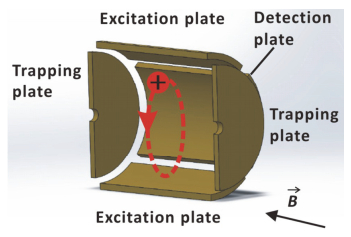
图 1.
磁场中的离子回旋运动及离子回旋共振池
Figure 1.
Ion cyclotron motion in a magnetic field and ion cyclotron resonance cell

1998年, 康奈尔大学的McLafferty教授等[1]在傅里叶变换离子回旋共振(Fourier transform ion cyclotron resonance, FTICR)质谱仪上观察到, 气相多电荷正离子可以通过捕获低能量电子而产生裂解碎片, 并将其命名为电子捕获裂解(electron capture dissociation, ECD).经过二十年的发展, ECD的基础理论、仪器方法、应用研究已经取得长足进步, 成为现代质谱分析中最重要且富有生命力的研究前沿, 为后基因组时代的生命分析问题提供新思路和新方法[2~5].
ECD的本质是有机分子离子自由基化学[6].与早期电子轰击电离(electron impact ionization, EI)方法不同, 经典的ECD方法通常采用低能量电子(<0.2 eV)与离子发生反应.随着电喷雾(electrospray ionization, ESI)和基质辅助激光解离(matrix-assisted laser desorption ionization, MALDI)等软电离技术的出现, 研究闭壳层偶电子离子成为可能[7~11].在这种条件下, 典型的生物大分子(如蛋白质和多肽)的ECD反应主要发生主链N-Cα裂解, 并保留翻译后修饰基团及分子的弱相互作用(如氢键).与其他“慢热”裂解方式不同, ECD谱可以提供丰富的碎片离子信息, 为生物分子分析提供新的视角[12~15].
与ECD类似, 电子转运裂解(electron transfer disscociation, ETD)是另一种实现离子与低能量电子反应的裂解方法[16]. Hunt等[16]发现, 多电荷的蛋白质或多肽正离子可以与负离子自由基反应, 其裂解行为与ECD类似.二者的区别在于: (1)机理上, ETD中的负离子自由基可以产生额外的反应途径, 例如蛋白质或多肽离子中质子向负离子自由基转移反应(proton-transfer reaction, PTR)等[17~19]; (2)仪器上, 待分析的正离子束与ETD负离子云更容易重叠, 因此ETD可以很方便的在四级杆离子阱或线性离子阱中实现[16, 20, 21].目前大多数商业质谱仪器配备有ETD模块, 国内外许多研究小组在此基础上做了大量有关生物分子分析的研究工作, 感兴趣的读者可以参考相关的综述文献[22~25].
国际上有关ECD的研究是质谱学、物理化学、蛋白质组学、结构生物学等多领域共同关注的热点问题.我国在此领域的研究相对滞后, 相关的成果较少在国际会议及期刊上发表[22, 23].本文详细总结了ECD技术的基本概念及原理, 同时结合本课题组的研究, 展望ECD的研究热点和发展前沿.希望这篇文章可以方便我国学者了解该领域研究现状并理清源头.
迄今为止, ECD主要是在FTICR质谱仪上实现的.现今的FTICR质谱具有超高分辨率以及多样化的离子裂解方式, 是现代生物分子分析的重要工具之一[26~30].鉴于其在生物分子分析中的重要作用, 尤其是对ECD的重要性, 本节简要介绍FTICR质谱的基本概念及相关的实验技术.
FTICR质谱的基础是离子回旋运动.在均匀的磁场中, 处于真空的带电离子由于受到洛伦兹力的作用, 将在垂直磁场方向的平面内进行匀速圆周运动(图 1).根据经典电磁学理论, 离子运动的频率f由下式确定:
|
$ f{\rm{ = }}\frac{B}{{2\pi }}/\frac{m}{q} $ |
其中, B为磁场强度, m/q为带电离子的质荷比.由这个基本式子可以看出: (1)离子的质荷比可以由其回旋频率测定; (2)离子回旋频率与离子动能无关.这是FTICR测量离子质荷比的物理基础, 同时也是FTICR具有超高分辨率的原因之一[28].
FTICR质谱的核心部件是离子回旋共振池, 共由三组电极板构成, 分别为检测电极、激发电极和囚禁电极(图 1).在FTICR质谱中, 离子回旋频率是通过检测电极上的镜像电流来测定的(图 1).当离子回旋经过电极板时, 由于静电效应会在靠近的电极板感应出相反电荷[28].随着离子的周期运动, 这一对检测电极将会在其检测电路中产生相同频率的镜像电流.该信号进一步放大处理就可以得到离子回旋频率f.
然而测定离子回旋运动的频率有两个难点:第一, 离子在垂直磁场平面内的初相位是均匀分布的, 离子包(ion package)的感应电流互相叠加的统计结果为零; 第二, 在室温下, 离子回旋半径的数量级约为10-1 mm, 距离检测电极很远不足以产生可检测的镜像电流[28].由此可见, 初始状态下离子回旋运动的频率几乎无法直接测量, 而激发电极可以很好的解决这两个问题.在激发电极之间的交流电场作用下, 离子变为受迫运动.当外加射频电场频率与离子固有频率一致时, 离子发生相干共振, 其运动半径增加, 且相位一致.一般来说, 激发电极会发出一组射频, 包含数目众多的频率信号, 回旋池内相应频率的离子均被激发、测量.最后, 通过傅里叶变换就可以将检测电极上的复合信号转换为相应的频率, 从而得到各个离子的质荷比.
离子在磁场中所受的洛伦兹力, 以及囚禁电极产生的静电势阱可以将离子约束在回旋池内往复运动, 以便进行质荷比测量以及串联质谱实验.在现代FTICR质谱中, 回旋池部分的真空度可达10-7 Pa, 在测量时间内, 离子与背景气体分子的碰撞可以忽略不计.因此可以认为回旋池是离子的贮存器, 在回旋池内离子运动频率的测量是无损的.在不同的实验条件下, 回旋池内可以发生丰富的离子反应[31~33].典型的FTICR串联质谱实验包括碰撞诱导解离(collision induced dissociation, CID)和ECD两种方式.在典型的CID实验中, 离子中化学键的活化与断裂是通过“慢热”的方式进行的.适当提高背景中性气体的压力, 并给激发电极一个频率很接近离子共振频率的射频电压, 离子动能反复增加, 与背景气体多次碰撞并转化为离子内能.当达到特定化学键的键能阈值, 就会发生裂解反应.与CID不同, ECD的机理较为复杂, 一般认为是通过低能量电子与质子化多电荷离子的非遍历性(non-ergodic)放热过程导致的化学键断裂[12].本文将在第四部分深入讨论目前主要的几种ECD机理.
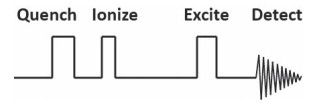
最后需要注意, 与其他类型质谱不同, FTICR质谱的所有实验均在回旋池中通过一组时间脉冲序列(pulse train)控制完成.典型的FTICR脉冲序列包括离子猝灭、离子引入、离子激发以及离子检测等四部分(图 2)[28].离子猝灭主要作用是清空回旋池内残留的离子, 一般通过升高囚禁电极一端电压实现.离子生成阶段, 外离子源产生的离子经由离子传输系统, 如六极杆或八极杆导引至回旋池.接下来的离子激发过程可以根据具体需要进行离子碎裂实验.离子检测阶段将精确记录所有离子回旋运动的时间域信号, 并通过傅里叶变换得到质荷比, 进一步分析可以得到分子结构的相关信息.
现代FTICR质谱的准确度一般小于0.5 mDa (分子量为500 Da), 分辨率可以达到几百万, 是解决生物分子分析的理想工具.目前, FTICR已经被广泛应用于完整蛋白质结构分析, 蛋白质非共价作用复合体分析, 代谢产物异构体结构阐明, 多肽分子精确组成的确定等诸多具有挑战性的问题[34, 35].
ECD是FTICR质谱的重要技术之一, 目前广泛应用于蛋白质组学、蛋白质翻译后修饰等重要生物分析领域[36].除了少数实验室在线性Paul阱上的尝试[37, 38], 一般而言, ECD实验都是在FTICR质谱上进行的.这是因为离子回旋共振池中的电磁场可以让电子稳定运动, 为电子捕获过程提供充足的反应时间.与此相反, Paul阱中的射频电场有明显的低质量滤过(low mass cut-off)效应, 很难有效地囚禁ECD所需的电子.
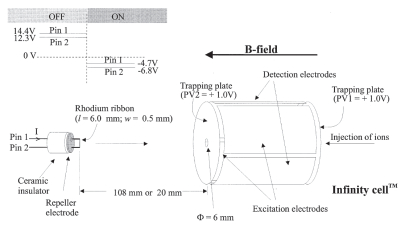
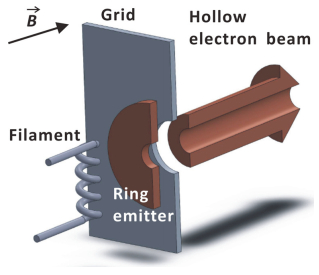
在McLafferty等[1]第一次观察到ECD现象的实验中, 低能量的自由电子是由紫外光辐射下的质谱仪腔内电极板所产生的.随后的实验中, 激光被换成灯丝电子枪, 并安置在回旋池后方(图 3).硬件优化实验表明, ECD效率与电子和离子间相对动能有关, 相对动能越小, ECD捕获效率越高[39].
作为早期ECD研究课题组之一, 我们对ECD的灯丝电子枪硬件因素进行了详尽的研究(图 3)[40].通过对商用FTICR质谱(4.7 T)的硬件升级改造, 我们发现: (1)灯丝偏置电压、电流均对ECD效率有影响; (2)在约50 ms的低能量电子(3~6 eV)辐射下, ECD效率较高; (3)向回旋池中泵入一定的惰性气体可以增加ECD碎片离子强度; (4)由于强磁场对电子运动的影响, ECD效率与电子枪到回旋池的距离位置无关.同时, 我们的实验也表明, 碎片离子对电子的二次捕获可能是影响碎片离子强度的主要因素之一.
在早期的ECD实验中, 由于灯丝电子枪产生的电子束很窄, 导致回旋池中离子捕获电子的效率很低, 且反应时间较长.现在ECD实验一般采用扩散阴极灯作为电子枪(图 4)[41], 其优点有几个方面:第一, 电子束截面大大增加, 因此ECD反应速率与效率大幅提高.第二, 回旋池内的电子束可以在其周围产生一个势阱, 有效地将正离子囚禁在电子束周围进行ECD反应.第三, 电子束势阱可以通过静电场进一步约束ECD反应过程中产生的碎片离子, 增加碎片离子丰度.实验表明, 扩散阴极灯电子枪可以将ECD反应时间缩短至几个毫秒, 使得在线HPLC联用成为可能[41, 42].
目前商用的ECD电子枪一般为空心扩散阴极灯, 其电子束的截面呈圆环形状(图 4), 中空部分没有电子[43].这样的电子枪设计可以在保持较大电子束截面积的情况下, 从中空位置引入其他类型的离子活化技术, 例如红外多光子解离(infrared multiphoton photodissociation, IRMPD)或紫外光子解离(ultraviolet photodissociation, UVPD)[43, 44].实验表明, 这些辅助的离子活化技术与ECD在时间序列上配合使用, 可以有效削弱生物大分子的分子间弱相互作用, 产生丰富的碎片离子. McLafferty等[45]观察到, 活化离子ECD技术可以将分子量为42 kDa左右的完整蛋白质分子离子去折叠, 生成大量多肽碎片离子, 获得完整蛋白质分子的结构信息.这种基于ECD的“自顶向下”质谱技术正在为大型完整蛋白质、蛋白质复合体的结构解析提供全新的分析视角[46, 47].
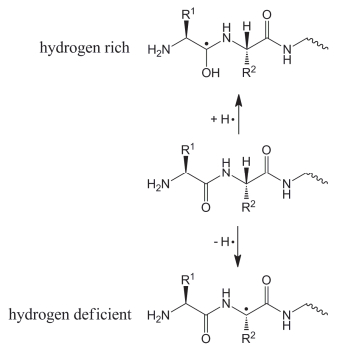
ECD反应得到的多肽自由基离子可以分为富氢自由基和贫氢自由基两类[12]. “富氢”指中性的多肽中额外增加了一个氢原子[M+H]•形成的自由基.与之相反, “贫氢”则指中性的多肽丢失一个氢原子[M-H]•而形成的自由基.对于多肽自由基离子[M+nH]n+(n是电荷数)而言, “富氢”表示在多电荷多肽离子上增加了一个电子, 记作[M+nH](n-1)+•, 其中n≥2. “贫氢”则表示质子化多肽离子减少了一个氢原子、自由基或者电子, 通常记作[M+(n-1)H]n+•, 其中n≥1, 当n=1时, 也可以写作M+•(图 5).
贫氢自由基可以由中性或者质子化的多肽电离产生, 也可以由多电荷的多肽-金属复合物裂解产生[48], 而富氢自由基则是由多电荷正离子捕获电子还原产生[1, 49].这两种自由基的电子状态及化学性质完全不同, 需要区别对待.由于ECD反应中富氢自由基离子较为常见, 因此本部分主要讨论这类离子的性质与反应机理.
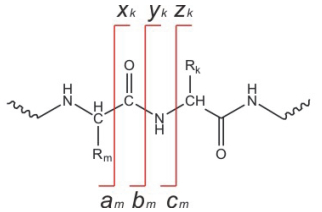
为了描述多肽自由基离子, Roepstorff, Fohlman和Biemann等[50, 51]提出多肽碎片的一般命名规则.如图 6所示, N端碎片通常标记为am, bm, cm, 其中m表示氨基酸残基α碳的数目.与之互补的C端碎片记为xk, yk, zk, 其中n=k+m是母离子氨基酸残基的数目.需要注意, 图 6所标记的并不一定是碎片离子的真实结构.一般情况下, 偶电子多肽离子裂解通常会产生b-和y-型碎片离子, 因为其熵变和焓变更为有利[52, 53].但是对于含有脯氨酸和天冬氨酸残基的多肽, 则更容易发生侧链断裂、抑制肽键裂解, 造成测序所需要信息的丢失.另外, 多肽正离子的骨架链断裂也经常伴有成环和重排反应[54~56], 这也限制了序列信息的提取.
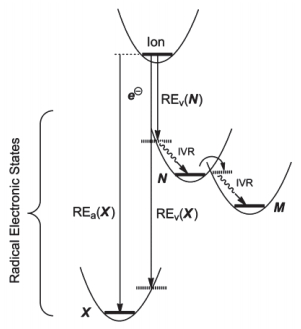
电子捕获过程是电子与多肽正离子静电场相互作用的结果.这种相互作用会产生一连串不同的电子状态, 其中第N个电子状态的绝热重组能REa=E(离子)-E(N)(图 7).绝热重组能也是基态到第N个状态的垂直重组能REv(N)与Frank-Codon能EFC之和.通过分子间振动, 重组能将重新分布耗散于第N个状态的每一个振动自由度上.如果存在足够的振动耦合, 电子状态也可以直接跨越到更低的能态, 并且与电荷还原产物的解离过程相互竞争(图 7)[57, 58].在一系列复杂的电荷跨越和振动能重新分布之后, 整个体系最终处于基态, 即离子重组能被转化成振动激发的状态[58].
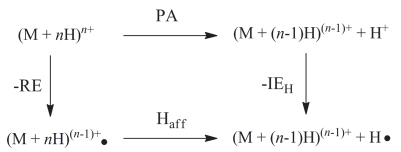
由于在实验时间内, FTICR回旋池中反应物的内能是守恒的, 因此, 重组能可以用一个热力学循环来估算[12, 59].如图 8所示, RE表示电子捕获所释放的热, 即离子-电子重组能, PA和Haff分别表示低价态的质子和氢原子亲和势, IEH则表示氢原子电离能(13.59 eV).计算表明[60~64]: (1)单质子化氨基酸、二肽和三肽的绝热重组能范围非常窄(3.2~3.7 eV); (2)质子化位点的化学环境, 如精氨酸残基上的胍基, 组氨酸残基上的咪唑环, 赖氨酸残基上的ε-氨基基团等, 并不影响绝热重组能; (3) REa随多肽离子的大小而减弱; (4)双质子化五肽的重组能通常在4.5~5.7 eV, 且与多肽的序列、组成及构象有关.
电子的捕获会使多肽自由基离子发生诸多反应, 主要包括:主链N—Cα键断裂、二硫键断裂、氢原子丢失、氨基丢失等.这些过程相互串、并联, 形成竞争或者连串反应, 与重排过程一起生成最终的产物离子, 形成ECD谱(图 9)[45].由于主链断裂在蛋白质序列与结构鉴定、翻译后修饰分析方面的重要意义, 我们首先讨论主链N—Cα键断裂的机理.
多肽ECD的一个主要特征是主链N—Cα键发生裂解, 氢原子转移到酰胺基, 生成N-端偶电子碎片(c离子)和C-端贫氢自由基(z离子)[1].同时, 产物离子可能进一步发生侧链丢失或环裂解, 生成a或x离子. ECD裂解通道主要取决于离子的电荷状态、离子内能、相对碱性等.关于ECD的研究可以归纳为两条主线, 即关注热力学、动力学、电子结构的唯理学讨论和侧重多肽主链氨基酸残基裂解频次的唯象学研究[12, 58].
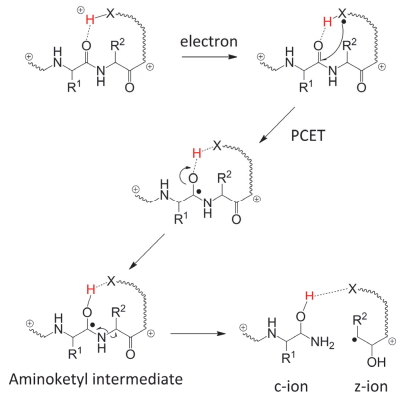
N—Cα键裂解的机理主要有康奈尔、犹他-华盛顿以及自由基级联三个模型.早期McLafferty等[1, 49]提出的康奈尔模型认为, 低能量电子被吸附到带正电的氨基酸碱性基团(赖氨酸、精氨酸或组氨酸)上, 形成自由基中间体(图 10).该中间体不稳定, 经过解离产生化学性质活跃的“热”氢原子, 并被酰胺基团捕获形成氨基羰游基(aminoketyl)自由基中间体, 从而发生N—Cα键断裂.也有部分研究者认为, 捕获电子通过高Rydberg态进入σ反键轨道也是主链N—Cα键断裂的原因[65].进一步的量子化学计算和实验表明, “热”氢原子的产生与再俘获过程的反应速率常数很小, 而质子耦合的电子转移(proton-coupled electron transfer, PCET)过程可能是多肽自由基裂解反应的关键步骤(图 10).康奈尔模型的要点是碱性氨基酸的侧基作为正电荷中心捕获电子, 产生“热”氢原子, C=O进一步进攻“热”氢原子并导致N—Cα键的断裂.可以看出, 康奈尔模型依赖于多肽上碱性氨基酸侧基, 因此, 对于不含有碱性氨基酸或碱性氨基酸侧基被保护的多肽离子, 其裂解行为就很难用该模型解释.
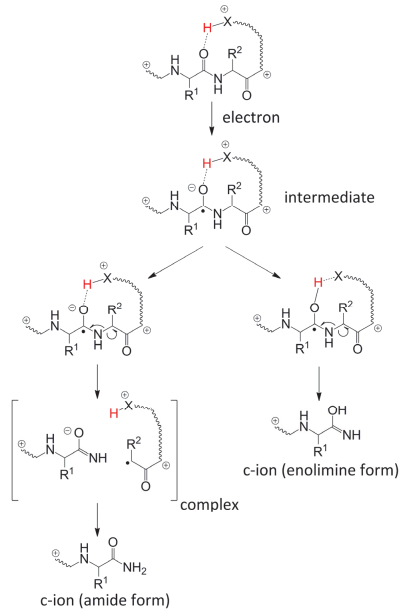
实验表明, 对于不含有氨基质子的化学修饰多肽, 主链N—Cα键也会发生裂解[61, 66, 67], 这与康奈尔模型不符.为了解释这些实验数据, 犹他大学的Simons教授[68~70]和华盛顿大学的Tureček教授[60, 65]研究小组分别独立提出犹他-华盛顿模型(图 11).这一模型认为, 在电子与多肽正离子之间的库仑作用影响下, 主链上酰胺键π*轨道的电子亲和势会增加, 并捕获ECD电子, 形成一个π*中间态.计算表明, 这个中间态的激发电子态与多肽电荷还原产物的过渡态一致.密度泛函理论计算发现, 富氢多肽正离子自由基的电子激发态与其离子-电子重组能一致.可以认为, 由高Rydberg态、氨基π*组成的电子激发态是下游ECD反应的关键步骤.事实上, 华盛顿学派认为, 接下来的反应中, 正电或中性位点的质子进攻强碱性烯醇亚胺酯(enol-imidate)负离子自由基, 形成氨基羰游基自由基, 并进而造成主链N—Cα键断裂(图 11).而犹他学派则认为, 化学性质活泼的烯醇亚胺酯负离子自由基会首先发生β诱导裂解, 使主链N—Cα键断裂, 生成的c和z型碎片.其中z型碎片上的质子进攻c型碎片强碱性烯醇亚胺酯基团, 得到ECD反应产物(图 11).犹他-华盛顿模型的两种形式的区别在于质子转移与N—Cα键断裂先后顺序不同, 但都认为酰胺的π*轨道捕获了电子.与康奈尔模型不同, 这种模型可以更好地解释不含有氨基质子的多肽离子裂解行为.
自由基级联模型是2003年O'Connor教授[71]提出的.他们在实验中观察到, 环状多肽正离子捕获电子以后, 会发生多肽主链多点位断裂. ECD的自由基级联模型认为, 电子捕获会首先造成主链N—Cα键断裂, 形成一个Cα自由基.这个活性中心进一步发生重排反应, 导致邻近N—Cα键断裂, 并形成一个新的Cα自由基.这一链式过程持续进行, 从而导致多肽主链上多点位发生裂解. Tureček研究小组[72, 73]最近在实验中也观察到类似的现象, 进一步验证了该学说.自由基级联模型弱化了c/z复合体分子内或分子间氢迁移的差异, 突出了z型碎片的自由基特性, 认为级联过程、侧基丢失、中性丢失都是自由基反应的结果.该模型可以更好地解释环状多肽离子的裂解规律, 如微囊藻素的结构分析[74].
蛋白质正离子中的二硫键可以与ECD电子发生反应并断裂.有学者注意到, 多电荷多肽离子的二硫键断裂后, 氢原子保留在电荷较多的碎片上[75], 即:
|
$ (m{{\rm{H}}^ + }){\rm{A}} - {\rm{S}} - {\rm{S}} - {\rm{B}}(n{{\rm{H}}^ + }) + {{\rm{e}}^ - } \to {\rm{A}}{{\rm{S}}^m}^ + \bullet + {\rm{BS}}{{\rm{H}}^{\left( {n - 1} \right) + }} $ |
其中n>m.一种机理认为, 氢原子被二硫键捕获后形成过渡态RS(H•)SR'自由基.该自由基发生断裂时, 氢原子的分配会受到极化作用影响.然而, 二硫键的质子亲和势小于一般氨基酸, 因此上述机理很难解释二硫键的裂解[76]. O'Connor等[77]设计了一组含有Se和S的多肽, 并且发现其ECD裂解行为可以用电子亲和势来解释.根据这一模型, 二硫键捕获电子后产生巯基自由基和巯醇负离子, 进一步可以与氢原子及质子反应得到电荷还原离子, 导致二硫键裂解[78, 79].
ECD过程中还会发生氢原子丢失和氨分子丢失[80, 81].氢原子丢失主要发生于赖氨酸、N-端氨基、脯氨酸等一系列基团, 而氨分子丢失主要是在N-氨基、精氨酸等基团上进行.热力学计算表明, 中性丢失的能垒很小, 反应较容易进行, 是ECD和电子捕获碰撞诱导解离(electron capture-induced dissociation, ECID)的常见反应, 但通常不能给出蛋白质结构方面的信息.
ECD实验可以为蛋白质结构分析提供丰富的数据, 因此有必要对多肽主链裂解频次进行详细研究.一般认为, 主链的裂解频率取决于母离子的电荷状态.例如, 对于十六个氨基酸构成的肽Ac-NH-(Ala-Lys-Ala-Ala- Lys)3-Ala-NH2进行ECD实验, +2电荷状态主要产生c7~c15和z9~z15碎片离子, +5主要是z2~z6碎片离子, 而+3和+4的电荷状态下, z与c碎片离子基本均等[82].
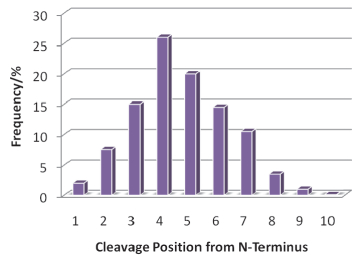
Zubarev研究小组[83]用胰蛋白酶解产生约15000多肽, 并用ECD质谱研究了主链不同位置的裂解频率.他们计算了多肽z离子和z+H离子的数目, 并以主链上断裂位置与N-端的距离为横坐标, 统计出ECD断裂频率(图 12).可以看出, ECD断裂频率的分布较宽, 其最大频率在距离N-端3到6个氨基酸的位置上.该小组还合成了一系列含有15种氨基酸各种组合的多肽, 包括丙氨酸、天冬酰胺、天冬氨酸、谷氨酸、谷氨酰胺、甘氨酸、组氨酸、异亮氨酸、亮氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、酪氨酸和缬氨酸.利用这些多肽, 他们进一步研究了主链裂解规律, 并发现Asp-Ala, Asp- Asn, Glu-Asn, Glu-His和Gln-Asn氨基酸之间的断裂频率较高.同时, 他们的数据也验证了“脯氨酸效应”[49], 即脯氨酸的N—Cα键不能发生ECD裂解.
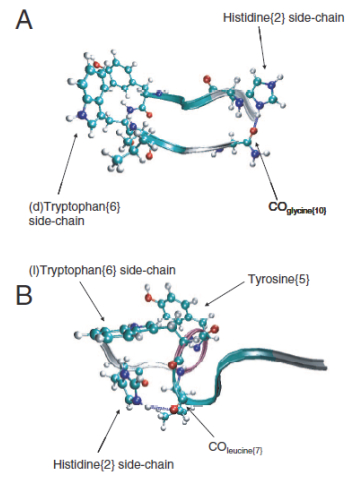
ECD的立体化学效应是一个十分有趣的问题.一般而言, 对映异构体的裂解碎片并无质量差异, 无法用其质荷比区分.因此, D-/L-氨基酸的区分需要考虑碎片离子的相对丰度.有研究者发现, 多肽离子中的L-被D-氨基酸取代时, ECD的产物离子丰度会发生变化[84~86].例如, 促性腺素释放素(pyro-Glu-His-Trp-Ser-Tyr-Xxx- Leu-Arg-Pro-Gly, Xxx=L-/D-色氨酸)的z型离子的强度与Xxx有很大关系[86]. Polfer等[86]通过分子动力学计算发现, 当Xxx=D-色氨酸时, 多肽构象为β-转角, 而Xxx=L-色氨酸时则是伸展构象(图 13). Polfer等[86]认为, 促性腺素释放素二级结构的区别可能是造成其ECD碎片离子丰度差异的原因.
更有趣的是, Tsybin等观察到, 对于三价的M2蛋白跨膜结构域衍生肽(Ser-Ser-Asp-Pro-Leu-Val-Val-Ala- Ala-Ser-Ile-Ile-Gly-Ile-Leu-His-Leu-Ile-Leu-Trp-Ile-Leu-Asp-Arg-Leu), 其ECD产物c/z离子的丰度随主链N-Cα键位置而发生周期性变化, 并且在Val6-Val7、Ala9- Ser10、Ile12-Gly13、His16-Leu17、Leu19-Trp20、Asp23- Arg24之间达到峰值.尽管暂时还没有气相构象的实验数据, 但是这种两亲多肽是以α-螺旋构象嵌入脂质膜的, 并且其螺旋周期为3.6个残基/转. Tsybin等[87]通过分子动力学模拟搜索了其稳定构象, 认为ECD辐射并没有改变溶液构象, 观察到的ECD周期性裂解现象与构象中未配对电子密度的酰胺基团有关.
O'Connor等[88, 89]通过时间分辨实验、红外多光子解离ECD技术研究了气相多肽离子的折叠与去折叠过程.他们认为, “慢热”裂解方式的特征时间是几百毫秒, 远大于主链N—Cα键断裂(小于10-5 s)[90]和蛋白质折叠时间(约102 ns).通过双共振时间分辨ECD实验, O'Connor等发现, 血纤维蛋白肽-B(Glu-Gly-Val-Asn-Asp-Asn- Glu-Glu-Gly-Phe-Phe-Ser-Ala-Arg)的产物离子z7、z8的丰度衰减符合一级动力学方程, 半衰期为1.8~2.1 ms.研究者认为, 这一现象是由于多肽Glu7和邻近精氨酸之间的分子内氢键作用造成的.这一方法也为检测气相多肽离子的构象奠定了基础.
近年来, 在ECD基础上产生了一系列衍生技术, 并且在糖蛋白、代谢物及糖类衍生物分析中取得一些新的成就.这些裂解反应的本质都是电子与离子相互作用, 因此有些文献将其统称为ExD技术[13, 91].在本小节, 我们主要讨论热电子捕获裂解(hot electron capture dissociation, HECD)、电子诱导解离(electron induced dissociation, EID)、负离子电子捕获解离(negative ion electron capture dissociation, niECD), 以及电子分离解离(electron detachment dissociation, EDD).
ECD反应所需的电子能量很小, 只有小于0.2 eV.然而, 当电子能量提高到10 eV左右时, 裂解产物中除了c/z型离子, 还有b/y型以及二次碎裂d/w型离子[92]. HECD过程中, 电子首先与母离子发生碰撞, 导致电子冷却及母离子活化.接下来, 如果母离子捕获到冷却电子, 就会发生ECD反应, 主链N—Cα键断裂; 如果捕获不成功, 激发态的母离子则会将其能量分布在分子的各个振动自由度上, 并发生C—N键断裂.实际上, 上述反应同时进行, 并且由于ECD反应很快, 因此主链N—Cα键断裂的离子会进一步发生二次裂解, 生成其他类型的离子. HECD中二次裂解离子可以为异构体分析提供关键信息, 现已成功用于氨基酸异构体分析[93, 94].
Cody和Freiser[95]发现, 当电子能量提高到大于10 eV时, +1电荷的离子会与电子发生作用并产生很多裂解碎片.这一观察与通常的ECD实验完全不同, 因为由于电子捕获的还原作用, 单电荷离子的ECD反应产物是无法观测的.随后, 许多小组对这一现象进行了研究, 逐步发展了有机分子电子诱导激发解离(electron-induced excitation of ions from organics, EIEIO)、电子激发裂解(electron excitation dissociation, EED)以及电子电离解离(electron ionization dissociation, EID)等反应[13, 95, 96]. Wolff和O'Hair[97, 98]指出, 由于这些反应中电子能量范围较宽, 电子激发、电离、二次电离、电子诱导裂解、电荷诱导裂解等过程交织进行, 因此这些反应的机理复杂、很难用统一的模型描述.现在一般将上述反应统称为电子诱导解离(electron induced dissociation, EID)[13].多肽的EID产物包括a, b, c, x, y, z型离子, 以及氨基酸侧链碎片[96].
2011年, H kansson研究组[99]发现, 多肽负离子可以捕获具有中等能量的电子(3.5~6.5 eV)并发生自由基诱导的裂解反应.这种负离子电子捕获反应(niECD)的产物类型与普通ECD类似, 并且可以保留蛋白质上的修饰基团.进一步研究表明, 不含有强碱性基团或去质子化的多肽不会发生niECD反应.因此, niECD反应机理可能是通过两性离子结构发生的, 多肽离子中正电荷位点捕获电子, 进而发生ECD反应. niECD现已成功用于磷酸化蛋白及O-糖肽的分析, 其较高的序列覆盖度为结构鉴定提供了重要依据[100].
另一种分析负离子结构的技术是EDD[101].在高能量电子(大于10 eV)的辐射下, 多电荷负离子会电离产生一个正电荷空穴, 并且发放出一个电子.由于库仑作用, 多肽负离子上的正电荷空穴会向着负电荷中心移动, 并与之发生氧化还原反应.这一过程释放的能量会使母离子进入激发态, 进而发生Cα—C键裂解. EDD的产物包括a•, x, c, z型离子, 但是由于静电排斥作用, EDD的裂解效率低于ECD[102, 103].
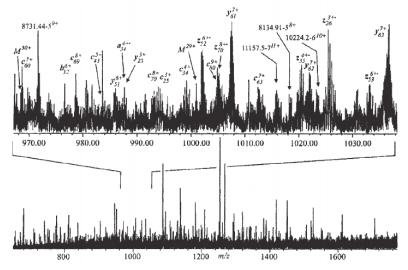
对于正离子来说, ECD反应会产生丰度较高的电荷还原产物离子及丰度较低的裂解碎片离子.如果研究对象是分子量较大的蛋白质, 其ECD谱将明显分为两个区域(图 14)[104].一般来说, 碎片离子区域可以提供丰富的结构信息.
根据ECD机理, 蛋白质及多肽离子的产物c离子通常为偶电子离子(c*+), 而z离子一般为自由基离子(z•+).但实验中经常观察到自由基型的c离子(c•+)与偶电子型的z离子(z*+)[105].一些研究人员[89, 105~107]认为ECD反应中生成的c*+与z•+会在非共价作用下以复合体的形式存在一段时间, 而z•+有可能夺取c*+的氢原子, 从而产生z*+和c•+.如图 15所示, 单电荷的c*+丢失氢原子会让谱图向较低m/z方向偏移约1 Da, 而单电荷的z•+获取氢原子会让谱图向较高m/z方向偏移约1 Da.实际实验中观察到的同位素结构会表现为红色与蓝色谱图的重叠(图 15)[108].显然, 这种氢重排过程会使ECD谱的解析变得更为复杂.
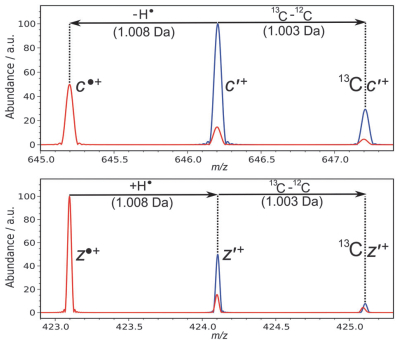
Coon等[109]注意到, 偶电子型的z*+离子含有偶数个化合价为奇数的原子(例如氮原子), 而自由基型的c•+离子则含有奇数个化合价为奇数的原子.利用这种“氮规则”可以在一定程度上化简ECD谱图的解析.另一方面, 注意到单电荷的c*+的同位素结构中, 第一个峰与第二个峰的距离应为1.0033 Da(对应于碳-12与碳-13同位素相对质量差), 而c•+的同位素结构中前两个峰的则等于1.0078 Da(对应于氢相对原子量)[108]. z离子的情况类似.这一规律也可以辅助ECD谱图解析.
对于蛋白质分子的“自顶向下”ECD质谱分析来说, 图谱解析需要大量时间, 并且由于氢重排等原因, 其同位素峰的分析往往很复杂.目前ECD数据的解析算法包括蛋白质鉴定(identification)[110~113]与图谱反卷积(deconvolution)[114]两大类.蛋白质鉴定算法通常是在图谱反卷积算法基础上, 通过与数据库挖掘或谱比对实现蛋白质或多肽的结构鉴定.有关这方面的一些最新进展读者可以参考文献[110~113].
反卷积算法可以将ECD谱图中的质谱峰分组成各个碎片离子的同位素峰, 进一步可以在同位素结构基础上解析各个碎片离子的电荷及离子精确相对分子量值. Horn等[114]开发的THRASH是反卷积算法中使用最普遍的方法之一. THRASH通过一个质荷比窗口对全谱进行步进扫描, 并且利用傅里叶变换/Patterson方法计算窗口中同位素峰的电荷数及单一同位素质量(monoisotopic mass)[114].同时, THRASH使用最小二乘法解析复杂的同位素结构, 增加了谱图解析的可靠度.最近, 一些新的算法如Decon2LS和MASH Suite等优化了TRHASH, 提高了“自顶向下”解谱正确率[115, 116].然而需要指出, 由于“自顶向下”ECD质谱数据的复杂性, 其最终数据往往需要人工逐一比对验证, 因此一些实验室往往采用自主编程结合人工验证的复合方式解谱[117~119].
本研究团队属于我国较早开展研究多肽电子捕获裂解基础研究的课题组, 在金属离子电荷载体效应, 多肽ECD侧链丢失, 二硫键断裂等方面发表了一系列的论文, 并于2006年承办了第4届国际ECD会议.本节将结合实验室相关工作介绍ECD的前沿进展.
Fung等[120]合成了一系列长度不同的模型多肽KG3WG3K、NG3WG3N、RG7R、RG3WG3R、RG3DG3R、RG3EG3R、RG3FG3R和RG5WG5R, 通过和CID的比较进行了ECD的机理研究.实验数据支持了氢键网络模型, 认为ECD的裂解模式受到由氢键维系的多肽离子空间构象影响.但实验中发现, 模型肽中色氨酸附近的ECD裂解频次并没有明显增加, 这一点与McLafferty等的工作相反.可以认为, 多肽ECD裂解频次规律与序列有相关性.同时, 实验也验证了CID的质子迁移模型, 认为多肽的CID裂解途径主要由氢键和迁移质子控制.
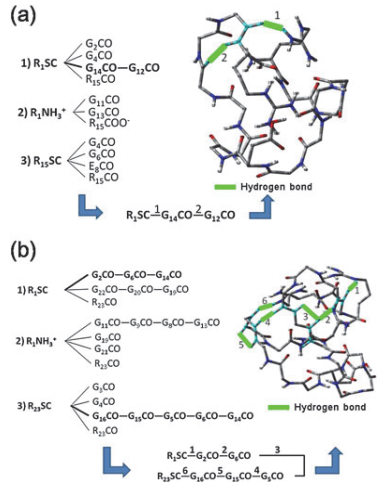
通过合成模型多肽的方法, Chan等[121]发现, 二价多肽的ECD裂解与谷氨酸、天冬氨酸的数目密切相关.当这两种氨基酸数目增多时, 主链N—Cα键裂解及氢原子丢失反应被抑制, 当二者数目达到6时, 裂解通道被强烈抑制.与此相反, 含5个谷氨酸和天冬氨酸的血纤维蛋白肽-B却有大量的ECD碎片产生.分析这种有趣的ECD抑制反应的机理可能会对ECD模型提供进一步的验证和补充.通过比较分析可以发现, 合成的模型肽可能含有分子内氢键, 从而稳定了母离子的电荷还原产物, 抑制了进一步的ECD碎裂反应. Wong等[122]进一步合成了长度、种类不同的模型肽, 通过ECD实验并结合理论计算发现了模型肽的“氢键梯”结构, 验证了分子内氢键稳定母离子的作用(图 16).
Fung等[123]还进一步利用模型肽系统研究了不同氨基酸的侧链在ECD裂解中的行为.模型肽RGGGXGGGR(其中X为二十个天然氨基酸中的任意一个)的ECD实验显示, 多肽正离子首先发生主链N—Cα键裂解, 产生Cα自由基, 并进一步发生侧链二次裂解.其中有13个氨基酸丢失了奇电子中性碎片, 9个丢失了偶电子中性碎片.利用从头计算方法, Fung等[123]对侧链断裂的各种可能反应机理做了详细研究并发现: (1) N-端Cα自由基进攻氨基酸Cα侧链基团上的氢原子, 形成α-β碳碳双键, 产生奇电子中性丢失, 反应的能垒为89 kJ/mol; (2) N-端Cα自由基进攻侧链γ-氢原子, 形成β-γ碳碳双键, 得到偶电子中性碎片, 反应能垒为50 kJ/mol.
Fung等[124]研究了二价碱金属元素对多肽ECD反应的影响.实验发现, 大部分碱金属离子化的多肽, ECD结果非常相似, 主要生成c和z型碎片离子, 而二价镁离子略有不同.与质子化多肽相比, 二价镁离子化多肽c型碎片有所差异.理论计算表明, 金属离子会增强酰胺基团的酸性, 其他碱性基团或N-端氨基酸会夺取其质子, 形成双性离子.由于可迁移质子的存在, ECD电子首先被该质子捕获, 进而发生ECD反应.显然, 这种双性离子模型决定了金属离子不被ECD电子还原, 因此最终会观察到二价碱金属离子化多肽碎片.
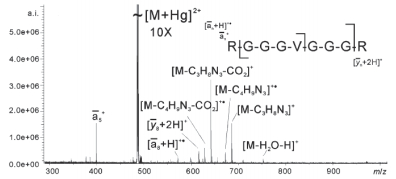
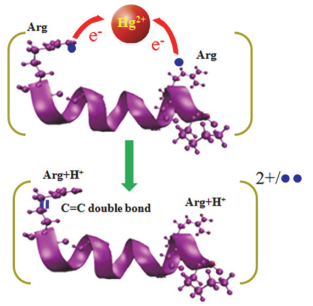
Chen等[125]发现, 与碱金属不同, IIB二价金属元素(Zn2+, Cd2+, Hg2+)对多肽ECD有很大的影响. Zn2+离子化的多肽主要产生非金属离子化的c和z型离子; Cd2+和Hg2+离子化的多肽则主要生成a型离子及侧链裂解碎片(图 17)[125].深入研究发现, 多肽与二价金属离子主要有三种离子化结构: (a) [多肽+金属]2+, (b)[(多肽+金属-H)+H]2+, 以及(c)[(多肽+金属-2H)+2H]2+.具有较小离子半径的Zn2+主要与多肽形成(b)和(c)型结构, 而较大半径的Cd2+和Hg2+主要形成(a)结构.根据双性离子模型, (b)和(c)型结构主要发生迁移质子与ECD电子的捕获反应, 生成c和z型离子.而(a)型结构没有可迁移质子, 因此ECD电子的捕获由金属离子完成.在这一裂解通道中, 二价金属被还原, 但同时电子又从多肽上相应基团转移到金属离子上, 最终形成a型碎片离子. Wong等[126]发现, Hg2+离子化的多肽还可以通过“慢热”裂解方式产生M2+双自由基贫氢离子(图 18).由于多肽中的碱性氨基酸及双性离子结构, 激发态的双自由基离子可以经过重排反应生成可迁移质子, 进而产生大量的c/z、(c-2)/(z-2)碎片.
Chen等[127]还发现, 二价过渡金属Mn2+, Fe2+, Co2+, Ni2+和Cu2+离子化的多肽会发生不同的ECD反应.其中Mn2+的效应与Zn2+类似, Fe2+, Co2+, Ni2+主要产生a/y型碎片, Cu2+主要生成b/y型离子[128].理论计算表明, 造成这种差异的原因可能与过渡金属离子的电子构型有关, d轨道半满和全满的金属离子(Mn2+, Zn2+)不能捕获ECD电子, 而其他金属离子则参与ECD电子捕获, 改变了普通的ECD裂解反应, 产生a, b, y型离子.实验也发现, 多肽上的碱性氨基酸以及主链长度也会对金属离子化ECD反应造成影响.除此以外, 对IIIB三价金属(Al3+, Ga3+, In3+, Rh3+)的研究表明, 大部分金属离子化多肽主要产生c和z型离子, 而Rh3+则通过捕获ECD电子主要生成金属离子化a/b型、质子化y型碎片离子[129].
蛋白质分子中二硫键的选择性裂解一直是串联质谱的热点和难点问题[75]. CID裂解模式以振动激发方式活化母离子, 很难造成二硫键断裂.传统ECD虽然可以选择性的裂解二硫键, 但同时会造成母离子电荷还原, 因此2+以下的多肽离子会因为ECD还原生成质谱无法观测的碎片离子. Fung等[130]通过157 nm的紫外光辅助, 成功将多肽和蛋白质的二硫键选择性的裂解, 并且保留母离子电荷状态.这一技术与ECD结合可以很好的对二硫键蛋白质进行结构分析.
Chan等[131]研究了寡聚核苷酸的ECD裂解反应会产生类核苷离子、核碱基离子、丢失一个核苷酸的w/d型离子、没有中性丢失的z/a离子, 以及少量z/a+H自由基离子.这些产物的形成可能与母离子自由基的二次裂解有关, 同时, 实验也证明了寡聚核苷酸的电子捕获主要发生于质子化的核碱基.
由糖环和其他小分子如生物碱、黄酮、脂类形成的糖复合物是重要的次生代谢产物, 具有抗氧化、抗炎等药用活性[132, 133].但是由于糖链的结构复杂性和不均一性, 这类化合物的结构分析一直富有挑战性[134, 135].由于分子量很小, 这类分子电离后通常产生单电荷母离子, 因此无法用传统的ECD进行结构研究. Wong等[136]利用EID方法对质子化和钠离子化人参皂苷进行研究, 发现高能量电子辐射可以阻断质子参与的长程糖环迁移反应, 并且生成结构特异性离子B2和C2, 可以对人参皂苷异构体进行快速有效区分(图 19).同时, Wong等[137]通过糖生物碱的EID实验发现, CID反应主要造成中性碎片丢失和糖苷键断裂, 有价值的碎片离子很少, 而EID可以在糖链和非糖链部分均产生大量化学键裂解, 并且生成很多跨环碎裂产物离子. EID反应中多次裂解得到的跨环碎裂产物离子如C11H18NO+, C8H16N+, C6H12N+可以有效地对结构异构体进行区分.
ECD的另一个广泛应用是“自顶向下”(top down)蛋白质结构分析[138~140].由于ECD反应的特点, 蛋白质分子在发生裂解的时候可以保留其化学修饰基团, 甚至一些弱相互作用.实际上, ECD方法已经成功用于诸多蛋白质翻译后修饰的分析, 例如磷酸位点的分析.但是由于糖链的结构复杂性, 蛋白质后修饰中一类重要的分子糖蛋白的“自顶向下”分析, 尤其是糖环部分结构分析一直是蛋白质质谱的难点问题[117, 141].这类分析通常需要使用较为复杂的FTICR时间序列, 将“慢热”活化和ECD反应结合, 从而得到最大的序列覆盖度[117]. Wang等[119]利用核糖核酸酶B为研究对象, 对其ECD反应特征进行了研究, 发现即使不用复合时间序列, ECD和CID实验可以产生很高的序列覆盖度(大于90%), 从而精确测定糖基修饰位点(图 20).另外, Wang等[119]发现, “自顶向下”的CID反应可以提供完整糖蛋白的糖环部分结构信息, 并建立了核糖核酸酶B糖环侧链结构分析的实验方法.
综上所述, ECD是FTICR质谱的重要裂解模式之一.可以看出, 随着对ECD机理的深入阐明, 其硬件设计也有了很大改进.空心扩散阴极电子枪使得ECD的反应效率更高、反应时间更短、反应选择性更多.在新的仪器技术基础上, 产生了大量实验数据, 促使人们深入挖掘ECD反应机理.目前, 康奈尔、犹他-华盛顿、自由基级联三种模型可以很好地描述大部分的实验结果, 包括模型多肽、完整蛋白分子、金属电荷载体效应等.进一步地, 仪器技术和反应机理的基础研究又推动了ECD技术在生物分子分析中的应用.在后基因组时代, ECD已经成为研究蛋白质结构、蛋白质翻译后修饰, 蛋白质相互作用等重要问题的关键方法.可以说ECD技术的发展是现代生物质谱的一个缩影, 机理阐明、仪器方法、应用研究三者缺一不可.
经过二十年发展, ECD的研究已经在生物分子分析中显示出巨大应用潜力, 然而需要指出的是, 该领域目前仍然存在许多挑战性的问题.对于分子量极大的蛋白质分子, ECD反应通常无法观察到其中心区域的裂解碎片, 导致分子结构信息的缺失.另一方面, 对于分子量很小的生物小分子, 利用ECD对其结构异构体的阐明仍然充满困难.我们认为, 该领域未来发展的挑战和机遇在于: (1)通过理论计算, 对ECD的三种机理进行进一步阐明; (2)通过电荷载体诱导离子产生新的ECD裂解方式; (3)结合ECD机理, 通过新的仪器技术实现生物大分子体系的深度活化裂解; (4)积极扩展ExD技术在生物小分子方面的应用研究.
Zubarev, R. A.; Kelleher, N. L.; McLafferty, F. W. J. Am. Chem. Soc. 1998, 120, 3265. doi: 10.1021/ja973478k
Lin, Z.; Guo, F.; Gregorich, Z. R.; Sun, R.; Zhang, H.; Hu, Y.; Shanmuganayagam, D.; Ge, Y. J. Am. Soc. Mass Spectrom. 2018, 29, 1284. doi: 10.1007/s13361-018-1925-y
Floris, F.; Chiron, L.; Lynch, A. M.; Barrow, M. P.; Delsuc, M.-A.; O'Connor, P. B. Anal. Chem. 2018, 90, 7302. doi: 10.1021/acs.analchem.8b00500
Wongkongkathep, P.; Han, J. Y.; Choi, T. S.; Yin, S.; Kim, H. I.; Loo, J. A. J. Am. Soc. Mass Spectrom. 2018, 29, 1870. doi: 10.1007/s13361-018-2002-2
Shaw, J. B.; Malhan, N.; Vasil'ev, Y. V.; Lopez, N. I.; Makarov, A. A.; Beckman, J. S.; Voinov, V. G. Anal. Chem. 2018, 90, 10819. doi: 10.1021/acs.analchem.8b01901
McLafferty, F. W.; Tureček, F. Interpretation of Mass Spectra, University Science Books, Mill Valley, 1993.
Fenn, J. B.; Mann, M.; Meng, C. K.; Wong, S. F.; Whitehouse, C. M. Science 1989, 246, 64. doi: 10.1126/science.2675315
Tanaka, K.; Waki, H.; Ido, Y.; Akita, S.; Yoshida, Y.; Yoshida, T.; Matsuo, T. Rapid Commun. Mass Spectrom. 1988, 2, 151. doi: 10.1002/(ISSN)1097-0231
Karas, M.; Hillenkamp, F. Anal. Chem. 1988, 60, 2299. doi: 10.1021/ac00171a028
宋凤瑞, 闫存玉, 刘宁, 刘志强, 刘淑莹, 化学学报, 2009, 67, 1103. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract328531.shtmlSong, F.; Yan, C.; Liu, N.; Liu, Z.; Liu, S. Acta Chim. Sinica 2009, 67, 1103. http://manu19.magtech.com.cn/Jwk_hxxb/CN/abstract/abstract328531.shtml
李卉卉, 郑波, 叶蕴华, 袁谷, 化学学报, 2009, 67, 1869. doi: 10.3321/j.issn:0567-7351.2009.16.009Li, H.; Zheng, B.; Ye, Y.; Yuan, G. Acta Chim. Sinica 2009, 67, 1869. doi: 10.3321/j.issn:0567-7351.2009.16.009
Zubarev, R. A. Mass Spectrom. Rev. 2003, 22, 57. doi: 10.1002/(ISSN)1098-2787
Chen, X.; Wang, Z.; Wong, Y. L. E.; Wu, R.; Zhang, F.; Chan, T. W. D. Mass Spectrom. Rev. 2018, 37, 793. doi: 10.1002/mas.v37.6
Peng, Y.; Ayaz-Guner, S.; Yu, D.; Ge, Y. Proteomics Clin. Appl. 2014, 8, 554. doi: 10.1002/prca.v8.7-8
Lermyte, F.; Valkenborg, D.; Loo, J. A.; Sobott, F. Mass Spectrom. Rev. 2018, 37, 750. doi: 10.1002/mas.21560
Syka, J. E. P.; Coon, J. J.; Schroeder, M. J.; Shabanowitz, J.; Hunt, D. F. Proc. Natl. Acad. Sci., U. S. A. 2004, 101, 9528. doi: 10.1073/pnas.0402700101
Mentinova, M.; Crizer, D. M.; Baba, T.; McGee, W. M.; Glish, G. L.; McLuckey, S. A. J. Am. Soc. Mass Spectrom. 2013, 24, 1676. doi: 10.1007/s13361-013-0606-0
Gunawardena, H. P.; He, M.; Chrisman, P. A.; Pitteri, S. J.; Hogan, J. M.; Hodges, B. D. M.; McLuckey, S. A. J. Am. Chem. Soc. 2005, 127, 12627. doi: 10.1021/ja0526057
Pitteri, S. J.; McLuckey, S. A. Mass Spectrom. Rev. 2005, 24, 931. doi: 10.1002/(ISSN)1098-2787
Coon, J. J.; Ueberheide, B.; Syka, J. E. P.; Dryhurst, D. D.; Ausio, J.; Shabanowitz, J.; Hunt, D. F. Proc. Natl. Acad. Sci., U. S. A. 2005, 102, 9463. doi: 10.1073/pnas.0503189102
Pitteri, S. J.; Chrisman, P. A.; Hogan, J. M.; McLuckey, S. A. Anal. Chem. 2005, 77, 1831. doi: 10.1021/ac0483872
孙瑞祥, 董梦秋, 迟浩, 杨兵, 秀丽蕴, 王乐珩, 付岩, 贺思敏, 生物化学与生物物理进展, 2010, 37, 94. http://www.cnki.com.cn/Article/CJFDTotal-SHSW201001016.htmSun, R.; Dong, M.-Q.; Chi, H.; Yang, B.; Xiu, L.-Y.; Wang, L.-H.; Fu, Y.; He, S.-M. Progress in Biochemistry and Biophysics 2010, 37, 94. http://www.cnki.com.cn/Article/CJFDTotal-SHSW201001016.htm
贾伟, 应万涛, 钱小红, 质谱学报, 2007, 28, 55. doi: 10.3969/j.issn.1004-2997.2007.01.012Jia, W.; Ying, W.-T.; Qian, X.-H. J. Chin. Mass Spectrom. Soc. 2007, 28, 55. doi: 10.3969/j.issn.1004-2997.2007.01.012
Riley, N. M.; Coon, J. J. Anal. Chem. 2018, 90, 40. doi: 10.1021/acs.analchem.7b04810
Nie, A.; Lu, H.; Yang, P.; He, F. Chin. J. Chem. 2011, 29, 171. doi: 10.1002/cjoc.v29.1
Nagornov, K. O.; Gorshkov, M. V.; Kozhinov, A. N.; Tsybin, Y. O. Anal. Chem. 2014, 86, 9020. doi: 10.1021/ac501579h
Nicolardi, S.; Deelder, A. M.; Palmblad, M.; van der Burgt, Y. E. M. Anal. Chem. 2014, 86, 5376. doi: 10.1021/ac500383c
Scigelova, M.; Hornshaw, M.; Giannakopulos, A.; Makarov, A. Mol. Cell. Proteomics 2011, 10, M111.009431. doi: 10.1074/mcp.M111.009431
骆丹, 辛培勇, 闫吉军, 房爽, 孙晓红, 闫静, 褚金芳, 闫存玉, 质谱学报, 2013, 34, 263. doi: 10.7538/zpxb.2013.34.05.0263Luo, D.; Xin, P.-Y.; Yan, J.-J.; Fang, S.; Sun, X.-H.; Yan, J.; Chu, J.-F.; Yan, C.-Y. J. Chin. Mass Spectrom. Soc. 2013, 34, 263. doi: 10.7538/zpxb.2013.34.05.0263
Zhang, J.; Chai, Y.; Wang, W.; Shang, W.; Pan, Y. Chin. J. Chem. 2012, 30, 2383. doi: 10.1002/cjoc.201200610
Li, B.; An, H. J.; Hedrick, J. L.; Lebrilla, C. B. In Glycomics: Methods and Protocols, Humana Press, Totowa, New Jersey, 2009, p. 133.
McDonald, L. A.; Barbieri, L. R.; Carter, G. T.; Kruppa, G.; Feng, X.; Lotvin, J. A.; Siegel, M. M. Anal. Chem. 2003, 75, 2730. doi: 10.1021/ac0264731
Li, B.; An, H. J.; Hedrick, J. L.; Lebrilla, C. B. In Glycomics: Methods and Protocols, Humana Press, Totowa, New Jersey, 2009, p. 23.
Li, H.; Wolff, J. J.; Van Orden, S. L.; Loo, J. A. Anal. Chem. 2014, 86, 317. doi: 10.1021/ac4033214
Ohta, D.; Kanaya, S.; Suzuki, H. Curr. Opin. Biotech. 2010, 21, 35. doi: 10.1016/j.copbio.2010.01.012
Cooper, H. J.; H kansson, K.; Marshall, A. G. Mass Spectrom. Rev. 2004, 24, 201. http://www.ncbi.nlm.nih.gov/pubmed/12939500
Baba, T.; Hashimoto, Y.; Hasegawa, H.; Hirabayashi, A.; Waki, I. Anal. Chem. 2004, 76, 4263. doi: 10.1021/ac049309h
Satake, H.; Hasegawa, H.; Hirabayashi, A.; Hashimoto, Y.; Baba, T.; Masuda, K. Anal. Chem. 2007, 79, 8755. doi: 10.1021/ac071462z
Zubarev, R. A.; Nielsen, M. L.; Budnik, B. A. Eur. J. Mass Spectrom. 2000, 6, 235. doi: 10.1255/ejms.351
Chan, T. W. D.; Ip, W. H. H. J. Am. Soc. Mass Spectrom. 2002, 13, 1396. doi: 10.1016/S1044-0305(02)00703-1
Tsybin, Y. O.; H kansson, P.; Budnik, B. A.; Haselmann, K. F.; Kjeldsen, F.; Gorshkov, M.; Zubarev, R. A. Rapid Commun. Mass Spectrom. 2001, 15, 1849. doi: 10.1002/rcm.v15:19
Haselmann, K. F.; Budnik, B. A.; Olsen, J. V.; Nielsen, M. L.; Reis, C. A.; Clausen, H.; Johnsen, A. H.; Zubarev, R. A. Anal. Chem. 2001, 73, 2998. doi: 10.1021/ac0015523
Tsybin, Y. O.; Witt, M.; Baykut, G.; Kjeldsen, F.; H kansson, P. Rapid Commun. Mass Spectrom. 2003, 17, 1759. doi: 10.1002/rcm.1118
Shaw, J. B.; Robinson, E. W.; Paša-Tolić, L. Anal. Chem. 2016, 88, 3019. doi: 10.1021/acs.analchem.6b00148
Horn, D. M.; Ge, Y.; McLafferty, F. W. Anal. Chem. 2000, 72, 4778. doi: 10.1021/ac000494i
Erba, E. B. Proteomics 2014, 14, 1259. doi: 10.1002/pmic.201300333
Lanucara, F.; Eyers, C. E. Mass Spectrom. Rev. 2012, 32, 27.
Hao, Q.; Song, T.; Ng, D. C. M.; Quan, Q.; Siu, C.-K.; Chu, I. K. J. Phys. Chem. B 2012, 116, 7627. doi: 10.1021/jp301882p
Zubarev, R. A.; Horn, D. M.; Fridriksson, E. K.; Kelleher, N. L.; Kruger, N. A.; Lewis, M. A.; Carpenter, B. K.; McLafferty, F. W. Anal. Chem. 2000, 72, 563. doi: 10.1021/ac990811p
Roepstorff, P.; Fohlman, J. Biomed. Mass Spectrom. 1984, 11, 601. doi: 10.1002/(ISSN)1096-9888
Biemann, K. Biomed. Environ. Mass Spectrom. 1988, 16, 99. doi: 10.1002/(ISSN)1096-9888
Paizs, B.; Suhai, S. Mass Spectrom. Rev. 2004, 24, 508.
Laskin, J.; Yang, Z.; Song, T.; Lam, C.; Chu, I. K. J. Am. Chem. Soc. 2010, 132, 16006. doi: 10.1021/ja104438z
Yu, L.; Tan, Y.; Tsai, Y.; Goodlett, D. R.; Polfer, N. C. J. Proteome Res. 2011, 10, 2409. doi: 10.1021/pr101235w
Harrison, A. G.; Young, A. B.; Bleiholder, C.; Suhai, S.; Paizs, B. J. Am. Chem. Soc. 2006, 128, 10364. doi: 10.1021/ja062440h
Bleiholder, C.; Osburn, S.; Williams, T. D.; Suhai, S.; Van Stipdonk, M.; Harrison, A. G.; Paizs, B. J. Am. Chem. Soc. 2008, 130, 17774. doi: 10.1021/ja805074d
Tureček, F.; Reid, P. J. Int. J. Mass Spectrom. 2003, 222, 49. doi: 10.1016/S1387-3806(02)00983-1
Tureček, F.; Julian, R. R. Chem. Rev. 2013, 113, 6691. doi: 10.1021/cr400043s
Zubarev, R. A.; Haselmann, K. F.; Budnik, B.; Kjeldsen, F.; Jensen, F. Eur. J. Mass Spectrom. 2002, 8, 337. doi: 10.1255/ejms.517
Tureček, F.; Chen, X.; Hao, C. J. Am. Chem. Soc. 2008, 130, 8818. doi: 10.1021/ja8019005
Chen, X.; Tureček, F. J. Am. Chem. Soc. 2006, 128, 12520. doi: 10.1021/ja063676o
Tureček, F.; Yao, C.; Fung, Y. M. E.; Hayakawa, S.; Hashimoto, M.; Matsubara, H. J. Phys. Chem. B 2009, 113, 7347. doi: 10.1021/jp900719n
Tureček, F.; Chung, T. W.; Moss, C. L.; Wyer, J. A.; Ehlerding, A.; Holm, A. I. S.; Zettergren, H.; Nielsen, S. B.; Hvelplund, P.; Chamot-Rooke, J.; Bythell, B.; Paizs, B. J. Am. Chem. Soc. 2010, 132, 10728. doi: 10.1021/ja907808h
Tureček, F.; Jones, J. W.; Towle, T.; Panja, S.; Nielsen, S. B.; Hvelplund, P.; Paizs, B. J. Am. Chem. Soc. 2008, 130, 14584. doi: 10.1021/ja8036367
Syrstad, E. A.; Tureček, F. J. Am. Soc. Mass Spectrom. 2005, 16, 208. doi: 10.1016/j.jasms.2004.11.001
Panja, S.; Nielsen, S. B.; Hvelplund, P.; Tureček, F. J. Am. Soc. Mass Spectrom. 2008, 19, 1726. doi: 10.1016/j.jasms.2008.08.001
Tureček, F.; Panja, S.; Wyer, J. A.; Ehlerding, A.; Zettergren, H.; Nielsen, S. B.; Hvelplund, P.; Bythell, B.; Paizs, B. J. Am. Chem. Soc. 2009, 131, 16472. doi: 10.1021/ja9050229
Sawicka, A.; Skurski, P.; Hudgins, R. R.; Simons, J. J. Phys. Chem. B 2003, 107, 13505. doi: 10.1021/jp035675d
Sobczyk, M.; Anusiewicz, I.; Berdys-Kochanska, J.; Sawicka, A.; Skurski, P.; Simons, J. J. Phys. Chem. A 2005, 109, 250. doi: 10.1021/jp0463114
Sobczyk, M.; Simons, J. J. Phys. Chem. B 2006, 110, 7519. doi: 10.1021/jp0604701
Leymarie, N.; Costello, C. E.; O'Connor, P. B. J. Am. Chem. Soc. 2003, 125, 8949. doi: 10.1021/ja028831n
Chung, T. W.; Hui, R.; Ledvina, A.; Coon, J. J.; Tureček, F. J. Am. Soc. Mass Spectrom. 2012, 23, 1336. doi: 10.1007/s13361-012-0408-9
Ledvina, A. R.; Chung, T. W.; Hui, R.; Coon, J. J.; Tureček, F. J. Am. Soc. Mass Spectrom. 2012, 23, 1351. doi: 10.1007/s13361-012-0409-8
Qi, Y.; Bortoli, S.; Volmer, D. A. J. Am. Soc. Mass Spectrom. 2014, 25, 1253. doi: 10.1007/s13361-014-0893-0
Wongkongkathep, P.; Li, H.; Zhang, X.; Loo, R. R. O.; Julian, R. R.; Loo, J. A. Int. J. Mass Spectrom. 2015, 390, 137. doi: 10.1016/j.ijms.2015.07.008
Uggerud, E. Int. J. Mass Spectrom. 2004, 234, 45. doi: 10.1016/j.ijms.2004.01.020
Li, H.; O'Connor, P. B. J. Am. Soc. Mass Spectrom. 2012, 23, 2001. doi: 10.1007/s13361-012-0473-0
Cole, S. R.; Ma, X.; Zhang, X.; Xia, Y. J. Am. Soc. Mass Spectrom. 2012, 23, 310. doi: 10.1007/s13361-011-0300-z
Mentinova, M.; Han, H.; McLuckey, S. A. Rapid Commun. Mass Spectrom. 2009, 23, 2647. doi: 10.1002/rcm.v23:17
O'Connor, P. B.; Lin, C.; Cournoyer, J. J.; Pittman, J. L.; Belyayev, M.; Budnik, B. A. J. Am. Soc. Mass Spectrom. 2006, 17, 576. doi: 10.1016/j.jasms.2005.12.015
Holm, A. I. S.; Hvelplund, P.; Kadhane, U.; Larsen, M. K.; Liu, B.; Nielsen, S. B.; Panja, S.; Pedersen, J. M.; Skrydstrup, T.; Støchkel, K.; Williams, E. R.; Worm, E. S. J. Phys. Chem. A 2007, 111, 9641. doi: 10.1021/jp075943y
Iavarone, A. T.; Paech, K.; Williams, E. R. Anal. Chem. 2004, 76, 2231. doi: 10.1021/ac035431p
Savitski, M. M.; Kjeldsen, F.; Nielsen, M. L.; Zubarev, R. A. Angew. Chem., Int. Ed. 2006, 45, 5301. doi: 10.1002/(ISSN)1521-3773
Adams, C. M.; Kjeldsen, F.; Zubarev, R. A.; Budnik, B. A.; Haselmann, K. F. J. Am. Soc. Mass Spectrom. 2004, 15, 1087. doi: 10.1016/j.jasms.2004.04.026
Adams, C. M.; Zubarev, R. A. Anal. Chem. 2005, 77, 4571. doi: 10.1021/ac0503963
Polfer, N. C.; Haselmann, K. F.; Langridge-Smith, P. R. R.; Barran, P. E. Mol. Phys. 2005, 103, 1481. doi: 10.1080/00268970500095998
Hamidane, H. B.; He, H.; Tsybin, O. Y.; Emmett, M. R.; Hendrickson, C. L.; Marshall, A. G.; Tsybin, Y. O. J. Am. Soc. Mass Spectrom. 2009, 20, 1182. doi: 10.1016/j.jasms.2009.02.015
Lin, C.; Cournoyer, J. J.; O'Connor, P. B. J. Am. Soc. Mass Spectrom. 2006, 17, 1605. doi: 10.1016/j.jasms.2006.07.007
Lin, C.; Cournoyer, J. J.; O'Connor, P. B. J. Am. Soc. Mass Spectrom. 2008, 19, 780. doi: 10.1016/j.jasms.2008.01.001
Tureček, F.; Syrstad, E. A.; Seymour, J. L.; Chen, X.; Yao, C. J. Mass Spectrom. 2003, 38, 1093. doi: 10.1002/(ISSN)1096-9888
Qi, Y.; Volmer, D. A. Analyst 2016, 141, 794. doi: 10.1039/C5AN02171E
Kjeldsen, F.; Haselmann, K. F.; Budnik, B. A.; Jensen, F.; Zubarev, R. A. Chem. Phys. Lett. 2002, 356, 201. doi: 10.1016/S0009-2614(02)00149-5
Kjeldsen, F.; Haselmann, K. F.; Sørensen, E. S.; Zubarev, R. A. Anal. Chem. 2003, 75, 1267. doi: 10.1021/ac020422m
Yu, X.; Zhong, W. Anal. Chem. 2016, 88, 5914. doi: 10.1021/acs.analchem.6b00823
Cody, R. B.; Freiser, B. S. Anal. Chem. 1979, 51, 547. doi: 10.1021/ac50040a022
Fung, Y. M. E.; Adams, C. M.; Zubarev, R. A. J. Am. Chem. Soc. 2009, 131, 9977. doi: 10.1021/ja8087407
Wolff, J. J.; Laremore, T. N.; Aslam, H.; Linhardt, R. J.; Amster, I. J. J. Am. Soc. Mass Spectrom. 2008, 19, 1449. doi: 10.1016/j.jasms.2008.06.024
Lioe, H.; O'Hair, R. A. J. Anal. Bioanal. Chem. 2007, 389, 1429. doi: 10.1007/s00216-007-1535-1
Yoo, H. J.; Wang, N.; Zhuang, S.; Song, H.; H kansson, K. J. Am. Chem. Soc. 2011, 133, 16790. doi: 10.1021/ja207736y
Hersberger, K. E.; H kansson, K. Anal. Chem. 2012, 84, 6370. doi: 10.1021/ac301536r
Budnik, B. A.; Haselmann, K. F.; Zubarev, R. A. Chem. Phys. Lett. 2001, 342, 299. doi: 10.1016/S0009-2614(01)00501-2
Larraillet, V.; Vorobyev, A.; Brunet, C.; Lemoine, J.; Tsybin, Y. O.; Antoine, R.; Dugourd, P. J. Am. Soc. Mass Spectrom. 2010, 21, 670. doi: 10.1016/j.jasms.2010.01.015
Kjeldsen, F.; Silivra, O. A.; Ivonin, I. A.; Haselmann, K. F.; Gorshkov, M.; Zubarev, R. A. Chem.-Eur. J. 2005, 11, 1803. doi: 10.1002/(ISSN)1521-3765
Zhang, H.; Cui, W.; Wen, J.; Blankenship, R. E.; Gross, M. L. Anal. Chem. 2011, 83, 5598. doi: 10.1021/ac200695d
Savitski, M. M.; Kjeldsen, F.; Nielsen, M. L.; Zubarev, R. A. J. Am. Soc. Mass Spectrom. 2007, 18, 113. doi: 10.1016/j.jasms.2006.09.008
Ledvina, A. R.; Beauchene, N. A.; McAlister, G. C.; Syka, J. E. P.; Schwartz, J. C.; Griep-Raming, J.; Westphall, M. S.; Coon, J. J. Anal. Chem. 2010, 82, 10068. doi: 10.1021/ac1020358
Swaney, D. L.; McAlister, G. C.; Wirtala, M.; Schwartz, J. C.; Syka, J. E. P.; Coon, J. J. Anal. Chem. 2007, 79, 477. doi: 10.1021/ac061457f
Zhurov, K. O.; Fornelli, L.; Wodrich, M. D.; Laskay, . A.; Tsybin, Y. O. Chem. Soc. Rev. 2013, 42, 5014. doi: 10.1039/c3cs35477f
Hubler, S. L.; Jue, A.; Keith, J.; McAlister, G. C.; Craciun, G.; Coon, J. J. J. Am. Chem. Soc. 2008, 130, 6388. doi: 10.1021/ja7099985
Tsur, D.; Tanner, S.; Zandi, E.; Bafna, V.; Pevzner, P. A. Nat. Biotechnol. 2005, 23, 1562. doi: 10.1038/nbt1168
Fu, Y.; Yang, Q.; Sun, R.; Li, D.; Zeng, R.; Ling, C. X.; Gao, W. Bioinformatics 2004, 20, 1948. doi: 10.1093/bioinformatics/bth186
Li, D.; Fu, Y.; Sun, R.; Ling, C. X.; Wei, Y.; Zhou, H.; Zeng, R.; Yang, Q.; He, S.; Gao, W. Bioinformatics 2005, 21, 3049. doi: 10.1093/bioinformatics/bti439
Wang, L.-H.; Li, D.-Q.; Fu, Y.; Wang, H.-P.; Zhang, J.-F.; Yuan, Z.-F.; Sun, R.-X.; Zeng, R.; He, S.-M.; Gao, W. Rapid Commun. Mass Spectrom. 2007, 21, 2985. doi: 10.1002/(ISSN)1097-0231
Horn, D. M.; Zubarev, R. A.; McLafferty, F. W. J. Am. Soc. Mass Spectrom. 2000, 11, 320. doi: 10.1016/S1044-0305(99)00157-9
Jaitly, N.; Mayampurath, A.; Littlefield, K.; Adkins, J. N.; Anderson, G. A.; Smith, R. D. BMC Bioinformatics 2009, 10, 87. doi: 10.1186/1471-2105-10-87
Guner, H.; Close, P. L.; Cai, W.; Zhang, H.; Peng, Y.; Gregorich, Z. R.; Ge, Y. J. Am. Soc. Mass Spectrom. 2014, 25, 464. doi: 10.1007/s13361-013-0789-4
Bourgoin-Voillard, S.; Leymarie, N.; Costello, C. E. Proteomics 2014, 14, 1174. doi: 10.1002/pmic.201300433
Li, H.; Wongkongkathep, P.; Van Orden, S. L.; Loo, R. R. O.; Loo, J. A. J. Am. Soc. Mass Spectrom. 2014, 25, 2060. doi: 10.1007/s13361-014-0928-6
Wang, Z.; Chen, X.; Deng, L.; Li, W.; Wong, Y. L. E.; Chan, T. W. D. Eur. J. Mass Spectrom. 2015, 21, 707. doi: 10.1255/ejms.1386
Fung, Y. M. E.; Duan, L.; Chan, T. W. D. Eur. J. Mass Spectrom. 2004, 10, 449. doi: 10.1255/ejms.648
Chan, W. Y. K.; Chan, T. W. D. J. Am. Soc. Mass Spectrom. 2010, 21, 1235. doi: 10.1016/j.jasms.2010.03.034
Wong, P. S. J.; Chen, X.; Deng, L.; Wang, Z.; Li, W.; Wong, Y. L. E.; Chan, T. W. D. Rapid Commun. Mass Spectrom. 2015, 29, 1757. doi: 10.1002/rcm.7275
Fung, Y. M. E.; Chan, T. W. D. J. Am. Soc. Mass Spectrom. 2005, 16, 1523. doi: 10.1016/j.jasms.2005.05.001
Fung, Y. M. E.; Liu, H.; Chan, T. W. D. J. Am. Soc. Mass Spectrom. 2006, 17, 757. doi: 10.1016/j.jasms.2006.01.014
Chen, X.; Chan, W. Y. K.; Wong, P. S.; Yeung, H. S.; Chan, T. W. D. J. Am. Soc. Mass Spectrom. 2011, 22, 233. doi: 10.1007/s13361-010-0035-2
Wong, Y. L. E.; Chen, X.; Wu, R.; Hung, Y. L. W.; Yeung, H. S.; Chan, T. W. D. Anal. Chem. 2017, 89, 7773. doi: 10.1021/acs.analchem.7b01808
Chen, X.; Fung, Y. M. E.; Chan, W. Y. K.; Wong, P. S.; Yeung, H. S.; Chan, T. W. D. J. Am. Soc. Mass Spectrom. 2011, 22, 2232. doi: 10.1007/s13361-011-0246-1
Chen, X.; Wang, Z.; Li, W.; Wong, Y. L. E.; Chan, T. W. D. Eur. J. Mass Spectrom. 2015, 21, 649. doi: 10.1255/ejms.1382
Chen, X.; Liu, G.; Elaine Wong, Y. L.; Deng, L.; Wang, Z.; Li, W.; Chan, T. W. D. Rapid Commun. Mass Spectrom. 2016, 30, 705. doi: 10.1002/rcm.7502
Fung, Y. M. E.; Kjeldsen, F.; Silivra, O. A.; Chan, T. W. D.; Zubarev, R. A. Angew. Chem., Int. Ed. 2005, 44, 6399. doi: 10.1002/(ISSN)1521-3773
Chan, T. W. D.; Choy, M. F.; Chan, W. Y. K.; Fung, Y. M. E. J. Am. Soc. Mass Spectrom. 2009, 20, 213. doi: 10.1016/j.jasms.2008.08.018
Stobiecki, M. Phytochemistry 2000, 54, 237. doi: 10.1016/S0031-9422(00)00091-1
曲峰, 李英霞, 张一纯, 臧静, 有机化学, 2003, 23, 249. http://www.cnki.com.cn/Article/CJFDTotal-YJHU200303004.htmQu, F.; Li, Y.-X.; Zhang, Y.-C.; Zang, J. Chin. J. Org. Chem. 2003, 23, 249. http://www.cnki.com.cn/Article/CJFDTotal-YJHU200303004.htm
Ni onuevo, M. R.; Lebrilla, C. B. Nutr. Rev. 2009, 67, S216. doi: 10.1111/nure.2009.67.issue-s2
王耀君, 黄纯翠, 高枫, 张敬玮, 李岩, 卜东波, 孙世伟, 生物化学与生物物理进展, 2017, 44, 830. http://www.cnki.com.cn/Article/CJFDTotal-SHSW201710003.htmWang, Y.-J.; Huang, C.-C.; Gao, F.; Zhang, J.-W.; Li, Y.; Bu, D.-B.; Sun, S.-W. Prog. Biochem. Biophys. 2017, 44, 830. http://www.cnki.com.cn/Article/CJFDTotal-SHSW201710003.htm
Wong, Y. L. E.; Chen, X.; Li, W.; Wang, Z.; Hung, Y. L. W.; Wu, R.; Chan, T. W. D. Anal. Chem. 2016, 88, 5590. doi: 10.1021/acs.analchem.6b00908
Wong, Y. L. E.; Chen, X.; Wu, R.; Hung, Y. L. W.; Chan, T. W. D. Anal. Chem. 2017, 89, 10111. doi: 10.1021/acs.analchem.7b03128
Fischle, W.; Tseng, B. S.; Dormann, H. L.; Ueberheide, B. M.; Garcia, B. A.; Shabanowitz, J.; Hunt, D. F.; Funabiki, H.; Allis, C. D. Nature 2005, 438, 1116. doi: 10.1038/nature04219
Ge, Y.; Lawhorn, B. G.; ElNaggar, M.; Strauss, E.; Park, J.-H.; Begley, T. P.; McLafferty, F. W. J. Am. Chem. Soc. 2002, 124, 672. doi: 10.1021/ja011335z
Pesavento, J. J.; Mizzen, C. A.; Kelleher, N. L. Anal. Chem. 2006, 78, 4271. doi: 10.1021/ac0600050
Reid, G. E.; Stephenson, J. L.; McLuckey, S. A. Anal. Chem. 2002, 74, 577. doi: 10.1021/ac015618l

图 1 磁场中的离子回旋运动及离子回旋共振池
Figure 1 Ion cyclotron motion in a magnetic field and ion cyclotron resonance cell

图 8 热力学循环与离子-电子重组能
Figure 8 Thermochemical cycle and ion-electron recombination energy



 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:















