图 1.
CdSe量子点的TEM图(a)和荧光光谱图(b)
Figure 1.
Representative TEM image (a) and photoluminescence spectrum (b) of CdSe QDs

莱克多巴胺和克伦特罗都是胺类药物, 俗称“瘦肉精”, 能抑制脂肪沉积、提高机体的瘦肉率和饲料转化率.大量或长期使用, 则会在动物组织中蓄积, 人食用这种动物产品后会引发中毒甚至死亡.目前, 检测瘦肉精的方法有气相色谱法[1]、高效液相色谱法[2]、酶联免疫法[3]、电化学发光法[4]等.
随着纳米科技的迅速发展, 纳米粒子以其比表面积大、易于合成、优良的稳定性以及良好的生物亲和性等优点而被广泛用于分析检测中[5].量子点, 又称半导体纳米晶体, 因其独特的荧光特性而广泛应用于发光装置与生物标记领域[6], 其荧光特性通常通过光致发光、电致化学发光等进行研究[7].电致化学发光在量子点的基础研究和分析应用中是一种非常有用的技术, 量子点电化学发光通过电化学控制, 不需激发光源, 背景小, 灵敏度高. Dong等[8]通过化学氧化活性炭制备了直径3~4 nm的石墨烯量子点, 具有很强的电化学发光特性. Ma等[9]用氨基脲作为共反应剂增强CdTe量子点的电化学发光强度检测凝血酶. Jie等[10]研究了树枝状的CdSe-ZnS量子点纳米簇用于电化学发光检测肿瘤细胞. Zhang等[11]构建了ZnO@C纳米球量子点检测K562白血病细胞, 检出限为46细胞•mL−1.杨淑平等[12]制备了高性能的CdTe/CdS核壳型量子点用于小麦面粉中呕吐毒素的荧光免疫检测研究, 检出限达0.038 ng•mL-1.毛细管电泳(CE)量子点(QD)电化学发光(ECL)结合了CE的高分离效率和QD’s ECL检测的高灵敏度的优点, 在复杂样品分析中很具潜力.然而, 到目前为止, CE分离和QD’s ECL检测联用的研究还未见文献报道, 主要原因可能是常规毛细管电泳进样量太小, 样品经CE分离后对检测池中量子点的发光强度的影响不太明显.
毛细管电泳由于进样量小, 使检测灵敏度比较低, 近年来出现了大量在线富集方法如场放大进样[13]、等速电泳[14]、固相萃取[15]等富集技术来提高检测灵敏度.作者也在毛细管电泳预富集方面做了大量的工作, 研究了离子交换预富集[16]、顺序富集法[17]等.控制电渗流胶束扫集预富集可使灵敏度提高四个数量级[18].
本文研究了莱克多巴胺和克伦特罗两种胺分子对CdSe量子点电化学发光的增强作用, 结合胶束扫集预富集技术, 发展了一种基于胶束反向扫集的CE分离QD’s ECL检测的新方法.采用SDS胶束反向扫集, 带正电荷的样品离子在进样过程中被带负电荷的SDS胶束捕获并富集, 控制毛细管内的电渗流, 使胶束-样品结合物在毛细管内处于静止状态, 可使进样时间延长到50 min, 经CE分离后, 对检测池中的QD’s ECL强度起到极大的增强作用.
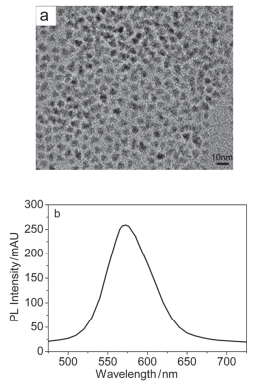
图 1为CdSe量子点的透射电镜(TEM)图(a)和荧光光谱图(b), 从图中可以看出, CdSe量子点的尺寸比较均匀, 晶体结构较良好, 颗粒分散, 粒径约为5~6 nm(图 1a); 荧光发射峰在573 nm处, 并且峰强度非常高(图 1b), 表明此量子点具有良好的光学特性.
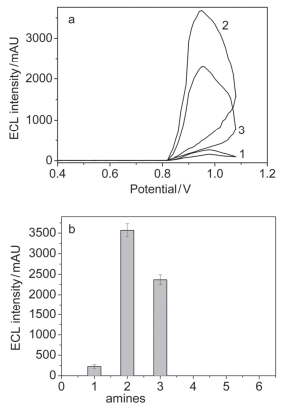
从图 2a可以看出, CdSe量子点在0.95 V左右有明显的氧化峰(曲线1), 当在量子点的溶液中加入莱克多巴胺和克伦特罗时, 氧化峰增大(曲线2和3), 说明莱克多巴胺和克伦特罗也被氧化.从图 2b也可以清楚地看出, CdSe QD’s ECL传感器放入含有莱克多巴胺和克伦特罗的溶液中时, QD’s ECL强度明显增强, 其最佳检测电势为0.95 V(S1, 支持信息).胺(amine)分子作为共反应剂, 可能的电致化学发光机理如下所示:
|
$ {\rm{QD - }}{{\rm{e}}^{\rm{ - }}} \to {\rm{Q}}{{\rm{D}}^{ + \bullet }} $ |
(1) |
|
$ {\rm{Amine - }}{{\rm{e}}^{\rm{ - }}} \to {\rm{Amin}}{{\rm{e}}^{{\rm{ + \bullet }}}} $ |
(2) |
|
$ {\rm{Amin}}{{\rm{e}}^{{\rm{ + \bullet }}}}{\rm{ - }}{{\rm{H}}^ + } \to {\rm{Amin}}{{\rm{e}}^{\rm{ \bullet }}} $ |
(3) |
|
$ {\rm{Q}}{{\rm{D}}^{{\rm{ + \bullet }}}}{\rm{ + Amin}}{{\rm{e}}^{\rm{ \bullet }}} \to {\rm{Q}}{{\rm{D}}^{\rm{*}}}{\rm{ + Amine}}\left( {{\rm{fragments}}} \right) $ |
(4) |
|
$ {\rm{Q}}{{\rm{D}}^{\rm{*}}} \to {\rm{QD}} + h\nu $ |
(5) |
组装到电极上的CdSe QD氧化为纳米晶自由基(QD+•), 同时共反应物Amine被氧化为Amine+•, 接着Amine+•失去质子(H+)生成强还原剂Amine•, 由于Amine•的还原电势比QD+•更负, QD+•与Amine•反应过程中, QD+•得到电子生成激发态的量子点(QD*)[19], 激发态的QD*回到基态时发光.
由于本实验中莱克多巴胺和克伦特罗在QD’s ECL检测前先进行胶束反向扫集预富集, 因此莱克多巴胺和克伦特罗进入检测池后都被富集在SDS胶束上.研究了SDS胶束对CdSe量子点的ECL性能的影响, 取两份完全相同的量子点溶液, 在其中一份中加入50 mmol•L-1的SDS胶束, 分别进行ECL强度-扫描电位实验, 并测其ECL强度, 发现SDS胶束对QD’s ECL性能没有影响.
研究了该QD’s ECL传感器的重现性(图S2, 支持信息), 连续循环电位扫描13次, ECL强度的相对标准偏差小于5.2%.
分别研究了影响CE分离莱克多巴胺和克伦特罗的条件, 包括缓冲溶液的种类、浓度、pH及分离电压, 结果表明, 最佳分离条件为50 mmol•L-1磷酸盐缓冲液, pH 7.4, 分离电压18 kV (S2: CE分离缓冲溶液种类及浓度的选择, S3: CE分离缓冲溶液pH的选择, S4: CE分离电压的选择, 支持信息).
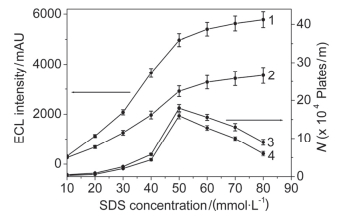
常规电动进样进样量非常小, 经CE分离后进入检测池的样品量很少, 对QD’s ECL强度的影响很弱.本实验采用胶束反向扫集预富集技术, 增大进样量, 提高检测灵敏度.由于样品离子进入毛细管后被SDS胶束捕获, 因此SDS胶束的浓度影响样品离子的富集效果.将不同浓度(10~80 mmol•L-1)的SDS胶束加入到50 mmol•L-1的磷酸盐缓冲溶液中, 如图 3所示, 当SDS胶束浓度为10 mmol•L-1时, 胶束浓度太低, 不足以将样品离子全部捕获, 样品区带宽, 分离效率很差, 随着SDS浓度的增大, QD’s ECL强度增大, 分离效率升高, 当SDS胶束浓度为50 mmol•L-1时, 分离效率达最大值, 继续增大SDS胶束浓度, 虽然QD’s ECL强度继续增大, 但由于高浓度的SDS胶束产生大量的焦耳热, 使分离效率迅速下降, 因此, 富集缓冲溶液中SDS胶束浓度选择50 mmol•L-1.
电动进样过程中, SDS胶束带负电荷, 其在毛细管中的迁移速率(VSDS)与其电泳速率(Vep, SDS)及电渗流速率(Veo)的关系为:
|
$ {V_{{\rm{SDS}}}} = {V_{{\rm{eo}}}} - {V_{{\rm{ep,SDS}}}} $ |
(1) |
带正电荷的样品离子进入毛细管后被SDS胶束捕获形成胶束-样品结合物, 使胶束的电泳速率减小而将样品离子堆积在毛细管入口端, 结合物的迁移速率(VSDS-S)可表示为:
|
$ {V_{{\rm{SDS - S}}}} = {V_{{\rm{eo}}}} - {V_{{\rm{ep,SDS - S}}}} $ |
(2) |
从式(2)可以看出, 当毛细管中的电渗流等于结合物的电泳速率时, 结合物在毛细管中的净迁移速率为0, 可以无限延长进样时间, 极大地增大进样量, 从而增大QD’s ECL强度.而电渗流随缓冲溶液pH的改变而变化, 研究了不同pH(8~3)条件下胶束-样品结合物的迁移时间, 发现随着pH的减小, 电渗流速减小, 结合物的迁移时间延长, 当pH=5时, 80 min后结合物才被检测到, 而当pH=4.5时, 160 min时还未检测到结合物, 说明此时结合物的迁移速率(即堆积界面的迁移速率)很小, 接近于0, 因此富集缓冲液的pH选择4.5.
尽管pH=4.5时胶束-样品结合物的净迁移速率接近于0, 可以大大延长进样时间而保持堆积界面处于毛细管入口端, 不会缩短后续分离毛细管的有效长度.但进样时间还受样品在胶束中的保留因子(k)的限制[20]:
|
$ {l_{{\rm{sweep}}}} = \left( {\frac{1}{{1 + k}}} \right){l_{{\rm{inj}}}} $ |
(3) |
lsweep和linj分别是扫集后样品区带和进样溶液区带长度.因此, 进样时间还需通过实验确定.研究了进样时间(5~70 min)对QD’s ECL强度和分离效率的影响(图S6, 支持信息), 随着进样时间的延长, QD’s ECL强度增大, 当进样时间超过50 min后, ECL强度增大的趋势变小, 分离效率降低, 说明进样时间50 min时, SDS胶束接近饱和, 继续增大进样时间时, 部分样品不再被SDS胶束捕获, 导致富集区带展宽.因此, 进样时间选择50 min.
同常规电动进样(15 kV×20 s)相比, 采用胶束反向富集技术、同时控制电渗流使胶束-样品结合物在富集过程中处于准静止状态, 极大地增加进样量(20 kV×50 min), 提高QD’s ECL强度, 使检测灵敏度提高6000倍(峰高比×样品稀释因子)(图 4).
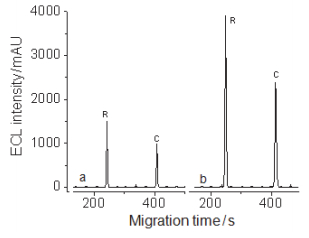
Sample injection: a, 15 kV×20 s; b, 20 kV×50 min. The concentration of ractopamine and clenbuterol: a, 100 mg•L-1; b, 50 µg•L-1. Sweeping buffer: 50 mmol•L-1 phosphate+50 mmol•L-1 SDS, pH 4.5. Separation buffer, 50 mmol•L-1 phosphate, pH=7.4. Detection potential, 0.95 V
在最佳实验条件下研究了胶束反向扫集CE分离QD’s ECL检测莱克多巴胺和克伦特罗的工作曲线、线性范围、检出限和精密度(相对标准偏差, RSD).测定了莱克多巴胺的浓度在0.5~3500 µg/L、克伦特罗的浓度在1.0~6000 µg/L范围内, 其峰高与浓度的关系(图S7, 支持信息), 结果表明, 在0.8~2960 µg/L范围内, 莱克多巴胺的峰高与浓度成良好的线性关系(图S7A, 支持信息), 回归方程为y=73x+32 (y为峰高, mAU; x为莱克多巴胺的浓度, µg/L), 线性相关系数R2为0.9987, 检出限为96.8 ng/L, 峰高和峰面积的RSD(n=5)分别为3.5%和5.1%;在3.0~5520 µg/L范围内, 克伦特罗的峰高与浓度成良好的线性关系(图S7B, 支持信息), 回归方程为y=43x+36 (y为峰高, mAU; x为克伦特罗的浓度, µg/L), 线性相关系数R为0.9926, 检出限为192.5 ng/L, 峰高和峰面积的RSD(n=5)分别为4.4%和6.3%.
将预处理好的猪肉样品(S4.2样品处理)分别采用本实验研究的胶束反向扫集CE分离QD’s ECL法和酶联免疫吸附法(ELISA)进行检测, 结果见表 1, 两种检测方法的结果一致, 说明本方法准确、可靠.
 下载:
导出CSV
下载:
导出CSV
| Sample No. | The proposed method | ELISA | |||
| Ractopamine (µg•L-1) |
Clenbuterol (µg•L-1) |
Ractopamine (µg•L-1) |
Clenbuterol (µg•L-1) |
||
| 1 | 4.1±1.6 | 3.9±1.2 | 3.8±0.9 | 4.5±1.7 | |
| 2 | 8.7±3.7 | 9.1±3.2 | 9.5±2.6 | 8.9±3.5 | |
| 3 | 19.2±4.3 | 18.7±3.9 | 17.9±3.1 | 18.1±4.5 | |
| 4 | 48.5±5.6 | 48.1±4.4 | 48.8±6.1 | 49.1±5.8 | |
为进一步评价方法的准确性, 进行了回收率实验, 在猪肉样品中按低、中、高三个浓度水平(10, 100, 500 µg/L)添加标准溶液进行回收实验和精密度实验, 发现莱克多巴胺和克伦特罗的回收率分别在91.5%~104.8%(RSD 3.6%~5.1%)和96.1%~107.5%(RSD 4.3%~6.8%)之间, 方法准确可靠, 可用于猪肉样品中莱克多巴胺和克伦特罗的同时分离测定.
本文研究了两种胺分子对CdSe量子点电化学发光的增强作用, 发展了CE分离QD’s ECL检测的新方法. CE分离和QD’s ECL检测的成功联用, 不仅扩展了CE的检测方式, 还扩大了QD’s ECL在复杂样品中的多组分同时检测的应用领域.由于常规CE进样量小, 经CE分离后的胺分子对QD’s ECL强度的增强作用有限, 本文在进样时采用胶束反向扫集预富集技术, 通过控制电渗流使胶束-样品结合物在进样过程中处于准静止状态, 可以大大延长进样时间, 对QD’s ECL强度起到极大的增强作用.通过实际样品的分析证明, 基于胶束反向扫集的CE分离QD’s ECL检测对复杂样品中微量不同胺分子的同时分离检测具有良好的应用前景.
CE分离缓冲溶液为50 mmol•L-1磷酸盐, pH=7.4;胶束反向扫集富集缓冲液为50 mmol•L-1磷酸盐+50 mmol•L-1 SDS, pH 4.5.其他试剂和仪器见支持信息(S 4.1试剂与仪器).
首先制备CdSe量子点:基于文献[10], 对量子点的制备进行适当改进, 在三口烧瓶中加入5 mL蒸馏水, 边通N2除氧边加入150 mg NaBH4、50 mg硒粉, 继续通氮10 min; 停止通氮, 塞住瓶口, 80 ℃反应30 min; 置于冰水浴中冷却, 得到无色澄清溶液, 即前驱体NaHSe.另取三口烧瓶, 在氮气保护下, 依次放入搅拌磁子、100 mL 2.5 mmol•L-1 CdC12溶液、0.1 mL巯基乙酸, 调节pH 9~10, 持续通氮30 min; 磁力搅拌下升温至90 ℃, 迅速加入3 mL NaHSe前驱体溶液, 继续于90 ℃下搅拌回流2~3 h, 得到黄色溶液, CdSe水溶性量子点.避光保存.
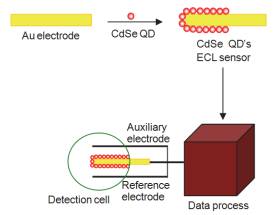
QD’s ECL传感器的制备(如图 5所示):取5 µL上述制备的CdSe量子点滴涂到金电极表面, 汞灯照射5 min, 即可将CdSe量子点修饰到金电极表面; 然后放入到检测池中, 与甘汞(SCE)参比电极, 铂丝对电极形成三电极体系, 进行电致化学发光的性能研究.
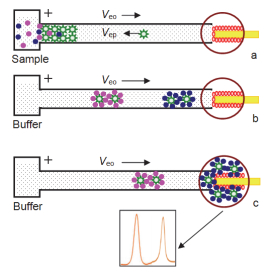
基于胶束反向扫集的CE分离QD’s ECL检测原理如图 6所示:毛细管内首先充满富集缓冲液(50 mmol• L-1磷酸盐+50 mmol•L-1 SDS, pH 4.5), 电动进样时, 样品离子快速迁移进入毛细管, 在入口端被SDS胶束捕获并富集(图 6a), SDS胶束带负电荷, 向毛细管正极端迁移, 捕获带正电荷的样品离子后, 电泳速率减慢, pH 4.5时, 胶束-样品结合物的电泳速率近似等于电渗流速率, 因此胶束-样品结合物处于准静止状态, 这种静止状态可以大大延长进样时间、增大进样体积; 20 kV进样50 min后, 毛细管两端换上pH=7.4的分离缓冲液, 电渗流增大, 被富集的样品离子经CE分离(图 6b)后顺次进入检测池, 被胶束富集的高浓度的分析物显著增强量子点的ECL电化学发光强度(图 6c), 实现对待测物的定量分析.
Regueiro, J. A. G.; Pérez, B.; Casademont, G. J. Chromatogr. A 1993, 655, 73. doi: 10.1016/0021-9673(93)87012-B
Josefsson, M.; Sabanovic, A. J. Chromatogr. A 2006, 1120, 1. doi: 10.1016/j.chroma.2006.03.013
Wei, L.; Chengbian, Z.; Yulan, Z. J. Agric. Food Chem. 2007, 55, 6829. doi: 10.1021/jf070620k
(a) Yao, X.; Yan, P.; Tang, Q.; Deng, A.; Li, J. Anal. Chim. Acta 2013, 798, 82. (b) Chen, X.; Wu, R.; Sun, L.; Yao, Q.; Chen, X. J. Electroanal. Chem. 2016, 781, 310.
(a) Zhang, Z. X.; Zhang, F.; Liu, Y. Acta Chim. Sinica 2012, 70, 2251(in Chinese). (张召香, 张飞, 刘营, 化学学报, 2012, 70, 2251.) (b) Zhang, Z. X.; Luan, W. X.; Zhang, C. Y.; Liu, Y. J. Acta Chim. Sinica 2017, 75, 403(in Chinese). (张召香, 栾文秀, 张超英, 刘玉洁, 化学学报, 2017, 75, 403.) (c) Zhang, Z. X.; Li, X. L.; Ge, A. Q.; Zhang, F.; Sun, X. M.; Li, X. M. Biosens. Bioelectron. 2013, 41, 452. (d) Zhang, Z. X.; Zhang, C. Y.; Luan, W. X.; Li, X. F.; Liu, Y.; Luo, X. L. Anal. Chim. Acta 2015, 888, 27. (e) Wang, Q.; Nie, Z.; Hu, Y.; Yao, S. Acta Chim. Sinica 2017, 75, 1109(in Chinese). (王琴, 聂舟, 胡宇芳, 姚守拙, 化学学报, 2017, 75, 1109.)
(a) Ponomarenko, L. A.; Schedin, F.; Katsnelson, M. I.; Yang, R.; Hill, E. W.; Novoselov, K. S.; Geim, A. K. Science 2008, 320, 356. (b) Hai, X.; Guo, Z.; Lin, X.; Chen, X.; Wang, J. ACS Appl. Mater. Interfaces 2018, 10, 5853. (c) Bacon, M.; Bradley, S. J.; Nann, T. Part. Part. Syst. Char. 2014, 31, 415.
(a) Zhu, H.; Wang, X.; Li, Y.; Wang, Z.; Yang, F.; Yang, X. Chem. Commun. 2009, 5118. (b) Loh, K. P.; Bao, Q.; Eda, G.; Chhowalla, M. Nature Chem. 2010, 2, 1015. (c) Lei, J.; Ju, H. Trends Anal. Chem. 2011, 30, 1351.
Dong, Y.; Zhou, N.; Lin, X.; Lin, J.; Chi, Y.; Chen, G. Chem. Mater. 2010, 22, 5895. doi: 10.1021/cm1018844
Ma, M. N.; Zhuo, Y.; Yuan, R.; Chai, Y. Q. Anal. Chem. 2015, 87, 11389. doi: 10.1021/acs.analchem.5b02848
Jie, G.; Wang, L.; Yuan, J.; Zhang, S. Anal. Chem. 2011, 83, 3873. doi: 10.1021/ac200383z
Zhang, M.; Liu, H.; Chen, L.; Yan, M.; Ge, L.; Ge, S.; Yu, J. Biosens. Bioelectron. 2013, 49, 79. doi: 10.1016/j.bios.2013.05.003
杨淑平, 金鑫, 郑佳, 陶丽华, 李在均, 柯叶芳, 刘俊康, 化学学报, 2011, 69, 687. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract339867.shtmlYang, S.; Jin, X.; Zheng, J.; Tao, L.; Li, Z.; Ke, Y.; Liu, J. Acta Chim. Sinica 2011, 69, 687(in Chinese). http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract339867.shtml
Zhu, Q.; Xu, X.; Huang, Y.; Xu, L.; Chen, G. J. Chromatogr. A 2012, 1246, 35. doi: 10.1016/j.chroma.2012.02.005
Bahga, S. S.; Chambers, R. D.; Santiago, J. G. Anal. Chem. 2011, 83, 6154. doi: 10.1021/ac200268f
Ibarra, I. S.; Rodriguez, J. A.; Miranda, J. M.; Vega, M.; Barrado, E. J. Chromatogr. A 2011, 1218, 2196. doi: 10.1016/j.chroma.2011.02.046
Zhang, Z. X.; He, Y. Z. J. Chromatogr. A 2005, 1066, 211. doi: 10.1016/j.chroma.2005.01.081
(a) Zhang, Z. X.; He, Y. Z.; Hu, Y. Y. J. Chromatogr. A 2006, 1109, 285. (b) Zhang, Z. X.; Zhang, X. W.; Wang, J. J.; Zhang, S. S. Anal. Bioanal. Chem. 2008, 390, 1645.
Zhang, X. W.; Zhang, Z. X. J. Pharmaceut. Biomed. 2011, 56, 330. doi: 10.1016/j.jpba.2011.05.016
Shin, I.; Kim, J.; Kwon, T.; Hong, J.; Lee, J.; Kim, H. J. Phys. Chem. C 2007, 111, 2280. doi: 10.1021/jp067644z
Quirino, J. P.; Terabe, S. Anal. Chem. 1999, 71, 1638. doi: 10.1021/ac9810866

图 1 CdSe量子点的TEM图(a)和荧光光谱图(b)
Figure 1 Representative TEM image (a) and photoluminescence spectrum (b) of CdSe QDs

图 2 莱克多巴胺和克伦特罗对CdSe量子点电化学发光强度的影响
Figure 2 Effect of ractopamine and clenbuterol on the ECL intensities of CdSe QDs. (1) No amines; (2) 2.0 mg•L-1 ractopamine; (3) 2.0 mg•L-1 clenbuterol. Measured in phosphate buffer (pH 7.4). (a) Scan rate: 100 mV•s-1; (b) detection potential, 0.95 V

图 3 SDS胶束浓度对QD’s ECL强度(曲线1, 2)和分离效率(3, 4)的影响
Figure 3 Effect of SDS micelles concentration on QD's ECL intensity (curve 1, 2) and separation efficiency (curve 3, 4). (curve 1 and 3, ractopamine; curve 2 and 4, clenbuterol). Sample injection, 20 kV for 10 min

图 4 莱克多巴胺(R)和克伦特罗(C)的CE QD’s ECL检测谱图
Figure 4 Electropherograms of ractopamine (R) and clenbuterol (C) with (b) and without (a) micellar reversed sweeping preconcentration
Sample injection: a, 15 kV×20 s; b, 20 kV×50 min. The concentration of ractopamine and clenbuterol: a, 100 mg•L-1; b, 50 µg•L-1. Sweeping buffer: 50 mmol•L-1 phosphate+50 mmol•L-1 SDS, pH 4.5. Separation buffer, 50 mmol•L-1 phosphate, pH=7.4. Detection potential, 0.95 V

图 6 基于胶束反向扫集的毛细管电泳量子点电化学发光示意图
Figure 6 Schematic protocol of CE separation and QD's ECL detection by micellar reversed sweeping
表 1 加标猪肉样品中莱克多巴胺和克伦特罗的检测结果
Table 1. Results of detection of ractopamine and clenbuterol in meat samples added standards using the proposed method and ELISA
| Sample No. | The proposed method | ELISA | |||
| Ractopamine (µg•L-1) |
Clenbuterol (µg•L-1) |
Ractopamine (µg•L-1) |
Clenbuterol (µg•L-1) |
||
| 1 | 4.1±1.6 | 3.9±1.2 | 3.8±0.9 | 4.5±1.7 | |
| 2 | 8.7±3.7 | 9.1±3.2 | 9.5±2.6 | 8.9±3.5 | |
| 3 | 19.2±4.3 | 18.7±3.9 | 17.9±3.1 | 18.1±4.5 | |
| 4 | 48.5±5.6 | 48.1±4.4 | 48.8±6.1 | 49.1±5.8 | |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们