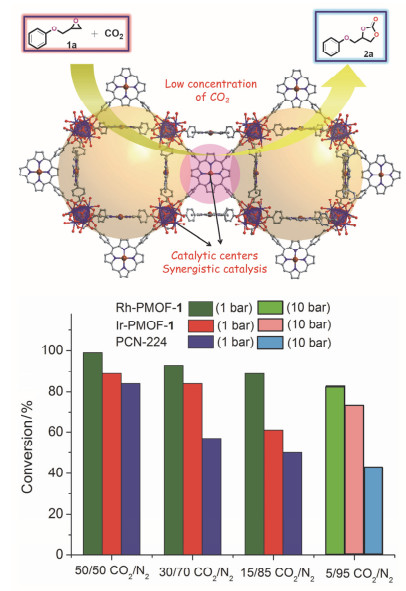
我们课题组[29]报道了PMOFs材料在低浓度CO2环加成反应中的应用(图 12).我们合成了两种同为she拓扑结构的Rh-PMOF-1和Ir-PMOF-1, 常压下以2-苯氧甲基环氧丙烷为底物、nBu4NBr为共催化剂时, 100 ℃下反应24 h后, Rh-PMOF-1和Ir-PMOF-1的转化率分别为99%和79%, Rh-PMOF-1能在此反应条件下进行10轮循环实验.温度下降至室温(25 ℃)时, Rh-PMOF-1和Ir-PMOF-1的转化率分别下降至65%和30%. CO2的浓度降低至5%(其余为N2)、反应温度为100 ℃、Rh-PMOF-1作为催化剂时, 72 h后转化率仍低于15%.利用高压反应器将反应压力从常压提高到10 bar时, 3 h后转化率达到83%.通过Rh-PMOF-1与均相催化剂Rh(TPP)Cl和具有相同she拓扑结构但卟啉中心没有金属中心的PCN-224的催化数据进行比较, Rh-PMOF-1具有明显的催化优势, 这主要得力于金属卟啉配体和Zr-O簇两种反应活性位点的协同作用, 以及框架中有序多孔结构对CO2的浓缩效应.
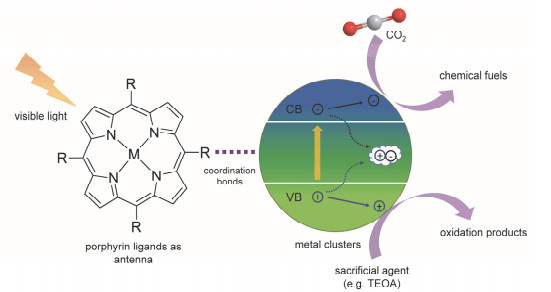
可见光驱动的CO2光催化反应具有以下优点:反应条件温和, 通常为常温常压; 可见光是可再生能源; 反应物CO2来源丰富; 反应生成的甲酸、甲醇和甲烷等为有机燃料, 可以缓解能源危机.因此, CO2光催化反应受到了广泛的关注.
传统上常用的光催化剂主要为无机半导体材料和均相金属有机配合物. TiO2是一种典型的无机半导体材料, 具有易得、无毒等优点, 但对CO2的吸附能力较差, 其催化效率也由于光生电子和空穴过快的重新结合而受到限制[60]. Ru、Re、Ir等金属的配合物具有良好的吸光性, 但很难从溶液中回收并循环利用[61]. PMOFs的金属卟啉配体作为吸光位点可有效促进光生电子的产生, 有序排列且大小适中的孔道可以吸附大量CO2, 异相催化的特征也令PMOF光催化剂具有可循环使用的优点(图 13).目前, 发展PMOFs及其复合材料作为CO2光催化剂的研究取得了极大关注.
卟啉中心的金属离子对CO2的化学吸附可有效提高PMOFs的光催化效率. 2013年, Huang等[62]通过后修饰的方法, 利用无限氧化铝长链的框架Sp与CuSO4•5H2O的溶剂热反应合成得到了卟啉中心插入了Cu2+的框架SCu. CO2吸附脱附实验中, SCu在三轮循环中曲线都没有闭合, 而具有相同框架结构的Sp则为闭合的循环曲线.作者由此推断, SCu对CO2具有化学吸附作用.作者进一步研究了SCu与Sp在CO2光催化反应中的性能并进行对比: SCu与Sp的反应主产物均为甲醇, 生成速率分别为262.6和37.5 ppm•g-1•h-1, 表明SCu对CO2的化学吸附使其光还原的效率提高了.
类似地, Ye等[63]同样使用后修饰策略在MOF-525的卟啉中心插入了Co3+和Zn2+, 并与未插入金属的MOF-525进行了比较.反应所得主要产物为CO和CH4, 插入金属的MOF-525(Co)和MOF-525(Zn)均能提高两种产物的生成速率, 且前者提高的程度更大, 反应对CH4的选择性也更高.由于MOF-525(Co)和MOF- 525(Zn)的比表面积以及对CO2的吸附性能相差无几, 作者通过入射光电转化效率、荧光猝灭等表征对两种催化剂的电荷分离效率、电子传递行为等进行研究, 证明了卟啉中心的Co能更有效地促进光生电子与空穴的分离, 进而更大程度地提高光催化反应速率.
Jiang和Zhang等[64]对同为基于Zr-O簇的PCN-222进行了研究, 卟啉中心未插入金属也能有良好的催化活性.通过Mott-Schottky与Tauc曲线, 作者先后计算得到PCN-222的LUMO (-0.40 V vs. NHE)、能带间隙(1.75 eV)和HOMO (1.35 V vs. NHE), 理论上证明了PCN-222是一种n型半导体并能进行CO2光催化(CO2生成甲酸根离子的还原电位为-0.28 V vs. NHE).催化实验中, 加入50 mg催化剂, 在可见光照射下反应10 h, 可以产生30 μmol的甲酸根离子.通过瞬态吸收光谱与光致发光光谱, 作者证明PCN-222中长时间存在的电子捕获态抑制了光生电子和空穴的重新结合, 由此大大增加了催化效率.
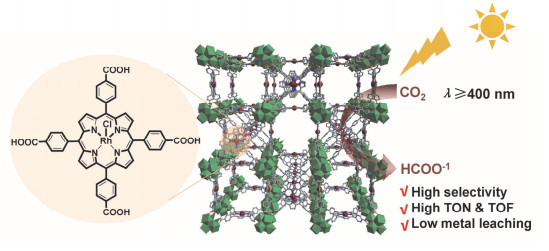
我们课题组[30]合成了与PCN-224[25]同构的Rh- PMOF-1并进行了研究(图 14).我们罕见地检测到铑卟啉的室温磷光, 证明Rh-PMOF-1是一种有效的光催化剂.在不额外添加光敏剂的情况下, Rh-PMOF-1光催化反应获得了较好的光催化性能, 生成的甲酸根离子(HCOO-)的量在18 h后达到6.1 μmol/μmolcat, 同时产物的选择性高达99%.我们使用蒙特卡洛(Canonical Monte Carlo)模拟得到了CO2在框架中的概率密度图, 发现由于CO2分子与Zr6O8团簇的µ3-O原子或配位水分子的O原子之间的非共价相互作用, 吸附在Rh-PMOF-1孔中的CO2分子主要分布在卟啉环和Zr6O8团簇周围.我们提出了可能的反应机理:在Rh-PMOF-1中, 铑卟啉单元既是吸收可见光的天线, 又是催化中心.一方面, 在可见光照射下, 金属卟啉配体表现为吸收可见光的天线, 产生激发态, 并将电子转CO2还原为甲酸盐.另一方面, 铑卟啉配体本身可以作为光催化中心并进行CO2还原.框架和CO2的弱作用以及两种催化模式, 使得Rh-PMOF-1获得了良好的CO2吸附和光催化性能.
Bu、Zhang和Lin等[65]使用多酚配体THPP (5, 10, 15, 20-四(3, 4, 5-三羟基苯基)卟啉)与THBPP (5, 10, 15, 20-四(3, 4, 5-三羟基联苯基)卟啉)和Zr4+分别合成了具有nbo拓扑结构的ZrPP-1和ZrPP-2. ZrPP-1表现出了非常高的化学稳定性, 在0.1 mol/L盐酸与饱和NaOH水溶液中都能稳定存在7天.此外, ZrPP-1-Co表现出优异的CO2吸附性与光催化活性.在可见光照射下, CO的生成量为207 μmol•g-1, 产物选择性高达96%.
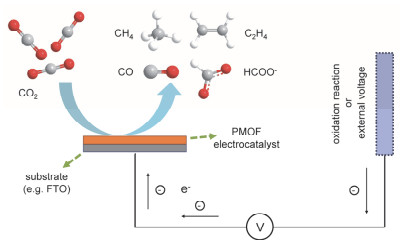
电催化CO2还原反应通常在CO2饱和的碳酸氢盐水溶液、有机溶液或离子液体中进行, 反应条件相对于使用强酸或强碱体系的析氢反应、析氧反应、氧还原反应温和[66, 67].异相CO2RR的产物为CO、HCOO-、CH4和C2H4等, 反应中需要重点关注对产物选择性的控制.使用异相催化剂能有效抑制二聚等破坏催化体系稳定性的副反应, 控制催化环境位点的化学环境以提高催化性能, 也不必考虑催化剂在所用溶剂的溶解度.但是目前, 受限于较低的催化效率与选择性, PMOFs在CO2电催化还原上的应用研究相对较少.
Hupp,Farha和Kubiak等[68]将MOF-525通过电泳沉积(Electrophoretic Deposition, EPD)的方式负载到FTO (FTO=Fluorine-Doped Tin oxide, 掺杂氟的SnO2)导电玻璃基底, 再通过后修饰法在卟啉中心插入Fe3+得到Fe_MOF-525(图 15).薄膜上负载的Fe_MOF-525颗粒大小约为300~500 nm.循环伏安法(CV)和恒电位电解(CPE)研究表明, 电流密度增加的Fe(I/0)氧化还原峰对应的电位即是CO2发生还原反应的电位.气相色谱表明还原所得产物为CO和H2, 法拉第效率分别为(54±2)%和(45±1)%.作者分析其中的H2可能来自溶剂乙腈中痕量水的电解, 也可能来自电解质四丁基六氟磷酸铵(TBAPF6)的Hofmann降解.在反应体系中加入弱酸, 例如2, 2, 2-三氟乙醇(TFE), 能对Fe-CO2加合物进行质子化, 促进C—O键断裂和产物CO的生成, 进而有效增大电流密度, 也能通过生成水的方式稳定体系中的O2-.加入TFE后产物CO的转化数(TON)由272上升至1520, 但CO的选择性略有下降, 两种产物的法拉第效率分别变为(41±8)%和(60±4)%.不同扫描速率下的CV图表明, Fe_MOF-525催化速率受电荷扩散的限制, 计时电流法计算所得电荷扩散常数的值为5×10-13 cm2•s-1.均相催化剂Fe-TPP的催化速率则主要取决于电位, TOF值比异相的Fe_MOF-525大了16倍.
Yaghi和Yang等[69]经过前期实验, 选取了无限Al-O长链SBU和Co-TCPP配体构筑的Al2(OH)2TCPP-Co用原子层沉积(ALD)的方法在碳盘电极上实现了负载.作者通过改变ALD的重复轮数来调整催化剂的负载厚度, 使催化剂在反应物扩散和电荷传输之间取得平衡, 力求催化效果最大化.实验结果表明进行50轮ALD(对应的催化剂厚度约为30~70 nm)时催化效果最好. CPE研究表明该催化剂能在反应条件下稳定7 h. Tafel斜率和原位光谱电化学研究表明, 反应的限速步骤可能为附着在Co(I)卟啉配体上的CO2经过一电子还原生成CO2˙自由基, 也可能为Co(I)-CO2加合物的一电子还原.
如上所述, PMOFs在CO2的捕获与转化上有着巨大的潜力, 各类应用已见诸报道.然而, PMOFs在实际应用中仍有不少难点亟待攻克: (1)从混合气体(尤其是废气)中选择性吸附CO2, 需要PMOFs在水蒸汽、CO、氮氧化物、硫氧化物等杂质气体存在的情况下能保持原有的稳定性, 同时对各种杂质气体的吸附性较差. (2)使用PMOFs催化CO2环加成反应已能得到相当高的转化率, 但催化反应完成时间通常超过24 h, 还有缩短的空间. (3) CO2也可作为环氧化物一步共聚反应的单体, 但目前还没有PMOFs催化剂应用于该聚合反应的报道.
光催化和电催化研究则有着相似的缺点: (1)大部分报道中产物的选择性不高, 副产物主要为H2, 尽管可以利用Fischer-Tropsch (FT)反应将CO和H2的混合物转化为各类碳氢化合物, 但若能一步生成碳氢化合物, 便不必消耗额外的能量. (2)截至目前还没有出现以CH4和高级烃为主产物的报道. (3) PMOFs的光催化和电催化效率还需进一步提高. (4) PMOFs的多孔性及卟啉中心的氮原子使其对CO2有浓缩效应, 在低浓度CO2光还原的应用中存在一定的优势, 但相关报道很少.
光电催化结合光催化和电催化的优势, 通过把CO2还原反应与H2O氧化反应分开在两个电极中进行, 使两个反应的效率都得到加强[70].发展基于PMOFs的光电催化剂也是一个极具潜力的研究方向.其中最理想的其中一种体系是, 在不加外压的情况下, 阳极上的光催化剂接受光照将水氧化为氧气, 电子通过电路传递给阴极上的电催化剂将二氧化碳还原为有机物.在上述体系中, 阳极和阴极都可使用PMOFs作为催化剂, 进而实现人工光合作用.
Feng, J.; Zeng, S.; Feng, J.; Dong, H.; Zhang, X. Chin. J. Chem. 2018, 36, 961. doi: 10.1002/cjoc.v36.10
常世磊, 梁风, 姚耀春, 马文会, 杨斌, 戴永年, 化学学报, 2018, 76, 515. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346606.shtmlChang, S.; Liang, F.; Yao, Y.; Ma, W.; Yang, B.; Dai, Y. Acta Chim. Sinica 2018, 76, 515(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346606.shtml
Du, P.; Su, T.; Luo, X.; Zhou, X.; Qin, Z.; Ji, H.; Chen, J. Chin. J. Chem. 2018, 36, 538. doi: 10.1002/cjoc.v36.6
闫婷婷, 邢国龙, 贲腾, 化学学报, 2018, 76, 366. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346538.shtmlYan, T.; Xing, G.; Ben, T. Acta Chim. Sinica 2018, 76, 366(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346538.shtml
徐佳伟, 张崇, 王迅昶, 蒋加兴, 汪锋, 化学学报, 2017, 75, 473. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346022.shtmlXu, J.; Zhang, C.; Wang, X.; Jiang, J.; Wang, F. Acta Chim. Sinica 2017, 75, 473(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346022.shtml
Li, Y.; Zou, B.; Xiao, A.; Zhang, H. Chin. J. Chem. 2017, 35, 1501. doi: 10.1002/cjoc.v35.10
黄刚, 陈玉贞, 江海龙, 化学学报, 2016, 74, 113. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345318.shtmlHuang, G.; Chen, Y.; Jiang, H. Acta Chim. Sinica 2016, 74, 113(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345318.shtml
Yang, Q.; Xu, Q.; Jiang, H.-L. Chem. Soc. Rev. 2017, 46, 4774. doi: 10.1039/C6CS00724D
Jiao, L.; Wang, Y.; Jiang, H.-L.; Xu, Q. Adv. Mater. 2018, 30, 1703663. doi: 10.1002/adma.v30.37
梁祥, 陈莲芬, 张利, 苏成勇, 科学通报, 2018, 63, 248.Liang, X.; Chen, L.; Zhang, L.; Su, C. Chin. Sci. Bull. 2018, 63, 248(in Chinese).
Haszeldine, R. S. Science 2009, 325, 1647. doi: 10.1126/science.1172246
Boot-Handford, M. E.; Abanades, J. C.; Anthony, E. J.; Blunt, M. J.; Brandani, S.; Mac Dowell, N.; Fernández, J. R.; Ferrari, M.-C.; Gross, R.; Hallett, J. P.; Haszeldine, R. S.; Heptonstall, P.; Lyngfelt, A.; Makuch, Z.; Mangano, E.; Porter, R. T. J.; Pourkashanian, M.; Rochelle, G. T.; Shah, N.; Yao, J. G.; Fennell, P. S. Energy Environ. Sci. 2014, 7, 130. doi: 10.1039/C3EE42350F
Cuéllar-Franca, R. M.; Azapagic, A. J. CO2 Util. 2015, 9, 82. doi: 10.1016/j.jcou.2014.12.001
Markewitz, P.; Kuckshinrichs, W.; Leitner, W.; Linssen, J.; Zapp, P.; Bongartz, R.; Schreiber, A.; Müller, T. E. Energy Environ. Sci. 2012, 5, 7281. doi: 10.1039/c2ee03403d
陈小明, 张杰鹏等, 金属-有机框架材料, 化学工业出版社, 2017, pp. 90~147.Chen, X. M.; Zhang, J. P. Metal-Organic Framework Materials, Chemical Industry Press, 2017, pp. 90~147(in Chinese).
Liu, J.; Chen, L.; Cui, H.; Zhang, J.; Zhang, L.; Su, C.-Y. Chem. Soc. Rev. 2014, 43, 6011. doi: 10.1039/C4CS00094C
Gao, W.; Chrzanowski, M.; Ma, S. Chem. Soc. Rev. 2014, 43, 5841. doi: 10.1039/C4CS00001C
陈华彬, 章小慧, 龚丽珍, 何婧, 许旋, 徐志广, 刘海洋, 物理化学学报, 2016, 32, 1983.Chen, H.; Zhang, X.; Gong, L.; He, J.; Xu, X.; Xu, Z.; Liu, H. Acta Phys.-Chim. Sin. 2016, 32, 1983(in Chinese).
Abrahams, B. F.; Hoskins, B. F.; Robson, R. J. Am. Chem. Soc. 1991, 113, 3606. doi: 10.1021/ja00009a065
Lin, K.-J. Angew. Chem., Int. Ed. 1999, 38, 2730. doi: 10.1002/(ISSN)1521-3773
Liu, T.; Feng, D.; Chen, Y.-P.; Zou, L.; Bosch, M.; Yuan, S.; Wei, Z.; Fordham, S.; Wang, K.; Zhou, H.-C. J. Am. Chem. Soc. 2015, 137, 413. doi: 10.1021/ja5111317
Wang, K.; Lv, X.; Feng, D.; Li, J.; Chen, S.; Sun, J.; Song, L.; Xie, Y.; Li, J.; Zhou, H.-C. J. Am. Chem. Soc. 2016, 138, 914. doi: 10.1021/jacs.5b10881
Lv, X.; Wang, K.; Wang, B.; Su, J.; Zou, X.; Xie, Y.; Li, J.; Zhou, H.-C. J. Am. Chem. Soc. 2017, 139, 211. doi: 10.1021/jacs.6b09463
Huang, N.; Wang, K.; Drake, H.; Cai, P.; Pang, J.; Li, J.; Che, S.; Huang, L.; Wang, Q.; Zhou, H.-C. J. Am. Chem. Soc. 2018, 140, 6383. doi: 10.1021/jacs.8b02710
Feng, D.; Chung, W.-C.; Wei, Z.; Gu, Z.; Jiang, H.; Chen, Y.; Darensbourg, D. J.; Zhou, H. J. Am. Chem. Soc. 2013, 135, 17105. doi: 10.1021/ja408084j
Cui, H.; Wang, Y.; Wang, Y.; Fan, Y.; Zhang L.; Su, C.-Y. CrystEngComm 2016, 18, 2203. doi: 10.1039/C6CE00358C
Wang, Y.; Cui, H.; Wei, Z.; Wang, H.-P.; Zhang, L.; Su, C.-Y. Chem. Sci. 2017, 8, 775. doi: 10.1039/C6SC03288E
Wang, Y.; Cui, H.; Zhang, L.; Su, C.-Y. ChemCatChem 2018, 10, 3901. doi: 10.1002/cctc.201800597
Liu, J.; Fan, Y.-Z.; Xu, Y.-W.; Zhang, L.; Su, C.-Y. ChemSusChem 2018, 11, 2340. doi: 10.1002/cssc.v11.14
Liu, J.; Fan, Y. Z.; Li, X.; Wei, Z.; Xu, Y.-W.; Zhang, L.; Su, C.-Y. Appl. Catal. B-Environ. 2018, 231, 173. doi: 10.1016/j.apcatb.2018.02.055
Chen, L.; Cui, H.; Wang, Y.; Liang, X.; Zhang, L.; Su, C.-Y. Dalton Trans. 2018, 47, 3940. doi: 10.1039/C8DT00434J
Choi, E.-Y.; Barron, P. M.; Novotny, R. W.; Son, H.-T.; Hu, C.; Choe, W. Inorg. Chem. 2009, 48, 426. doi: 10.1021/ic801677y
Farha, O. K.; Shultz, A. M.; Sarjeant, A. A.; Nguyen, S. T.; Hupp, J. T. J. Am. Chem. Soc. 2011, 133, 5652. doi: 10.1021/ja111042f
Wang, X.; Meng, L.; Cheng, Q.; Kim, C.; Wojtas, L.; Chrzanowski, M.; Chen, Y.; Zhang, X. P.; Ma, S. J. Am. Chem. Soc. 2011, 133, 16322. doi: 10.1021/ja204339e
Feng, D.; Jiang, H.; Chen, Y.; Gu, Z.; Wei, Z.; Zhou, H. Inorg. Chem. 2013, 52, 12661. doi: 10.1021/ic4018536
Fateeva, A.; Chater, P. A.; Ireland, C. P.; Tahir, A. A.; Khimyak, Y. Z.; Wiper, P. V.; Darwent, J. R.; Rosseinsky, M. J. Angew. Chem., Int. Ed. 2012, 51, 7440. doi: 10.1002/anie.201202471
Rhauderwiek, T.; Waitschat, S.; Wuttke, S.; Reinsch, H.; Bein, T.; Stock, N. Inorg. Chem. 2016, 55, 5312. doi: 10.1021/acs.inorgchem.6b00221
Leng, F.; Liu, H.; Ding, M.; Lin, Q.-P.; Jiang, H.-L. ACS Catal. 2018, 8, 4583. doi: 10.1021/acscatal.8b00764
Pearson, R. G. J. Am. Chem. Soc. 1963, 85, 3533. doi: 10.1021/ja00905a001
Yuan, S.; Feng, L.; Wang, K.; Pang, J.; Bosch, M.; Lollar, C.; Sun, Y.; Qin, J.; Yang, X.; Zhang, P.; Wang, Q.; Zou, L.; Zhang, Y.; Zhang, L.; Fang, Y.; Li, J.; Zhou, H.-C. Adv. Mater. 2018, 30, 1704303. doi: 10.1002/adma.201704303
Morris, W.; Volosskiy, B.; Demir, S.; Gándara, F.; McGrier, P. L.; Furukawa, H.; Cascio, D.; Stoddart, J. F.; Yaghi, O. M. Inorg. Chem. 2012, 51, 6443. doi: 10.1021/ic300825s
Feng, D.; Gu, Z.; Li, J.; Jiang, H.; Wei, Z.; Zhou, H. Angew. Chem., Int. Ed. 2012, 51, 10307. doi: 10.1002/anie.201204475
Jiang, H.; Feng, D.; Wang, K.; Gu, Z.; Wei, Z.; Chen, Y.; Zhou, H. J. Am. Chem. Soc. 2013, 135, 13934. doi: 10.1021/ja406844r
Feng, D.; Gu, Z.; Chen, Y.; Park, J.; Wei, Z.; Sun, Y.; Bosch, M.; Yuan, S.; Zhou, H.-C. J. Am. Chem. Soc. 2014, 136, 17714. doi: 10.1021/ja510525s
Chen, Y.; Hoang, T.; Ma, S. Inorg. Chem. 2012, 51, 12600. doi: 10.1021/ic301923x
Wang, T. C.; Bury, W.; Gómez-Gualdrón, D. A.; Vermeulen, N. A.; Mondloch, J. E.; Deria, P.; Zhang, K.; Moghadam, P. Z.; Sarjeant, A. A.; Snurr, R. Q.; Stoddart, J. F.; Hupp, J. T.; Farha, O. K. J. Am. Chem. Soc. 2015, 137, 3585. doi: 10.1021/ja512973b
Lin, Q.; Bu, X.; Kong, A.; Mao, C.; Zhao, X.; Bu, F.; Feng, P. J. Am. Chem. Soc. 2015, 137, 2235. doi: 10.1021/jacs.5b00076
Zheng, J.; Wu, M.; Jiang, F.; Su, W.; Hong, M. Chem. Sci. 2015, 6, 3466. doi: 10.1039/C5SC00213C
Xu, L.; Luo, Y.; Sun, L.; Xu, Y.; Cai, Z.; Fang, M.; Yuan, R.; Du, H. Chem. Eur. J. 2016, 22, 6268. doi: 10.1002/chem.v22.18
Wang, K.; Feng, D.; Liu, T.; Su, J.; Yuan, S.; Chen, Y.; Bosch, M.; Zou, X.; Zhou, H. J. Am. Chem. Soc. 2014, 136, 13983. doi: 10.1021/ja507269n
Yuan, S.; Liu, T.; Feng, D.; Tian, J.; Wang, K.; Qin, J.; Zhang, Q.; Chen, Y.; Bosch, M.; Zou, L.; Teat, S. J.; Dalgarno, S. J.; Zhou, H. Chem. Sci. 2015, 6, 3926. doi: 10.1039/C5SC00916B
Xu, L.; Zhai, M.-K.; Wang, F.; Sun, L.; Du, H.-B. Dalton Trans. 2016, 45, 17108. doi: 10.1039/C6DT03678C
Trickett, C. A.; Helal, A.; Al-Maythalony, B. A.; Yamani, Z. H.; Cordova, K. E.; Yaghi, O. M. Nat. Rev. Mater. 2017, 2, 17045. doi: 10.1038/natrevmats.2017.45
Li, J.; Yu, J.; Lu, W.; Sun, L. B.; Sculley J.; Balbuena, P. B.; Zhou, H.-C. Nat. Commun. 2013, 4, 1538. doi: 10.1038/ncomms2552
Johnson, J. A.; Chen, S.; Reeson, T. C.; Chen, Y.; Zeng, X. C.; Zhang, J. Chem. Eur. J. 2014, 20, 7632. doi: 10.1002/chem.201402006
Wang, X.; Chrzanowski, M.; Gao, W.; Wojtas, L.; Chen, Y.; Za-worotko, M. J.; Ma, S. Chem. Sci. 2012, 3, 2823. doi: 10.1039/c2sc20330h
Maina, J. W.; Pozo-Gonzalo, C.; Kong, L.; Schütz, J.; Hill, M.; Dumée, L. F. Mater. Horiz. 2017, 4, 345. doi: 10.1039/C6MH00484A
Gao, W.; Tsai, C.-Y.; Wojtas, L.; Thiounn, T.; Lin, C.-C.; Ma, S. Inorg. Chem. 2016, 55, 7291. doi: 10.1021/acs.inorgchem.6b00937
Zhang, L.; Yuan, S.; Feng, L.; Guo, B.; Qin, J.-S.; Xu, B.; Lollar, C.; Sun, D.; Zhou, H.-C. Angew. Chem., Int. Ed. 2018, 57, 5095. doi: 10.1002/anie.201802661
Tu, W.; Zhou, Y.; Zou, Z. Adv. Mater. 2014, 26, 4607. doi: 10.1002/adma.v26.27
Chen, Y.; Wang, D.; Deng, X.; Li, Z. Catal. Sci. Technol. 2017, 7, 4893. doi: 10.1039/C7CY01653K
Liu, Y.; Yang, Y.; Sun, Q.; Wang, Z.; Huang, B.; Dai, Y.; Qin, X.; Zhang, X. ACS Appl. Mater. Interfaces 2013, 5, 7654. doi: 10.1021/am4019675
Zhang, H.; Wei, J.; Dong, J.; Liu, G.; Shi, L.; An, P.; Zhao, G.; Kong, J.; Wang, X.; Meng, X.; Zhang, J.; Ye, J. Angew. Chem., Int. Ed. 2016, 55, 14310. doi: 10.1002/anie.v55.46
Xu, H.-Q.; Hu, J.; Wang, D.; Li, Z.; Zhang, Q.; Luo, Y.; Yu, S.-H.; Jiang, H.-L. J. Am. Chem. Soc. 2015, 137, 13440. doi: 10.1021/jacs.5b08773
Chen, E.-X.; Qiu, M.; Zhang, Y.-F.; Zhu, Y.-S.; Liu, L.-Y.; Sun, Y.-Y.; Bu, X.; Zhang, J.; Lin, Q. Adv. Mater. 2018, 30, 1704388. doi: 10.1002/adma.v30.2
Liao, P.-Q.; Shen, J.-Q.; Zhang, J.-P. Coord. Chem. Rev. 2018, 373, 22. doi: 10.1016/j.ccr.2017.09.001
Whipple, D. T.; Kenis, P. J. A. J. Phys. Chem. Lett. 2010, 1, 3451. doi: 10.1021/jz1012627
Hod, I.; Sampson, M. D.; Deria, P.; Kubiak, C. P.; Farha, O. K.; Hupp, J. T. ACS Catal. 2015, 5, 6302. doi: 10.1021/acscatal.5b01767
Kornienko, N.; Zhao, Y.; Kley, C. S.; Zhu, C.; Kim, D.; Lin, S.; Chang, C. J.; Yaghi, O. M.; Yang, P. J. Am. Chem. Soc. 2015, 137, 14129. doi: 10.1021/jacs.5b08212
Xie, S.; Zhang, Q.; Liu, G.; Wang, Y. Chem. Commun. 2016, 52, 35. doi: 10.1039/C5CC07613G

图 6 (a) MMPF-1的结构示意图和(b) MMPF-1中MOC的示意图
Figure 6 Schematics of MMPF-1 (a) and MOC in MMPF-1 (b)

图 8 (a) 金属-氧无限长链示意图和(b)基于金属-氧无限长链PMOFs的结构示意图
Figure 8 Schematics of infinite metal oxide chain (a) and structure of infinite metal oxide chain based PMOFs

图 9 PCN-224、PCN-222和MOF-525中Zr6簇的成键模式及框架结构
Figure 9 Schematics of Zr6 clusters and structures of PCN-224, PCN-222 and MOF-525

图 10 NUPF-2M的结构示意图: (a)三角形孔道; (b)菱形孔道
Figure 10 Schematics of NUPF-2M with triangular channels (a) and rhombus channels (b)

图 12 Rh-PMOF-1在CO2环加成反应中的应用示意图
Figure 12 Application of Rh-PMOF-1 in CO2 cycloaddition

图 14 Rh-PMOF-1在CO2光还原反应中的应用示意图
Figure 14 Application of Rh-PMOF-1 in CO2 photoreduction

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:







