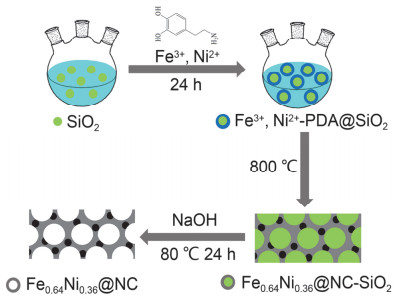
图 1.
Fe0.64Ni0.36@NC的合成示意图
Figure 1.
The synthetic process for Fe0.64Ni0.36@NC

经济和科技的持续发展使能源需求成为人们关注的热点.然而近年来传统化石能源大量开采, 储量日渐减少, 需求量却逐年增大, 同时传统化石能源燃烧产生的废气、废渣造成严重的环境污染、温室效应等问题, 使能源危机和环境问题日益加重[1~5].为了解决上述问题, 发展可持续的清洁新能源和不断提高能源转换效率成为现代社会最迫切的需求[6, 7].氢能作为一种重要的储存和转运能源的载体, 具有燃烧热高、能源转换过程无毒、无污染等优点[8~11].电解水制氢是氢能生产最便捷的途径之一, 可分解为析氢(HER)和析氧(OER)两个半反应, 其中OER为四电子转移过程, 动力学迟缓, 过电位高, 造成整体转换效率不高[12~17].目前广泛采用RuO2和IrO2等贵金属作为OER催化剂, 其活性较好, 但成本高、储备稀有、稳定性差, 无法得到广泛应用[18~21].因此, 开发高效耐用、储量丰富和高性能的替代催化剂是近年来大量研究工作的重点.研究表明过渡金属基材料, 例如Fe, Co, Ni, Mn等的化合物能够更多暴露d轨道电子[22~28], 提高催化活性, 有望成为可替代贵金属的析氧催化剂.
最近研究发现, Fe/Ni基催化剂对OER具有较高催化活性[29].例如Landon等[30]合成了Fe/Ni双层氢氧化物(Fe/Ni-LDH)催化剂并与多壁碳纳米管进行复合, 通过调控催化剂的局部电子结构提高OER性能[31~35].同时Fe/Ni基催化剂与导电基底相结合可加速电荷转移, 促进OER活性, 增强其稳定性[36, 37].例如, Yang等[38]采用氮掺杂石墨烯将二元合金或三元合金封装后进行退火处理, 所得的三元Fe/Co/Ni合金-石墨烯催化剂具有良好的OER性能, 包裹在金属合金上的薄碳层能够优化其吸附能力[39, 40].同时氮原子的掺杂不仅可以改变碳的电子状态, 调节表面电子结构以降低局部功函数, 还可以改变反应中间体的吸附强度并提供C-N活性位点[41, 42].此外大量研究表明, Fe/Ni基催化剂与掺杂碳壳之间存在协同效应, 高度分散的铁镍合金颗粒可以提供更多的活性位点, 同时具有3d轨道的金属合金可以通过有效调节电子结构和吸附能达到提升催化活性的目的.
但是, 在电化学循环过程中, 过渡金属催化剂经过多次循环反应后容易直接暴露于电解质溶液中, 发生溶解和颗粒团聚的现象, 造成活性快速下降.此外, 还存在铁镍合金颗粒被过厚碳层包裹, 不利于活性位点暴露等问题.
本项工作制备了铁镍合金纳米颗粒镶嵌的多级孔氮掺杂碳催化剂Fe0.64Ni0.36@NC.以SiO2纳米小球为大孔模板, 多巴胺(PDA)为氮碳源, 不同浓度比例的Fe3+, Ni2+为金属源, 通过原位吸附、聚合、焙烧、NaOH刻蚀等步骤制备得到Fe0.64Ni0.36嵌入的NC多级孔结构, 合成过程如图 1所示.测试结果表明, 将合金颗粒镶嵌进薄碳涂层中不仅降低了金属物种在反应过程中的溶解和团聚程度, 明显提高稳定性及电导率, 还克服了铁镍合金被包裹在过厚碳层中不利于暴露活性位点等问题.
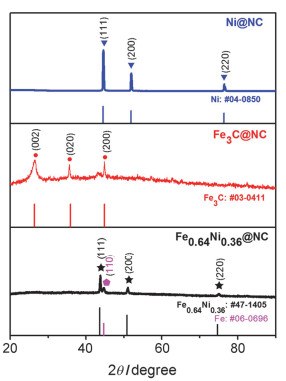
首先, 对加入不同比例Fe3+, Ni2+的样品进行了X射线衍射(XRD)测试(图 2).只加入Ni2+的样品在2θ=44.5º, 51.8º和76.3º出现衍射峰[43], 分别对应Ni(111), (200)和(220)晶面(JCPDS卡, #04-0850), 表明金属镍掺杂进入氮碳层, 命名为Ni@NC.只加入Fe3+的样品在2θ=26.4°, 35.8°和42.1°出现衍射峰, 分别对应Fe3C(002), (020)和(200)晶面(JCPDS卡, #03-0411), 命名为Fe3C@NC.同时加入Fe3+, Ni2+的样品呈现四个衍射峰, 其中三个(2θ=43.6°, 50.8°, 74.6°)可以归属到合金Fe0.64Ni0.36(JCPDS卡, #47-1405), 而处在2θ=44.6°的衍射峰来自于金属Fe(110)晶面衍射(JCPDS卡, #06-0696), 因此该样品中金属成分主要为Fe0.64Ni0.36合金, 同时包含少量单质铁, 命名为Fe0.64Ni0.36@NC. HYPERLINK "mailto:通过电感耦合等离子体发射光谱法(ICP)测定Fe0.64Ni0.36@NC"通过电感耦合等离子体发射光谱法(ICP)测定Fe0.64Ni0.36@NC催化剂中Fe:Ni原子比约为4:1, 表明催化剂中除了含有Fe0.64Ni0.36合金, 还含有少量Fe单质, 与XRD分析结果一致.
利用X射线光电子能谱(XPS) HYPERLINK "mailto:方法进一步探究Fe0.64Ni0.36@NC中的元素组成和表面化学态"方法进一步探究Fe0.64Ni0.36@NC中的元素组成和表面化学态. N元素高分辨图谱(图 3a)可以分解成四个峰, 分别位于397.5, 399.1, 400.1和401.3 eV, 对应于吡啶N, 吡咯N, 石墨N和氧化N[44]. Fe、Ni元素高分辨图谱(c)表明, Fe在表面呈现Fe0和Fe3+两种状态, Ni呈现Ni0, Ni2+两种状态, 其中Fe3+、Ni2+应为金属表面部分氧化所致.结合XRD分析表明, 所制备Fe0.64Ni0.36@NC材料的碳层内包裹了Fe0.64Ni0.36合金以及少量单质Fe纳米颗粒, 金属表面在空气中部分氧化, 同时元素N已成功掺杂到碳层中.
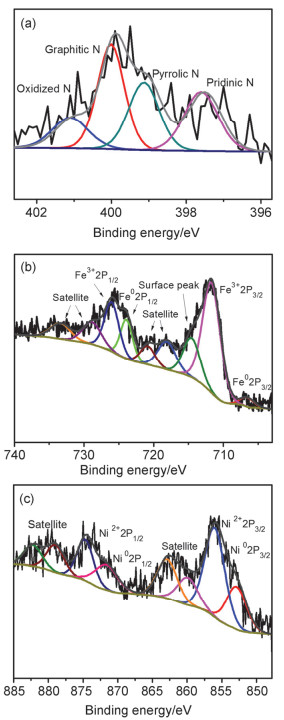
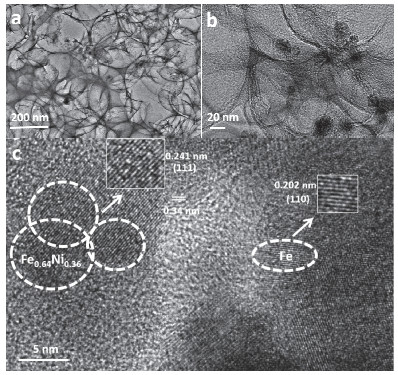
图 4的扫描电子显微镜(SEM)图片显示了所制备材料的微观形貌结构. 图 4a是大小均匀的二氧化硅微球, 直径约为300 nm.加入PDA和Fe3+, Ni2+充分反应后, 得到复合物Fe3+, Ni2+-PDA@SiO2 (图 4b), 从图中可以看出该复合物保留了二氧化硅球形形状且表面粗糙, 说明PDA已包覆在外层, 且没有出现明显的团聚现象. 图 4c是样品Fe0.64Ni0.36@NC, 发现经过NaOH水热处理24 h后, SiO2小球刻蚀完全, 材料呈现中空结构.为了了解催化剂材料的结构、尺寸大小和晶格参数等信息, 进一步进行了透射电子显微镜(TEM)的表征.从图 5中可以看出Fe0.64Ni0.36@NC合金颗粒高度分散在碳氮化物层中, 粒径约为20±2.2 nm.观察到间距为0.241 nm和0.202 nm的晶格, 分别归属于Fe0.64Ni0.36合金的(111)晶面[45]和Fe单质的(110)晶面.该结果进一步确认碳层内包含Fe0.64Ni0.36合金和单质Fe, 与XRD、XPS分析结果一致.

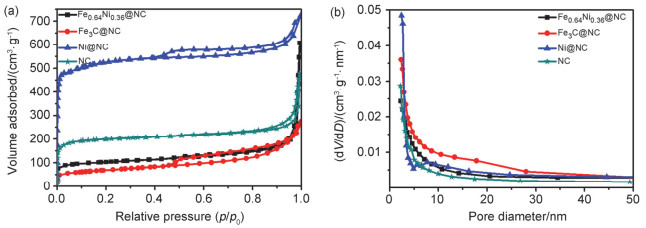
对所制备催化剂进行了氮气吸附脱附测试, 表征其孔道特征.如图 6a所示, 可以看出四种催化剂的等温吸附/脱附均属于Ⅳ型吸脱附等温线, 在超低压力区具有明显吸附, 在中压区p/p0=0.5~0.8具有明显的回滞环, 表明材料包含丰富的微孔和介孔. Fe0.64Ni0.36@NC催化剂的比表面积是386.4 m2·g-1, 孔径尺寸集中在7.0 nm以下(图 6b), 结合SiO2模板制造的300 nm大孔, 所制备材料应为包含微孔-介孔-大孔的多级孔结构.
所制得催化剂在1.0 mol/L KOH电解液中进行电化学性能测试表征(三电极体系), 相应的结果如图 7所示.相比于Fe3C@NC、Ni@NC和NC, HYPERLINK "mailto:Fe0.64Ni0.36@NC表现出更优异的OER活性" Fe0.64Ni0.36@NC表现出更优异的OER活性.从图 7a析氧极化曲线测试结果对比图可以看出, Fe0.64Ni0.36@NC只需要286和333 mV的过电位就可以达到10和100 mA·cm-2的电流密度, 显著优于贵金属RuO2催化剂在电流密度10 mA·cm-2时需要的380 mV的过电位[46, 47], 也明显优于Fe3C@NC、Ni@NC、NC在10 mA·cm-2的电流密度时的过电位.另外, Tafel斜率是评估OER动力学的重要指标[48].根据LSV曲线和Tafel斜率计算方程(η=b·log(j/j0), 其中ƞ, j和b分别是过电位、电流密度和Tafel斜率), 可以得到如图 7b所示的Tafel曲线.显示样品Fe0.64Ni0.36@NC, Fe3C@NC, Ni@NC, NC的Tafel斜率分别是55.8 mV·dec-1, 61.7 mV·dec-1, 92.3 mV·dec-1和124.5 mV·dec-1, 其中HYPERLINK "mailto:Fe0.64Ni0.36@NC的斜率最小,表示其析氧极化随电流密度增大的速度最慢," Fe0.64Ni0.36@NC的斜率最小, 表示其析氧极化随电流密度增大的速度最慢, OER动力学过程最快.

电化学阻抗谱(EIS)测试用来研究催化剂的电极反应动力学[49].在100 kHz至0.05 Hz的频率范围内(图 7c), 所有样品中Fe0.64Ni0.36@NC的电荷转移阻抗(8.79 Ω)最小, HYPERLINK "mailto:表明Fe0.64Ni0.36@NC具有最快的动力学过程,与Tafel数据结果一致"动力学过程最快, 与Tafel数据结果一致.进一步测试材料在不同扫速下的CV曲线(图S1), 并计算出电化学活性表面积(electrochemically active surface area, ECSA).如图 7d所示, HYPERLINK "mailto:Fe0.64Ni0.36@NC的ECSA为9.5" Fe0.64Ni0.36@NC的ECSA为9.5 mF·cm-2, 明显高于Fe3C@NC(1.9 mF·cm-2), Ni@NC(2.4 mF·cm-2)和NC(0.7 mF·cm-2). HYPERLINK "mailto:最后测试了Fe0.64Ni0.36@NC催化剂的稳定性,在1.3"最后测试了Fe0.64Ni0.36@NC催化剂的稳定性, 在1.3~1.6 V电位下连续扫描2000圈以后, 催化活性无明显衰减, 过电位仅有少量增加(图 7e).在电流密度10 mA·cm-2下连续测试30 h后, 电极输出非常稳定(图 7f).因此, HYPERLINK "mailto:Fe0.64Ni0.36@NC催化剂具有良好的电化学稳定性" Fe0.64Ni0.36@NC催化剂具有良好的电化学稳定性.此外进一步探究了不同Fe/Ni比例、不同多巴胺含量制备的催化剂在OER中的性能, 结果如图S2所示, 其中Fe0.64Ni0.36@NC性能最好, 体现了原料最优化配比.
上述测试结果表明, Fe0.64Ni0.36@NC催化剂具有优异的OER性能, 这来源于催化剂多重微观结构优势: (1)微孔-介孔-大孔的多级孔结构有利于传质及暴露活性位点, 提高活性位利用率; (2)嵌入的Fe0.64Ni0.36合金及单质Fe颗粒和NC层协同作用, 产生丰富活性位点; (3)金属颗粒由石墨化NC壳层包裹, 电化学反应过程中能很好地保持原始形态和催化活性, 进而提升了催化剂抗腐蚀能力和耐久性.
本文以尺寸约为300 nm二氧化硅微球为模板, 多巴胺为氮碳源, Fe3+, Ni2+为金属源, 通过原位吸附、聚合、焙烧、NaOH刻蚀得到铁镍合金纳米颗粒镶嵌的多级孔氮掺杂碳催化剂(HYPERLINK "mailto:Fe0.64Ni0.36@NC" Fe0.64Ni0.36@NC).在析氧反应中, 所制备的Fe0.64Ni0.36@NC催化剂相对于贵金属RuO2, 表现出优异的催化活性, 达到10 mA·cm-2的电流密度时所需过电位286 mV, 同时该材料在连续扫描2000圈后仍然保持较稳定的电极输出, 表现了良好的稳定性.嵌入的Fe0.64Ni0.36合金及Fe颗粒同NC外层协同作用, 产生丰富活性位点, 同时NC层的包覆大大提升了催化剂稳定性.本项工作设计的非贵金属催化剂廉价、高效、耐用, 为过渡金属催化剂在析氧方面的应用开辟了新的思路.
SiO2合成基于Stöber方法[50].首先, 将4.0 mL氨水溶液(25~28 wt%)、10.0 mL蒸馏水和60.0 mL乙醇混合, 并向其中滴加10.0 mL正硅酸乙酯(TEOS).将该反应溶液在30 ℃下连续搅拌4.5 h, 得到直径约为300 nm二氧化硅微球悬浮液.该悬浮液无需洗涤, 直接用于后续合成.
在所制备的分散有SiO2微球的悬浮液中加入10.0 g多巴胺作为碳氮源, 然后加入1.5 mmoL FeCl3·6H2O和1.5 mmoL NiCl2·6H2O作为铁源和镍源.该溶液在30 ℃下连续搅拌24 h后, 离心干燥得到Fe3+, Ni2+- PDA@SiO2复合物.将该样品在N2中以2 ℃/min升至200 ℃保持2 h, 再以5 ℃/min上升到800 ℃并保持2 h, 最后用3.0 mol/L NaOH溶液在80 ℃下水热24 h刻蚀除去SiO2模板, 用去离子水洗至中性, 并在60 ℃下真空干燥, HYPERLINK "mailto:得到样品Fe0.64Ni0.36@NC"得到样品Fe0.64Ni0.36@NC.此外, 我们采用相同方法, 改变Fe3+、Ni2+比例以及多巴胺加入量制得几种对比样品, 详细合成参数见支撑信息.
采用FEI Nova 400型扫描电子显微镜(SEM), 在20 kV电压条件下对样品尺寸和形貌进行分析; 采用Zeiss LIBRA 200型透射电子显微镜(TEM)在加速电压200 kV下观测样品的结构、晶格参数; 使用岛津XRD-6000型X射线衍射(XRD)设备表征晶体结构; 使用Micrometrics Tristar 2390型设备在77 K下测量N2吸附/脱附等温线, 在测量之前, 将样品在180 ℃下真空脱气10 h.
电化学数据采用三电极体系在电化学分析仪(CHI660D, CH Instruments, 中国上海)上进行测量, 对电极为石墨棒(直径10 mm), 参比电极为Hg/HgO, 电解质是1.0 mol/L KOH水溶液, 保持电解液达到O2饱和状态, 并确保所有实验在1.23 V vs. RHE下达到O2/H2O平衡.在1.3~1.7 V的电位下以5 mV·s-1的扫描速率测试得到线性扫描伏安曲线(LSV).恒电位状态下、1.0 mol/L KOH水溶液中, 在100 kHz至0.05 Hz频率范围内测量电化学阻抗谱(EIS).所有极化曲线均针对iR补偿进行校正, 计算公式如下:
|
${E_{{\rm{corrected}}}} = {E_{{\rm{uncorrected}}}} - i{R_{\rm{s}}} $ |
(1) |
其中i和Rs代表电流密度和溶液阻抗.
过电位计算如下:
|
$\eta = E\left( {{\rm{vs}}.{\rm{ RHE}}} \right) - 1.23 $ |
(2) |
在1.23 V vs. RHE下的O2/H2O平衡.
塔菲尔斜率根据塔菲尔方程进行计算:
|
$\eta = b \cdot {\rm{log}}(j/{j_0}) $ |
(3) |
其中, η, b, j和j0分别代表过电位, 塔菲尔斜率, 电流密度和交换电流密度.
加速老化测试(accelerated durability tests, ADT)采用循环伏安法, 在1.3~1.6 V高电位范围内进行2000次循环扫描, 以评估所制备催化剂在析氧反应中的电化学耐久性.
Armaroli, N.; Balzani, V. Angew. Chem., Int. Ed. 2007, 46, 52. doi: 10.1002/(ISSN)1521-3773
丁炜, 张雪, 李莉, 魏子栋, 电化学, 2014, 20, 426. http://www.cnki.com.cn/Article/CJFDTotal-DHXX201405005.htmDing, W.; Zhang, X.; Li, L.; Wei, Z. D. J. Electrochem. 2014, 20, 426 (in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-DHXX201405005.htm
彭立山, 魏子栋, 催化学报, 2018, 39, 1575. http://www.cnki.com.cn/Article/CJFDTotal-CHUA201810003.htmPeng, L. S.; Wei, Z. D. Chinese J. Catal. 2018, 39, 1575 (in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-CHUA201810003.htm
Villa, I.; Villa, C.; Monguzzi, A.; Babin, V.; Tervoort, E.; Nikl, M.; Niederberger, M.; Torrente, Y.; Vedda, A.; Lauria, A. Nanoscale 2018, 10, 7933. doi: 10.1039/C8NR00724A
Zhang, C.; Zhang, M. R.; Shi, H. Y.; Wang, J. P.; Niu, J. Y. Chem. Commun. 2018, 54, 5458. doi: 10.1039/C8CC01622D
郭宇, 姚宇, 李慧, 赫兰兰, 杨忠志, 赵东霞, 化学学报, 2017, 75, 903. doi: 10.3866/PKU.WHXB201702091Guo, Y.; Yao, Y.; Li, H.; He, L. L.; Yang, Z. Z.; Zhao, D. X. Acta Chim. Sinica 2017, 75, 903 (in Chinese). doi: 10.3866/PKU.WHXB201702091
Zuo, L. X.; Jiang, L.; Zhu, J. J. Chin. J. Chem. 2017, 35, 969. doi: 10.1002/cjoc.v35.6
Zou, X.; Zhang, Y. Chem. Soc. Rev. 2015, 44, 5148. doi: 10.1039/C4CS00448E
王森林, 王丽品, 张振洪, 物理化学学报, 2013, 29, 981. doi: 10.3866/PKU.WHXB201303071Wang, S. L.; Wang, L. P.; Zhang, Z. H. Acta Phys.-Chim. Sin.2013, 29, 981 (in Chinese). doi: 10.3866/PKU.WHXB201303071
王俊, 魏子栋, 物理化学学报, 2017, 33, 886. doi: 10.3866/PKU.WHXB201702092Wang, J.; Wei, Z. D. Acta Phys.-Chim. Sin. 2017, 33, 886 (in Chinese). doi: 10.3866/PKU.WHXB201702092
彭立山, 魏子栋, 化学进展, 2018, 30, 14. doi: 10.7536/PC170912Peng, L. S.; Wei, Z. D. Prog. Chem. 2018, 30, 14 (in Chinese). doi: 10.7536/PC170912
骆静利, 物理化学学报, 2018, 34, 7. doi: 10.3866/PKU.WHXB201707051Luo, J. L. Acta Phys.-Chim. Sin. 2018, 34, 7 (in Chinese). doi: 10.3866/PKU.WHXB201707051
Zhang, Y.; Shimoda, K.; Miyaoka, H. Int. J. Hydrogen Energy 2010, 35, 12405. doi: 10.1016/j.ijhydene.2010.08.018
Hinnemann, B.; Moses, P. G.; Bonde, J.; Jorgensen, K. P.; Nielsen, J. H. J. Am. Chem. Soc. 2005, 127, 5308. doi: 10.1021/ja0504690
Lee, Y.; Jin, S.; May, K. J.; Perry, E. E.; Yang, S. H. J. Phys. Chem. Lett. 2012, 3, 399. doi: 10.1021/jz2016507
Over, H. Chem. Rev. 2012, 43, 3356. https://www.ncbi.nlm.nih.gov/pubmed/22423981
Liu, T. T.; Xie, L. S.; Yang, J. H.; Kong, R. M.; Sun, X. P.; Chen, L. ChemElectroChem 2017, 4, 1840. doi: 10.1002/celc.v4.8
Fang, Y. H.; Liu, Z. P. J. Am. Chem. Soc. 2010, 132, 18214. doi: 10.1021/ja1069272
Fang, Z.; Peng, L.; Zhu, Y.; Yan, C.; Wang, S.; Kalyani, P.; Wu, X.; Yu, G. ACS Nano 2017, 11, 9550. doi: 10.1021/acsnano.7b05481
Feng, J. X.; Xu, H.; Dong, Y. T.; Ye, S. H.; Tong, Y. X.; Li, G. R. Angew. Chem., Int. Ed. 2016, 128, 3758. doi: 10.1002/ange.201511447
Ji, Y. Y.; Li, Y.; Ren, X.; Cui, G. W.; Xiong, X. L.; Sun, X. P. ACS Sustain. Chem. Eng. 2018, 6, 9555. doi: 10.1021/acssuschemeng.8b01841
Yang, J.; Zhu, G.; Liu, Y.; Xia, J.; Ji, Z.; Shen, X. Adv. Funct. Mater. 2016, 26, 4712. doi: 10.1002/adfm.v26.26
Gao, M.; Sheng, W.; Zhuang, Z.; Fang, Q.; Gu, S.; Jiang, J.; Yan, Y. J. Am. Chem. Soc. 2014, 136, 7077. doi: 10.1021/ja502128j
Chuan, T. Y.; Chun, Z. H. Chem. Commun. 2016, 52, 11591. doi: 10.1039/C6CC05699G
Hoare, J. P.; Schuldiner, S. J. Phys. Chem. 2002, 62, 229. doi: 10.1021/j150560a020
Tang, T.; Jiang, W. J.; Niu, S.; Liu, N.; Luo, H.; Chen, Y. Y.; Jin, S. F.; Gao, F.; Wan, L. J. J. Am. Chem. Soc. 2017, 139, 8320. doi: 10.1021/jacs.7b03507
Li, F. L.; Shao, Q.; Huang, X.; Lang, J. P. Angew. Chem., Int. Ed. 2018, 57, 1888. doi: 10.1002/anie.201711376
Chemelewski, W. D.; Rosenstock, J. R.; Mullins, C. B. J. Mater. Chem. 2014, 2, 14957. doi: 10.1039/C4TA03078H
何杨华, 徐金铭, 王楠, 化工进展, 2016, 35, 2057. http://www.cnki.com.cn/Article/CJFDTotal-HGJZ201607019.htmHe, Y. H.; Xu, J. M.; Wang, N. Chem. Eng. Prog. 2016, 35, 2057 (in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-HGJZ201607019.htm
Landon, J.; Demeter, E.; İnoğlu, N.; Keturakis, C.; Wachs, I. E.; Frenkel, A. I. ACS Catal. 2012, 2, 1793. doi: 10.1021/cs3002644
Gong, M.; Li, Y.; Wang, H.; Liang, Y.; Wu, J. Z.; Zhou, J.; Wang, J.; Regier, T.; Wei, F. J. Am. Chem. Soc. 2013, 135, 8452. doi: 10.1021/ja4027715
Trotochaud, L.; Young, S. L.; Ranney, J. K. J. Am. Chem. Soc. 2014, 136, 6744. doi: 10.1021/ja502379c
Smith, A. M.; Trotochaud, L.; Burke, M. S.; Boettcher, S. W. Chem. Commun. 2015, 51, 5261. doi: 10.1039/C4CC08670H
Yang, Y.; Zhuang, L.; Lin, R.; Li, M.; Xu, X.; Rufford, T. E.; Zhu, Z. J. Power Sources 2017, 349, 68. doi: 10.1016/j.jpowsour.2017.03.028
Liang, Y.; Liu, Q.; Asiri, A. M.; Sun, X.; He, Y. Int. J. Hydrogen Energy 2015, 40, 13258. doi: 10.1016/j.ijhydene.2015.07.165
Zhao, Y.; Chen, S.; Sun, B.; Su, D.; Huang, X.; Liu, H.; Yan, Y.; Sun, K.; Wang, G. Sci. Rep. 2015, 5, 7629. doi: 10.1038/srep07629
Ma, T. Y.; Dai, S.; Jaroniec, M.; Qiao, S. Z. J. Am. Chem. Soc. 2014, 136, 13925. doi: 10.1021/ja5082553
Yang, Y.; Lin, Z.; Gao, S.; Su, J.; Lun, Z.; Xia, G. ACS Catal. 2017, 7, 469. doi: 10.1021/acscatal.6b02573
Tavakkoli, M.; Kallio, T.; Reynaud, O.; Nasibulin, A. G.; Johans, C.; Sainio, J.; Jiang, H.; Kauppinen, E. I.; Laasonen, K. Angew. Chem., Int. Ed. 2015, 54, 4535. doi: 10.1002/anie.201411450
Tao, Z.; Wang, T.; Wang, X.; Zheng, J.; Li, X. ACS Appl. Mater. Interfaces 2016, 8, 35390. doi: 10.1021/acsami.6b13411
Liu, Z.; Sun, F.; Gu, L.; Chen, G.; Shang, T.; Liu, J.; Le, Z.; Li, X.; Wu, H. B.; Lu, Y. Adv. Energy Mater. 2017, 7, 1701154. doi: 10.1002/aenm.201701154
郭宇, 刘瑜, 戚娟娟, 李慧, 赫兰兰, 化学学报, 2017, 75, 914. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract346227.shtmlGuo, Y.; Liu, Y.; Qi, J. J.; Li, H.; He, L. L. Acta Chim. Sinica 2017, 75, 914 (in Chinese). http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract346227.shtml
鲍晋珍, 王森林, 物理化学学报, 2011, 27, 2849. doi: 10.3866/PKU.WHXB20112849Bao, J. Z.; Wang, S. L. Acta Phys.-Chim. Sin. 2011, 27, 2849 (in Chinese). doi: 10.3866/PKU.WHXB20112849
Yang, Q. Q.; Liu, L.; Xiao, L.; Wang, M. J.; Li, J.; Wei, Z. D. J. Mater. Chem. A 2018, 6, 14752. doi: 10.1039/C8TA03604G
Ma, Y. D.; Dai, X. P.; Liu, M. Z.; Zhang, X. ACS Appl. Mater. Interfaces 2016, 8, 34396. doi: 10.1021/acsami.6b11821
Wu, Y.; Chen, M.; Han, Y.; Luo, H.; Su, X.; Zhang, M. T.; Lin, X.; Sun, J.; Wang, L.; Deng, L.; Zhang, W.; Cao, R. Angew. Chem., Int. Ed. 2015, 54, 4870. doi: 10.1002/anie.201412389
Zhang, B.; Xiao, C.; Xie, S.; Liang, J.; Chen, X.; Tang, Y. Chem. Mater. 2016, 28, 6934. doi: 10.1021/acs.chemmater.6b02610
Han, X.; Yu, C.; Zhou, S.; Zhao, C.; Huang, H.; Yang, J.; Liu, Z.; Zhao, J.; Qiu, J. Adv. Energy Mater. 2017, 7, 1602148. doi: 10.1002/aenm.201602148
武刚, 李宁, 戴长松, 周德瑞, 催化学报, 2004, 25, 319. doi: 10.3321/j.issn:0253-9837.2004.04.016Wu, G.; Li, N.; Dai, C. S.; Zhou, D. R. Chinese J. Catal. 2004, 25, 319 (in Chinese). doi: 10.3321/j.issn:0253-9837.2004.04.016
Stöber, W.; Fink, A.; Bohn, E. J. Colloid Interface Sci. 1968, 26, 62. doi: 10.1016/0021-9797(68)90272-5

图 2 Fe0.64Ni0.36@NC, Fe3C@NC, Ni@NC的XRD图
Figure 2 XRD patterns of Fe0.64Ni0.36@NC, Fe3C@NC and Ni@NC

图 3 Fe0.64Ni0.36@NC的N 1s (a), Fe 2p (b)和Ni (c)的高分辨XPS谱图
Figure 3 The high resolution XPS spectra of N 1s (a), Fe 2p (b) and Ni 2p (c) for Fe0.64Ni0.36@NC

图 4 所制备催化剂的扫描电子显微镜图. (a) SiO2; (b) Fe3+, Ni2+- PDA@SiO2; (c) Fe0.64Ni0.36@NC
Figure 4 SEM images for the prepared products. (a) SiO2; (b) Fe3+, Ni2+-PDA@SiO2; (c) Fe0.64Ni0.36@NC

图 6 Fe0.64Ni0.36@NC, Fe3C@NC, Ni@NC和NC催化剂的N2吸附/脱附等温线(a)和孔尺寸分布曲线(b)
Figure 6 N2 adsorption-desorption isotherm (a) and pore size distribution plots (b) for Fe0.64Ni0.36@NC, Fe3C@NC, Ni@NC and NC products

图 7 Fe0.64Ni0.36@NC, Fe3C@NC, Ni@NC和NC催化剂电化学析氧性能测试. (a)极化曲线; (b)Tafel斜率; (c)电化学阻抗谱(EIS); 尼奎斯特曲线的等效电路(插图), (d)通过绘制1.0 mol/L KOH中的电流密度变化(Δj=(ja-jc)/2)来估算Cdl; (e, f) Fe0.64Ni0.36@NC催化剂的耐久性测试
Figure 7 The OER performance of the obtained catalysts: (a) polarization curves (with iR correction); (b) Tafel slopes for the prepared materials (measured at a scan rate of 5.0 mV·s-1 in O2-saturated 1.0 mol/L KOH solution); (c) Nyquist plots of the prepared catalysts; The inset shows the electrical equivalent circuit used to model the catalyst systems (inset); (d) An estimation of Cdl from plotting the current density variation (Δj=(ja-jc)/2) in 1.0 mol/L KOH; (e, f) Durability test of Fe0.64Ni0.36@NC catalyst

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
