图 1.
FLPs催化的醛酮的还原反应
Figure 1.
Reduction of carbonyl compounds catalyzed by FLPs

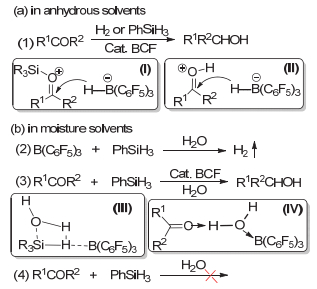
醛酮羰基还原为醇的研究一直以来都是有机化学研究的重要方向之一[1].其还原方法主要有催化氢化、金属还原、酶还原等[2~5].常用的催化氢化试剂大多是基于Ni、Pd、Pt、Rh等贵金属或有毒的过渡金属催化体系[6~11]; 近些年以发展新型绿色环保的非金属催化氢化试剂受到越来越多的重视[12~15], 其中受限路易斯对(FLPs)化学的发展最为突出.受限路易斯酸碱对在温和条件下对H2小分子的活化特性, 使FLPs在对亚胺、烯胺、醇醚、烯烃等非羰基化合物的催化氢化中都表现出了非常高的催化活性[16~19].但由于FLPs的结构特点, 在羰基化合物的氢化还原中路易斯酸硼烷如B(C6F5)3会与羰基氧形成较强的B―O键, 因此只能进行计量的还原反应[20].近年来, Stephan[21]及Ashley[22]课题组分别通过改变反应溶剂, 如以醚类乙醚、THF等配位溶剂为反应介质, 实现了FLPs催化的羰基化合物的氢化还原, 并且发现路易斯酸B(C6F5)3是一种耐水的催化剂, 其催化的醛酮的氢化还原可以在常规的溶剂中进行反应, 如图 1(a)所示.尽管这些针对醛酮的催化氢化研究的方法都取得了较多关注, 但不可回避的是, 在使用氢气作为还原剂时, 通常需要通过加热、加压等条件来实现较高的转化率.而且所谓的“有水”条件通常限于体系中水的含量是催化剂的3~5倍而已[23, 24].
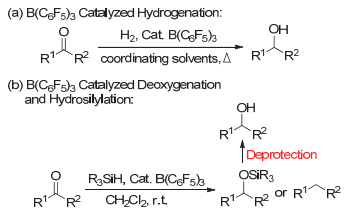
随着受限路易斯酸碱对对有机硅烷上Si―H键的活化作用及机理的研究, FLPs在以有机硅烷作为还原剂催化亚胺、醛酮的硅氢化反应以及含氧化合物的去氧化还原中也表现出了独特的反应活性[25~32].但是, 目前FLPs在催化醛酮羰基的直接还原为醇的方法上还没有很好的解决, 以硅烷作为还原剂所得到的硅氢化还原产物必须经过进一步的水解去保护才能转化成醇[33~35], 如图 1(b)所示.因此, 如何绿色、高效地实现醛酮直接还原成醇的方法研究仍然是化学工作者需要探索解决的课题之一.
近些年, 我们实验室对基于路易斯酸B(C6F5)3的FLPs体系的催化反应进行过较多的研究, 发现当向常规的有机胺FLPs体系中加入计量的Brønsted酸, 如HCl或H2O时, 在有机硅烷的作用下该体系可以生成具有还原活性的中间体“硼氢化胺盐[R2NH2][HB(C6F5)3]”, 如图 2(a)上式所示[36~40].并在醛酮与胺的还原胺化反应中进一步证明了路易斯酸B(C6F5)3不仅仅是一种“耐水”的催化剂[41, 42], 反应溶剂中的水或反应中所产生的水均可能直接影响着FLPs催化的反应历程[43, 44], 如图 2(a)下式所示.
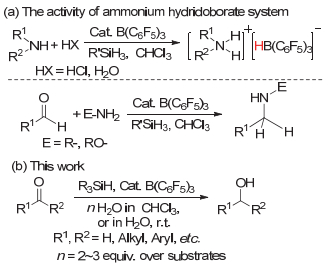
结合受限路易斯酸碱对对醛酮的催化反应研究, 以及实验室对有水条件下FLPs对醛酮的直接还原胺化反应机理的研究发现[45], 本文利用硅烷作为还原剂, 系统研究了有水条件下路易斯酸B(C6F5)3催化醛酮的羰基直接还原成醇的反应活性, 如图 2(b)所示.研究发现, 不同于以氢气作为还原剂时, FLPs对醛酮的催化氢化方法的较苛刻要求, 本文所报道的以硅烷作为还原剂, B(C6F5)3催化的醛酮直接还原成醇反应, 不仅实现了FLPs温和高效地催化醛酮转化成醇, 而且首次实现了FLPs在真正有水条件下的催化反应.
对反应机理的研究表明, 在有水条件下, H2O分子通过与催化剂B(C6F5)3及底物C=O之间的三组分相互作用, 不仅促使了羰基的活化, 而且还进一步抑制了硼烷与羰基氧的键合.研究表明在以水做反应溶剂时, B(C6F5)3催化的醛酮直接还原成醇反应也可顺利进行.
在研究以硅烷作为还原剂, B(C6F5)3催化的醛酮直接还原成醇反应时, 首先探索了在不同活性的有机硅烷作用下, 水的比例对苯甲醛的还原产物的影响.反应以氘代氯仿为溶剂, 通过核磁共振对反应进程和产物进行监测.考虑到在无水条件下, 硅烷对催化的醛酮的去氧化还原活性的影响, 因此在反应的加料顺序上, 硅烷会在水加入后最后加入反应体系. 表 1所示为不同条件下苯甲醛的还原产物及产率等数据.从表中可以看出, 由于PhSiH3具有较强的还原性, 只有当反应体系中水的量是苯甲醛的3倍时, 苯甲醛完全转化为苯甲醇, 见表 1 Entry 4;而反应活性较弱的Ph2SiH2及Et3SiH则分别在2 equiv.和3 equiv.过量水的作用下可使苯甲醛完全转化为苯甲醇, 见表 1中Entries 7~12.以PhSiH3作为还原剂, D2O作为反应介质的反应与氯仿中加入过量水类似, B(C6F5)3也可催化苯甲醛高效地转化为苯甲醇, 而该反应在不加催化剂的条件下不发生任何反应, 见表 1 Entries 5~6.
 下载:
导出CSV
下载:
导出CSV
 |
||||
| Entry | Water (n equiv.) |
Silane a (1 equiv.) |
Product 2a+3a |
Yield of 2a/% |
| 1 | 0 | PhSiH3 | 3a | 0 |
| 2 | 1 | PhSiH3 | 3a | 0 |
| 3 | 2 | PhSiH3 | 2a | 88.5 |
| 4 | 3 | PhSiH3 | 2a | 100 |
| 5 | D2O c | PhSiH3 | 2a | 100 |
| 6 | D2O d | PhSiH3 | 1a | 0 |
| 7 | 1 | Ph2SiH2 | 2a | (78.0) e |
| 8 | 2 | Ph2SiH2 | 2a | 91.0 |
| 9 | 3 | Ph2SiH2 | 2a | 94.0 |
| 10 | 1 | Et3SiH | 2a | 64.5 |
| 11 | 2 | Et3SiH | 2a | 84.0 |
| 12 | 3 | Et3SiH | 2a | 65.5 |
| a Reaction with PhSiH3 2 h/with Ph2SiH2 6 h/with Et3SiH 12 h, the 2a yield determined by 1H NMR of the crude mixture using C6Me6 as internal standard; b n H2O: n equiv. over silanes; c D2O as solvent, CDCl3 extracting; d catalyst free; e conversion. | ||||
由于在水相中研究醛酮的还原反应, 在反应结束时需要加入氯仿来提取有机产物, 而且水相中进行反应也不便对反应的进程进行监测; 考虑到在氘代氯仿中加入过量水的方法与水相中进行的反应产物相同, 但更便于观察反应的优点, 因此在研究B(C6F5)3催化的不同取代醛酮的直接还原成醇的反应时均以氘代氯仿为溶剂展开研究, 详细反应数据见表 2.从表 2的数据可以看出, 在2~3倍水的参与下, 对不同取代的七种芳醛和烷基醛2b~2g及不同取代的六种芳酮和烷基酮2h~2m均可被氢化硅烷完全转化成醇; 当苯环对位为给电子的4-甲氧基苯乙酮2n时, 即使是在3倍水的参与下也仅使少量酮转化成醇; 该方法目前对于二芳基取代酮类(如2o~2p)还不能实现其一步还原成醇.
下载:
导出CSV

|
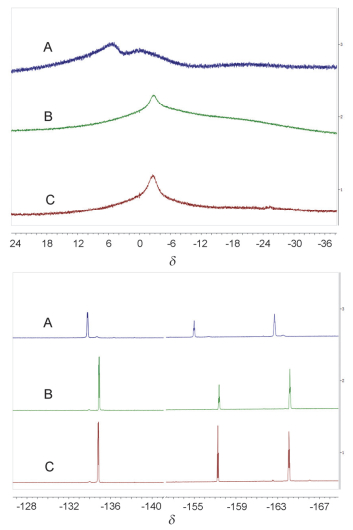
通过1H、11B和19F NMR等核磁共振数据对反应机理进行了详细的研究, 发现在大量水或重水中, 催化剂硼烷并没有分解, 也没有观察到硼烷与水的络合(H2O-B(C6F5)3)或其去质子化产物([HO-B(C6F5)3]-); 而是观察到对应的催化剂的对位和间位的δ 19F信号峰的化学位移存在较窄的间距约Δδp-m 7.0, 证明催化剂B(C6F5)3应该是以一种稳定的四配位形式存在, 其对应的11B NMR在δ -2.3处有一较宽的包峰.
图 4所示为通过核磁共振对催化剂B(C6F5)3在不同反应体系中的络合形式的研究, 从11B和19F NMR核磁共振数据可以看出, 在有水条件下硼烷催化的苯甲醛的还原, 催化剂硼烷的核磁共振信号峰化学位移(C)不同于硼烷与羰基氧直接络合的谱峰(A), 其四配位的核磁共振信号峰化学位移(C)与H2O、B(C6F5)3及PhCHO三组分的核磁共振信号峰位置吻合(B), 因此推断在催化反应过程中可能在底物羰基氧与催化剂之间存在以水介导的相互作用[46], 即可能存在“PhCH=O--- H-O(-H)---B(C6F5)3”的三组分络合形式.
在B3LYP[47, 48]/6-31+G(d)水平对六种不同路易斯碱(Et3N、THF、PhCH2OH、H2O、PhCHO和Ph3P)以水介导的“LB---H-O(-H)---B(C6F5)3”三组分复合物结构进行了结构优化和振动频率分析, 计算采用Gaussian 16程序包[49].相关键长或核间距数据如表 3所示.从表中数据可以看出, 在五种路易斯碱中“Et3N---H-O(-H) ---B(C6F5)3”三组分的复合物的H1―O核间距最长(1.628 Å), 而B―O键(1.509 Å)及LB―H1(1.072 Å)键最短(Entry 1), 事实上这与在实验条件下烷基胺类R3N在硼烷的作用下与水发生质子化作用, 生成“硼羟基化胺盐[R3NH][HO-B(C6F5)3]”的反应结果吻合[50]; 而THF、PhCH2OH、H2O、PhCHO和Ph3P五种路易斯碱对应复合物体系(Entries 2~6)的B―O间距(约1.61 Å)明显较H2O---BCF(Entry 8, 1.669 Å)短, 且对应三组分复合物H1―O键(1.00~1.02 Å)较H2O---BCF(Entry 8, 0.980 Å)长, 可以推测, 以水介导作用下, 硼烷也很易促使PhCHO质子化反应, 更有利于反应的进行.从(H2O)2---BCF(Entry 5)和H2O---BCF(Entry 8)的B―O及H1―O间距的比较可以看出, 当反应体系中存在较多水时或有路易斯碱LB(如有机磷、胺、醇、醚等)存在时, 体系的酸性也会明显增强, 这一计算数据也与表 1~2中实验结果相一致, 即当反应体系中仅有1 equiv.的水时其反应活性总是低于相应的2 equiv.或3 equiv.的反应.其实Parkin课题组在研究硼烷与水络合的Brønsted酸性时也表明在MeCN中H2O/B(C6F5)3体系具有与HCl近似的酸性[51, 52]. Entry 8中较长的B―O间距表明硼烷与水络合作用要明显弱于质子化作用, 该理论计算结果与核磁共振数据中没有观察到的H2O---BCF络合现象一致.
下载:
导出CSV
 |
|||||
| Entry | LB | B―O (Å) |
LB―H1 (Å) |
H1―O (Å) |
H2―O (Å) |
| 1 | Et3N | 1.509 | 1.072 | 1.628 | 0.969 |
| 2 | THF | 1.615 | 1.591 | 1.017 | 0.976 |
| 3 | PhCH2OH | 1.612 | 1.603 | 1.014 | 0.977 |
| 4 | PhCHO | 1.614 | 1.630 | 1.010 | 0.976 |
| 5 | H2O | 1.615 | 1.639 | 1.006 | 0.977 |
| 6 | Ph3P | 1.619 | 2.168 | 1.023 | 0.977 |
| 7 | PhCHO | 1.597 | ― | ― | ― |
| 8 | ― | 1.669 | ― | 0.980 | 0.981 |
Entry 7中较短的B―O间距说明PhCH=O---BCF存在较强络合作用, 在无水条件下该络合作用会影响反应进程; 但在有水条件下的醛酮还原反应的核磁共振数据中并没有观察到对应的PhCH=O---BCF的络合信号, 认为当体系中存在大量水时, 催化剂硼烷主要以三组分络合形式“PhCH=O---H-O(-H)---BCF”存在.
目前有关受限路易斯酸碱对(FLPs)催化的醛酮还原反应机理主要有两种推测, 如图 5中(a)所示: (1)在无水条件下, 以硅烷作为还原剂时, 催化剂硼烷对Si―H键活化所形成的硼氢化物[H-B(C6F5)3]-是促使底物官能团还原的关键(Ⅰ), 反应首先得到硅氢化产物; (2)在无水或常规乙醚、THF等配位溶剂中进行的醛酮氢化还原, 其关键反应为质子化的羰基与[H-B(C6F5)3]-的加成(Ⅱ), 羰基质子化的H+来源于被活化的H2, 而体系中少量水的存在既不参与催化反应也不影响催化剂的活性.
实验室前期在对有水条件下醛酮的直接还原胺化反应的研究中发现, 在以硅烷作为还原剂时, 体系中存在的水不仅会参与催化反应并会促使反应进行.在有水条件下醛酮的还原反应的研究中, 我们更进一步证明了水对FLPs催化反应的影响.如图 5中(b)所示, 我们发现, 在没有醛酮参与下, 硼烷可催化硅烷与水迅速反应并释放出氢气(Ⅲ); 但当反应体系中有醛酮存在时, 硅烷与水的直接反应被在很大程度上得到抑制, 因此我们推测体系中存在“R1R2C=O---H-O(-H)---B(C6F5)3”的三组分相互作用(Ⅳ).这种以水介导的相互作用不仅对催化剂硼烷具有络合作用, 同时又可以通过氢键活化羰基.较强B―O键之间的相互作用, 也避免了自由硼烷促使的硅烷与水的副反应发生, 也更适用于硅烷作为还原剂在有水条件下使用.
根据FLPs催化反应机理并结合实验和计算化学数据, 本文对有水条件下硼烷B(C6F5)3催化醛酮还原成醇的反应机理提出了如图 6所示的催化过程.以三乙基硅烷还原苯甲醛为例:在有水条件下, 催化剂硼烷和醛酮与水首先形成“PhCH=O---H-O(-H)---B(C6F5)3”三组分复合物; 然后是硅氢键的活化.对硅氢键的活化机理我们也提出了A, B两种不同的活化途径. A路径的活化过程与Piers等[25]提出的SN2机理类似, 亲核试剂OH从背面进攻硅烷与硼烷的加合物, 并产生硼氢化物[H-B(C6F5)3]-, 最后还原质子化的羰基成醇; B路径的活化过程与传统的氟离子活化硅氢键类似, 亲核试剂OH从背面进攻硅烷, 然后硅氢键直接加成到羰基碳原子上得到醇.由于在反应过程中极少看到特征的B―H信号峰, 因此我们推测对硅氢键的活化可能两种路径同时存在.
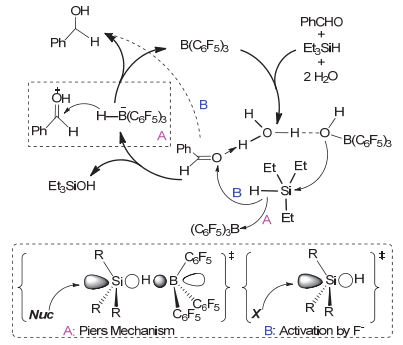
以硅烷作为还原剂对有水条件下FLPs对醛酮的还原研究, 不仅发展了一种温和条件下将醛酮转化醇的方法, 更重要的是对拓展受限路易斯酸碱对化学在水相中的应用至关重要.作为一个新兴的研究领域, FLPs化学还有很多未知的应用领域需要继续探索.对有水条件下FLPs催化的醛酮还原的选择性及其催化反应机理, 以及对其他反应的影响还需要进行更深入的研究.
醛酮的还原方法以PhSiH3对PhCHO的还原为例.
方法(1):向核磁管内加入B(C6F5)3 (0.01 mmol, 5.5 mg)以及内标六甲苯(0.01 mmol, 1.6 mg)后加入CDCl3使其溶解, 再加入PhCHO (0.1 mmol, 10.6 mg)及H2O (0.3 mmol, 5.4 mg), 震荡均匀后加入PhSiH3 (0.1 mmol, 10.8 mg), 室温反应2 h后进行核磁共振分析计算产率.
方法(2):向核磁管内加入B(C6F5)3 (0.01 mmol, 5.5 mg)后加入D2O震荡均匀, 向此悬浊液中再加入PhCHO (0.1 mmol, 10.6 mg)及PhSiH3 (0.1 mmol, 10.8 mg), 室温反应6 h后加入CDCl3提取并进行核磁共振分析.
de Vries, J. G.; Elsevier, C. J. The Handbook of Homogeneous Hydrogenation, Wiley-VCH, Weinheim, Germany, 2008.
Sorella, G. L.; Sperni, L.; Canton, P.; Coletti, L.; Fabris, F.; Strukul, G.; Scarso, A. J. Org. Chem. 2018, 83(14), 7438. doi: 10.1021/acs.joc.8b00314
Leischner, T.; Spannenberg, A.; Junge, K.; Beller, M. Organometallics 2018, DOI: 10.1021/acs.organomet.8b00410.
Cao, Y.; Ma, R.; Wang, N.; Wang, M.-Y.; Li, X.-D.; He, L.-N. J. CO2 Util. 2018, 24, 328. doi: 10.1016/j.jcou.2018.01.019
Call, A.; Lloret-Fillol, J. Chem. Commun. 2018, 54, 9643. doi: 10.1039/C8CC04239J
Zhang, J.; Qu, L.; Shi, G.; Liu, J.; Chen, J.; Dai, L. Angew. Chem., Int. Ed. 2016, 55, 2230. doi: 10.1002/anie.201510495
Chakraborty, S.; Bhattacharya, P.; Dai, H.; Guan, H. Acc. Chem. Res. 2015, 48, 1995. doi: 10.1021/acs.accounts.5b00055
Guo, J.; Chen, J.; Lu, Z. Chem. Commun. 2015, 51, 5725. doi: 10.1039/C5CC01084E
Dai, L.; Xue, Y.; Qu, L.; Choi, H. J.; Baek, J. B. Chem. Rev. 2015, 115, 4823. doi: 10.1021/cr5003563
Mahdi, T.; Stephan, D. W. Angew. Chem., Int. Ed. 2015, 54, 8511. doi: 10.1002/anie.201503087
Volkov, A.; Gustafson, K. P. J.; Tai, C. W.; Verho, O.; Baeckvall, J. E.; Adolfsson, H. Angew. Chem., Int. Ed. 2015, 54, 5122. doi: 10.1002/anie.v54.17
Parks, D. J.; Piers, W. E. J. Am. Chem. Soc. 1996, 118, 9440. doi: 10.1021/ja961536g
Stephan, D. W.; Erker, G. Angew. Chem., Int. Ed. 2010, 49, 46. doi: 10.1002/anie.200903708
刘勇兵, 杜海峰, 化学学报, 2014, 72, 771. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract344459.shtmlLiu, Y.-B.; Du, H.-F. Acta Chim. Sinica 2014, 72, 771(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract344459.shtml
Xuan, Q.; Zhao, C.; Song, Q. Org. Biomol. Chem. 2017, 15, 5140. doi: 10.1039/C7OB00820A
Wei, S.; Du, H. J. Am. Chem. Soc. 2014, 136, 12261. doi: 10.1021/ja507536n
Oestreich, M.; Hermeke, J.; Mohr, J. Chem. Soc. Rev. 2015, 44, 2202. doi: 10.1039/C4CS00451E
Stephan, D. W.; Erker, G. Angew. Chem., Int. Ed. 2015, 54, 6400. doi: 10.1002/anie.201409800
Ren, X.; Du, H. J. Am. Chem. Soc. 2016, 38, 810.
Stephan, D. W.; Greenberg, S.; Graham, T. W.; Chase, P.; Hastie, J. J.; Geier, S. J.; Farrell, J. M.; Brown, C. C.; Heiden, Z. M.; Welch, G. C.; Ullrich, M. Inorg. Chem. 2011, 50, 12338. doi: 10.1021/ic200663v
Mahdi, T. Stephan, D. W. J. Am. Chem. Soc. 2014, 136, 15809. doi: 10.1021/ja508829x
Scott, D. J.; Fuchter, M. J.; Ashley, A. E. J. Am. Chem. Soc. 2014, 136, 15813. doi: 10.1021/ja5088979
Scott, D. J.; Simmons, T. R.; Lawrence, E. J.; Wildgoose, G. G.; Fuchter, M. J.; Ashley, A. E. ACS Catal. 2015, 5, 5540. doi: 10.1021/acscatal.5b01417
Gyömöre, A.; Bakos, M.; Földes, T.; Papai, I.; Domja, N. A.; Soós, T. ACS Catal. 2015, 5, 5366. doi: 10.1021/acscatal.5b01299
Parks, D. J.; Blackwell, J. M.; Piers, W. E. J. Org. Chem. 2000, 65, 3090. doi: 10.1021/jo991828a
Piers, W. E.; Marwitz, A. J. V.; Mercier, L. G. Inorg. Chem. 2011, 50, 12252. doi: 10.1021/ic2006474
Nie, W.-L.; Klare, H. F. T.; Oestreich, M.; Froehlich, R.; Kehr, G.; Erker, G. Z. Naturforsch. 2012, 67b, 987.
Ermeke, J.; Mewald, M.; Oestreich, M. J. Am. Chem. Soc. 2013, 135, 17537. doi: 10.1021/ja409344w
Houghton, A. Y.; Hurmalainen, J.; Mansikkamaeki, A.; Piers, W. E.; Tuononen, H. M. Nat. Chem. 2014, 6, 983. doi: 10.1038/nchem.2063
Nimmagadda, R. D.; McRae, C. Tetrahedron Lett. 2006, 47, 5755. doi: 10.1016/j.tetlet.2006.06.007
Chatterjee, I.; Porwal, D.; Oestreich, M. Angew. Chem., Int. Ed. 2017, 56, 3389. doi: 10.1002/anie.201611813
Yang, W.-Y.; Gao, L.; Lu, J.; Song, Z.-L. Chem. Commun. 2018, 54, 4834. doi: 10.1039/C8CC01163J
Parks, D. J.; Piers, W. E. J. Am. Chem. Soc. 1996, 118, 9440. doi: 10.1021/ja961536g
Blackwell, J. M.; Foster, K. L.; Beck, V. H.; Piers, W. E. J. Org. Chem. 1999, 64, 4887. doi: 10.1021/jo9903003
Tahara, A.; Sunada, Y.; Takeshita, T.; Inoue, R.; Nagashima, H. Chem. Commun. 2018, 54, 11192. doi: 10.1039/C8CC04780D
胡茜, 田冲, Borzov Maxim, 聂万丽, 化学学报, 2015, 73, 1025. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345151.shtmlHu, X.; Tian, C.; Borzov, M.; Nie, W.-L. Acta Chim. Sinica, 2015, 73, 1025(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345151.shtml
田冲, 姜亚, Borzov Maxim, 聂万丽, 化学学报, 2015, 73, 1203). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345179.shtmlTian, C.; Jiang, Y.; Borzov, M.; Nie, W.-L. Acta Chim. Sinica 2015, 73, 1203(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract345179.shtml
温志国, 田冲, Borzov Maxim, 聂万丽, 化学学报, 2016, 74, 498).Wen, Z.-G.; Tian, C.; Borzov, M.; Nie, W.-L. Acta Chim. Sinica 2016, 74, 498(in Chinese).
Nie, W.-L.; Sun, G.-F.; Tian, C.; Borzov, M. Z. Naturforsch. 2016, 71(10)b, 1029.
张露文, 温志国, Borzov Maxim, 聂万丽, 化学学报, 2017, 75, 819). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346108.shtmlZhang, L.-W.; Wen, Z.-G.; Borzov, M.; Nie, W.-L. Acta Chim. Sinica 2017, 75, 819(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346108.shtml
Fasano, V.; Radcliffe, J. E.; Ingleson, M. J. ACS Catal. 2016, 6(3), 1793. doi: 10.1021/acscatal.5b02896
Fasano, V.; Ingleson, M. J. Chem. Eur. J. 2017, 23(9), 2217. doi: 10.1002/chem.201605466
孙国峰, 苏敏, 方洁, Borzov Maxim, 聂万丽, 化学学报, 2017, 75, 824). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346107.shtmlSun, G.-F.; Su, M.; Fang, J.; Borzov, M.; Nie, W.-L. Acta Chim. Sinica 2017, 75, 824(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346107.shtml
何云清, 腾金伟, 田冲, Borzov Maxim, 胡启山, 聂万丽, 化学学报, 2018, 76, 774). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346719.shtmlHe, Y.-Q.; Teng, J. W.; Tian, C.; Borzov, M.; Hu, Q. S.; Nie, W.-L. Acta Chim. Sinica 2018, 76, 774(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract346719.shtml
He, Y.-Q.; Zou, M.-Y.; Xue, Y.; Hu, Q.-S.; Borzov, M. V.; Nie, W.-L. Mechanism Aspects of the B(C6F5)3 Catalyzed Reductive Amination, Chem-Eur. J. 2018, Submitted.
Bergamaschi, G.; Lascialfari, L.; Pizzi, A.; Espinoza, M. I. M.; Demitri, N.; Milani, A.; Gori, A.; Metrangolo, P. Chem. Commun. 2018, DOI: 10.1039/C8CC06010.
Beck, A. D. J. Chem. Phys. 1993, 98, 5648. doi: 10.1063/1.464913
Parr, R. G.; Yang, W. Density Functional Theory of Atoms and Molecules, Oxford University Press, Oxford, 1989.
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O. Nakai, H. Vreven, T. Throssell, K. Montgomery, J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016.
The typical 11B NMR signal of[R3NH] [HO-B(C6F5)3] is located at δ-3.9; the corresponding 19F NMR signals of o-, p-and m-F in[HO-B(C6F5)3] are at δ -135.6, -160.1, -164.8, respectively.
Bergquist, C.; Bridgewater, B. M.; Harlan, C. J.; Norton, J. R.; Friesner, R. A.; Parkin, G. J. Am. Chem. Soc. 2000, 122, 10581. doi: 10.1021/ja001915g
Di Saverio, A.; Focante, F.; Camurati, I.; Resconi, L.; Beringhelli, T.; D'Alfonso, G.; Donghi, D.; Maggioni, D.; Mercandelli, P.; Sironi, A. Inorg. Chem. 2005, 44, 5030. doi: 10.1021/ic0502168

图 4 11B NMR(上图)和19F NMR(下图)核磁共振对氯仿中B(C6F5)3络合形式的研究: A: 1a与B(C6F5)3 (10 mmol%); B: 1a与B(C6F5)3及3 equiv. H2O; C: B(C6F5)3催化的1 equiv. 1a, 1 equiv. PhSiH3及3 equiv. H2O的反应
Figure 4 11B NMR (upper) and 19F NMR (down) complexation study of B(C6F5)3 in CDCl3, A: 1a with B(C6F5)3 (10 mmol%); B: 1a with B(C6F5)3 and 3 equiv. H2O; C: reaction of 1 equiv. 1a, 1 equiv. PhSiH3 with 3 equiv. H2O catalyzed by B(C6F5)3

图 6 有水条件下B(C6F5)3催化的醛酮还原反应机理
Figure 6 The mechanism of B(C6F5)3 catalyzed reduction of carbonyl compounds under water conditions
表 1 氢化硅烷参与下B(C6F5)3催化的苯甲醛的还原
Table 1. B(C6F5)3-catalyzed hydrogenation of PhCHO using hydridosilane
|
||||
| Entry | Water (n equiv.) |
Silane a (1 equiv.) |
Product 2a+3a |
Yield of 2a/% |
| 1 | 0 | PhSiH3 | 3a | 0 |
| 2 | 1 | PhSiH3 | 3a | 0 |
| 3 | 2 | PhSiH3 | 2a | 88.5 |
| 4 | 3 | PhSiH3 | 2a | 100 |
| 5 | D2O c | PhSiH3 | 2a | 100 |
| 6 | D2O d | PhSiH3 | 1a | 0 |
| 7 | 1 | Ph2SiH2 | 2a | (78.0) e |
| 8 | 2 | Ph2SiH2 | 2a | 91.0 |
| 9 | 3 | Ph2SiH2 | 2a | 94.0 |
| 10 | 1 | Et3SiH | 2a | 64.5 |
| 11 | 2 | Et3SiH | 2a | 84.0 |
| 12 | 3 | Et3SiH | 2a | 65.5 |
| a Reaction with PhSiH3 2 h/with Ph2SiH2 6 h/with Et3SiH 12 h, the 2a yield determined by 1H NMR of the crude mixture using C6Me6 as internal standard; b n H2O: n equiv. over silanes; c D2O as solvent, CDCl3 extracting; d catalyst free; e conversion. | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 氢化硅烷参与下B(C6F5)3催化的不同取代醛酮的还原
Table 2. B(C6F5)3-catalyzed hydrogenation of carbonyl functions using hydridosilane
|
|
下载: 导出CSV
表 3 “LB---H-O(-H)---B(C6F5)3”三组分复合物中原子间距优化数据
Table 3. The optimized interatomic distances for "LB---H-O(-H)--- B(C6F5)3" three component adducts
|
|||||
| Entry | LB | B―O (Å) |
LB―H1 (Å) |
H1―O (Å) |
H2―O (Å) |
| 1 | Et3N | 1.509 | 1.072 | 1.628 | 0.969 |
| 2 | THF | 1.615 | 1.591 | 1.017 | 0.976 |
| 3 | PhCH2OH | 1.612 | 1.603 | 1.014 | 0.977 |
| 4 | PhCHO | 1.614 | 1.630 | 1.010 | 0.976 |
| 5 | H2O | 1.615 | 1.639 | 1.006 | 0.977 |
| 6 | Ph3P | 1.619 | 2.168 | 1.023 | 0.977 |
| 7 | PhCHO | 1.597 | ― | ― | ― |
| 8 | ― | 1.669 | ― | 0.980 | 0.981 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们