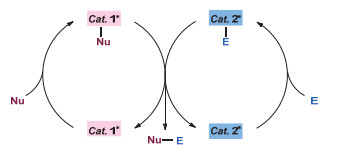
图 1.
协同催化的原理
Figure 1.
General mechanism of synergetic catalysis

过渡金属催化的烯丙基化反应在有机合成化学中占据着十分重要的地位, 是目前构建碳碳键和碳杂键最有效的方法之一[1]. 1965年, Tsuji等[2]实现了烯丙基氯化钯与丙二酸二乙酯钠盐化学计量的反应.随后, 钯催化的烯丙基化反应也被报道[3]. 1977年, Trost等[4]用手性的膦配体首次实现了钯催化的不对称烯丙基化反应.在之后的几十年中, 过渡金属催化的不对称烯丙基化反应得到快速的发展, 合成化学家也发展了许多手性的配体来控制该反应的对映选择性[5].尽管如此, 这种单一金属催化体系在构建立体多样性的产物方面, 特别是具有多个手性中心的化合物, 仍然面临一定的挑战.此外, 对于钯催化的烯丙基化反应而言, 亲核片段往往局限于软亲核试剂(pKa<25)[1, 6].如果硬亲核试剂(pKa>25)要想参与钯催化的烯丙基化反应, 往往需要使用预先或原位制备的金属试剂或者烯醇硅醚[7].这些反应不仅需要强碱性等苛刻的条件, 而且还会产生当量的废弃物.这就需要合成化学家们发展新的策略、新的催化体系来解决上述挑战性问题.
协同催化是指把两种或者多种不同的催化剂应用到一个反应体系中来实现化学转化的催化模式, 不同的催化剂需要负责活化不同的反应组分[8].对于过渡金属与有机小分子协同催化而言[8h], 如图 1所示, 它可以是有机催化剂cat. 1活化的亲核试剂Nu, 过渡金属催化剂cat. 2活化的亲电试剂E, 两者反应得到目标产物Nu-E.将这种催化模式应用到不对称烯丙基化反应可以有意义地拓宽亲核试剂的类型和烯丙基化反应的应用范围.任意一种催化剂具有手性, 都有可能实现反应的对映选择性控制; 如果两种催化剂都具有手性, 有可能提高反应的对映选择性, 甚至实现连续手性中心的非对映选择性控制.
早在2001年, 龚流柱课题组[9a]和Takemoto课题组[9b]通过使用非手性的钯催化剂和手性的相转移催化剂, 先后实现了氨基酸衍生物的不对称烯丙基化反应.之后, 过渡金属和有机小分子协同催化的不对称烯丙基化反应吸引了越来越多合成化学家的关注并取得了一批重要的研究成果[1d].本文将总结这一研究领域的进展, 重点探讨代表性反应的机理.根据手性的来源, 本文将这些烯丙基化反应归结为: (1)过渡金属催化剂控制手性的不对称烯丙基化反应, (2)有机小分子催化剂控制手性的不对称烯丙基化反应; (3)过渡金属催化剂和有机小分子催化剂共同控制手性的不对称烯丙基化反应.
二级胺或一级胺可以与醛酮原位缩合形成烯胺中间体, 从而提高羰基α位的亲核性[10]. 2006年, Córdova等[11]首次将这种原理(有机胺催化)和钯催化结合, 实现了醛、酮分子间羰基α位的烯丙基化反应(图 2).紧接着, Saicic等[12]在2007年用相同的策略实现了分子内的烯丙基化反应, 以较好的收率和非对映选择性得到五元环和六元环产物(图 3).同时, 作者对该反应的不对称过程也进行了初步的研究.在手性胺催化剂并没有给出理想对映选择性的情况下, 作者筛选了不同的磷配体, 最终发现以(R)-BINAP作为配体和在-20 ℃反应时, 可以以91%的对映选择性和中等的收率得到手性产物7a.后续的工作中, 作者发现当使用磷酸烯丙酯为烯丙基源, 以及使用Pd(OAc)2催化剂和富电子双膦配体L2时, 可以明显地提高该反应的收率和对映选择性[13].
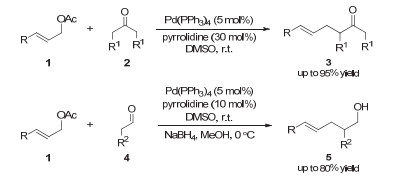

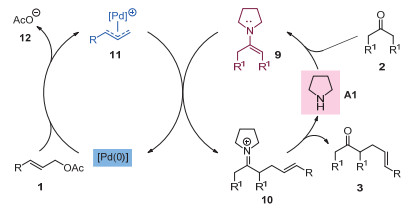
这类反应的原理如图 4所示:以酮和烯丙基醋酸酯的分子间烯丙基化反应为例, 酮2首先与吡咯烷催化剂缩合生成烯胺中间体9; 同时, 醋酸烯丙酯1与零价钯催化剂通过氧化加成形成烯丙基钯中间体11.然后, 具有亲核性的烯胺中间体9与亲电性物种π-烯丙基钯11作用生成亚胺离子中间体10.随后, 该中间体经过水解得到最终的酮α-烯丙基化产物3并重新产生零价钯催化剂和吡咯烷催化剂.
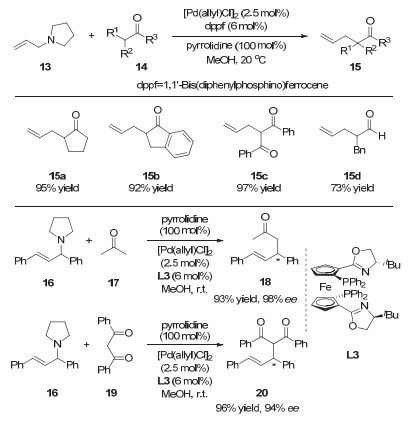
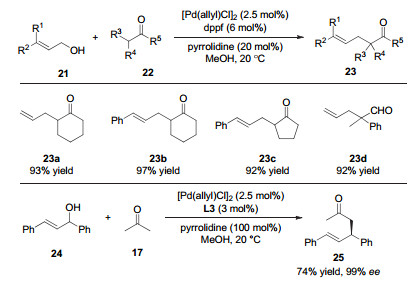
2011年, 张万斌课题组[14]以烯丙基胺为前体, 实现了钯催化的羰基α位的烯丙基化反应(图 5).作者发现, 当量吡咯烷催化剂和质子性溶剂的使用有利于反应的进行.同时, 作者还发现, 当使用二茂铁骨架的手性N, P配体L3时, 反应以很高的收率和对映选择性给出丙酮和1, 3-二酮的不对称烯丙基化产物.
2014年, 该课题组继续通过协同催化策略, 直接以烯丙醇为原料, 实现了羰基邻位的烯丙基化反应(图 6)[15].计算表明, 质子性溶剂与底物之间的氢键作用能使羟基离去的能垒降低25.9 kcal/mol, 从而有利于C—O键的断裂和π-烯丙基钯中间体的生成.紧接着, 作者用手性的N, P配体L3实现了该反应的不对称过程, 以99%的ee值和74%的收率得到手性产物.随后, 他们同样通过钯和胺协同催化的策略, 实现了烯丙基醚为烯丙基源的羰基α位烯丙基化反应[16].
除了有机胺催化剂外, Lewis酸或者Brønsted酸也被应用于协同催化中. 2006年, Trost小组[17]通过在反应体系中加入当量的硼烷, 实现了吲哚3位的不对称烯丙基化反应(图 7).作者认为, 硼烷作为Lewis酸, 可以屏蔽吲哚N的亲核性, 从而更加高选择性的得到3位进攻的产物; 另外, 硼烷也可能活化烯丙醇底物, 辅助羟基的离去.
2014年, 夏春谷和蒋高喜等[18]实现了吖内脂与烯丙醇的不对称烯丙基化反应(图 8).作者发现催化量的苯甲酸可以很好地活化反应底物, 得到直链的烯丙基化产物.该反应为手性氨基酸衍生物的合成提供了一个有效方法.
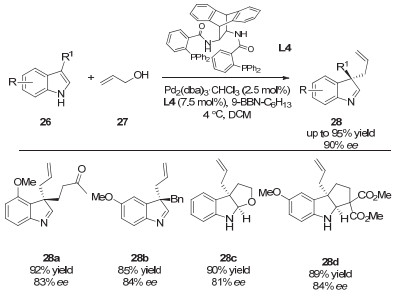
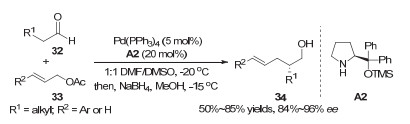
2012年, Córdova等[19]通过使用脯氨酸衍生的手性二级胺催化剂(A2)和非手性的Pd(PPh3)4实现了醛α位的不对称烯丙基化反应(图 9).该反应通过手性的烯胺中间体与非手性的π-烯丙基钯中间体作用, 实现了产物的对映选择性控制.
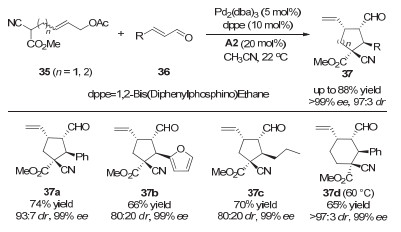
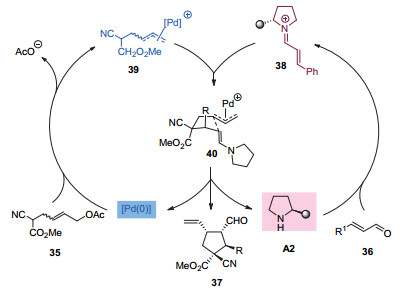
2013年, 该课题组用同一策略实现了分子间的环化反应, 构建了多取代的、包含一个手性季碳中心的环戊烷和环己烷产物(图 10)[20].该环化反应的机理如图 11所示:首先, 手性二级胺催化剂与α, β-不饱和醛36缩合形成亚胺正离子中间体38.之后, 该中间体与烯丙基底物35与钯催化剂生成的中间体39进行Michael加成并生成中间体40.最后, 该中间体经过分子内的亲核环化和水解过程给出目标环化产物37.同时, 手性二级胺催化剂和零价钯催化剂再生并参与下一个催化循环.该研究为发展基于烯丙基化过程的环化反应提供了一种新思路, 而关键在于过渡金属与有机小分子协同催化策略的巧妙应用.
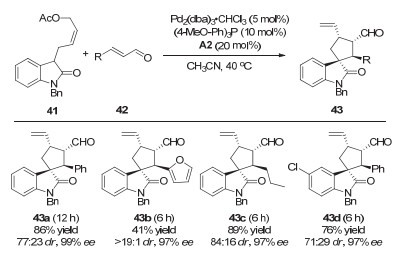
2015年, Córdova等[21]继续将协同的钯和二级胺催化策略应用到手性螺环氧化吲哚产物43的合成(图 12).该反应以2-氧化吲哚衍生的醋酸烯丙酯41和α, β-不饱和醛42作为底物, 以很好的产率、中等到较好的dr以及>99%的ee值得到目标产物.
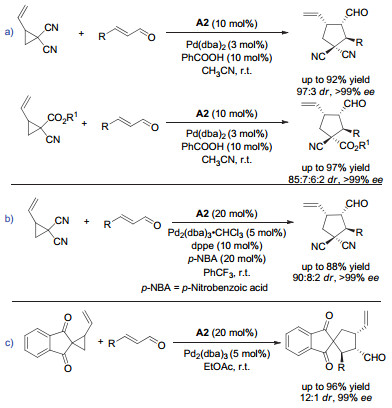
随后, Jrgensen等小组应用相似的策略, 从不同的底物出发, 通过形式上的不对称[3+2]环化反应构建了手性的环戊烷骨架(图 13)[22~24].这些反应极大地拓展了钯和手性胺协同催化策略的应用范围, 丰富了手性环戊烷产物的合成.
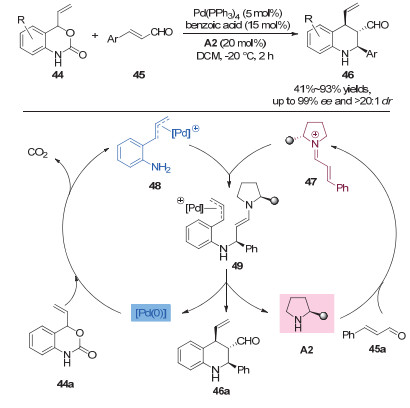
2016年, Jrgensen小组[25]应用协同的钯和二级胺催化, 发展不对称的形式上[4+2]环加成反应, 高效、高选择性地构建手性的四氢喹啉骨架(图 14).作者以烯基苯并噁嗪酮44和α, β-不饱和醛45为原料, 以Pd(PPh3)4和手性二级胺为催化剂, 最终以高达93%的产率以及>20:1的dr值、99%的ee值得到手性喹啉产物.同时, 作者提出了一个可能的反应机理:首先, 烯基苯并噁嗪酮44a在钯催化下脱去CO2形成π-烯丙基钯络合物49; 同时, 肉桂醛45a与手性胺催化剂A2反应生成共轭亚胺离子中间体47.随后, 两者通过分子间的氮杂Michael加成形成烯胺中间体49, 再通过分子内的烯丙基化和亚胺离子水解形成目标产物46a.同时, 钯催化剂和手性胺催化剂得以再生.
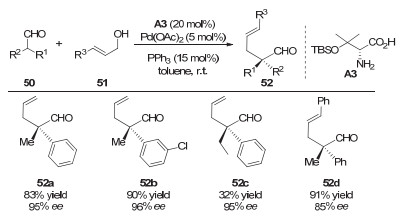
2014年, Yoshida等[26]将一级胺催化剂引入到了钯催化的不对称烯丙基化反应中, 进一步拓展了钯和手性胺协同催化策略的应用范围(图 15).作者通过结合手性的一级胺催化剂和非手性的钯催化剂, 实现了醛α位的不对称烯丙基化反应.该反应以烯丙醇为底物, 以氨基酸衍生物A3为手性伯胺催化剂, 最终以较高的收率和很好的对映选择性得到含有手性季碳中心的高烯丙基醛类产物.
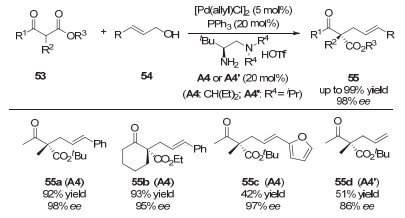
2015年, 罗三中研究团队通过使用自主研发的手性双胺催化剂, 实现了钯和手性胺协同催化β-酮酸酯55的不对称烯丙基化反应(图 16)[27].该反应同样为手性全碳季碳中心的不对称构建提供了一个温和而有效的方法.
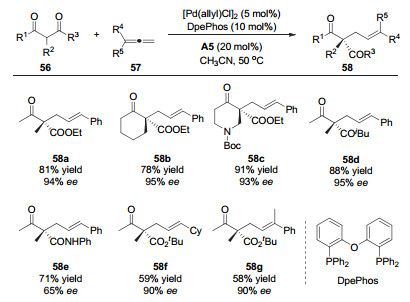
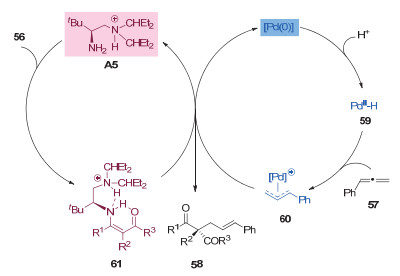
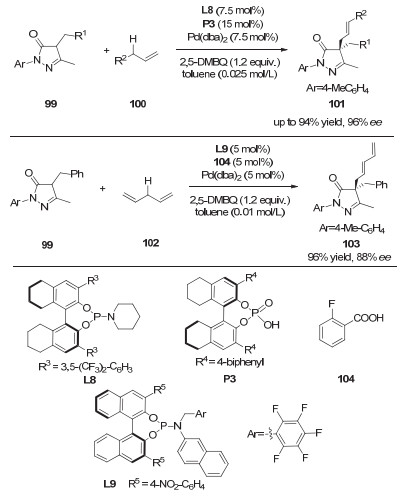
联烯作为一种更加原子经济性的烯丙基前体, 被广泛应用于有机反应中[28]. 2017年, 罗三中课题组[29]利用非手性的钯催化剂和手性的一级胺催化剂, 实现了β-酮酸酯的不对称烯丙基化反应, 得到了直链选择性的手性产物58(图 17).作者认为零价钯催化剂与酸作用生成钯氢物种59(图 18), 然后与联烯通过插入反应生成π-烯丙基钯中间体60.同时, 胺催化剂A5与β-羰基酯56缩合形成烯胺中间体61, 然后与中间体60发生烯丙基化反应得到直链选择性的产物58.
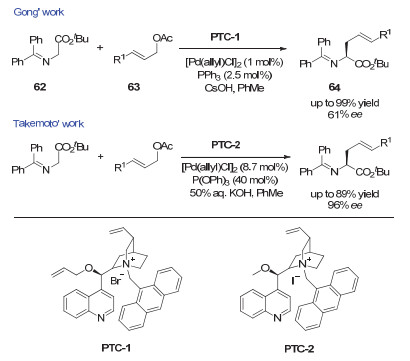
2001年, 龚流柱课题组[9a]和Takemoto课题组[9b]先后实现了手性相转移(PTC)催化剂和金属钯协同催化的席夫碱甘氨酸酯的不对称α-烯丙基化反应(图 19).
除了手性相转移催化剂和后来的手性胺催化剂外, 许多其它的手性质子酸催化剂、氢键催化剂和Lewis碱催化剂也被成功用于过渡金属和有机小分子协同催化的不对称烯丙基化反应.
在2004年, Terada和Akiyama等率先报道了手性联二萘酚(BINOL)衍生的手性磷酸型质子酸催化剂[30].这类催化剂可以通过氢键或酸碱对的模式提高底物的亲电性; 同时, 磷上的氧原子则可以作为Lewis碱来活化亲核试剂.因此, 手性磷酸催化剂可以表现出一定的双功能催化特性.最近, 手性磷酸催化剂和钯催化剂的联合使用, 大大扩展了手性磷酸催化的适用范围.
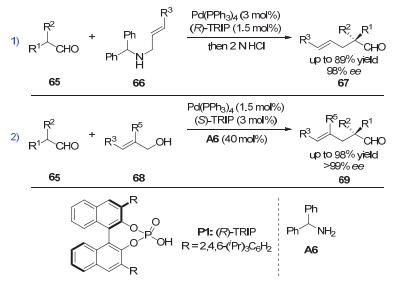
2007年, List等[31]首次通过手性磷酸和钯催化剂的协同催化, 实现了支链醛的α位烯丙基化反应, 成功构建了一个手性季碳中心(图 20, 式1).当底物醛上的两个取代基分别为烷基和芳基时, 对映选择性(ee值)可以高达97%, 但当底物换成双烷基取代的乙醛时, 只能得到70%的ee值. 2011年, 该课题组发现更加简单的烯丙醇也适用于手性磷酸和钯协同催化的不对称烯丙基化反应(图 20, 式2)[32].当反应以3 mol%的手性磷酸(S)-TRIP和1.5 mol%的Pd(PPh3)4作催化剂, 以很高的收率和对映选择性给出醛α位的不对称烯丙基化产物.
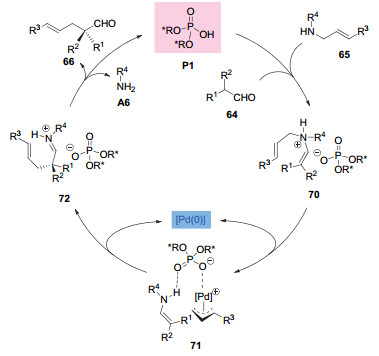
反应机理如图 21所示:首先, 支链醛和烯丙基胺在手性磷酸P1的催化作用下缩合产生磷酸盐中间体70, 之后金属钯与该中间体作用生成了含有π-烯丙基钯阳离子、烯胺以及手性磷酸阴离子的络合物71.烯胺与烯丙基钯的分子内烯丙基化以及后续的水解过程生成目标产物66并再生手性磷酸和非手性的钯催化剂.作者通过控制实验证明, 形成的手性离子对中间体70不能经过[3, 3]重排给出目标产物66.这说明了钯催化剂在反应中起到不可或缺的作用.手性磷酸不仅起到质子酸催化剂的作用, 而且其共轭碱还作为一种手性抗衡离子来控制烯丙基化过程的对映选择性.
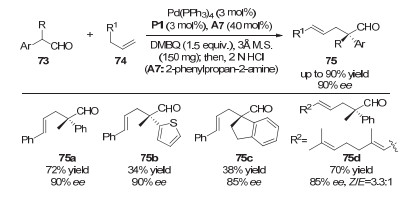
2014年, 龚流柱课题组通过使用催化量的手性磷酸和钯催化剂以及化学计量的醌类氧化剂DMBQ (2, 6-dimethylbenzoquinone), 率先实现了醛和端烯的不对称烯丙基化反应, 大大提高了反应的步骤经济性(图 22)[33].伯胺催化剂、手性磷酸催化剂、钯催化剂以及3 分子筛都对反应效率和立体选择性有着非常重要的影响, 且该催化体系广泛适用于α-位芳基和烷基取代的支链醛和端烯底物, 普遍给出中等到较好的收率以及很高的ee值.
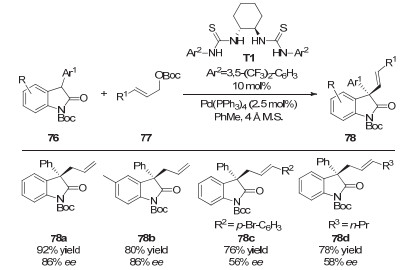
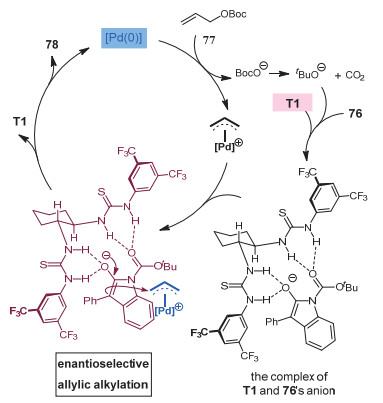
2016年, 陆良秋和肖文精等[34]通过协同的钯催化和氢键催化模式, 实现了3-芳基氧化吲哚的不对称烯丙基化反应, 成功在氧化吲哚的3位构建了一个手性季碳中心(图 23).环己二胺衍生的手性硫脲催化剂和非手性的Pd(PPh3)4可以给出最好的结果, 普遍以很好的收率和中等到较好的ee值得到目标产物.作者提出了一个可能的反应机理(图 24):底物77与零价钯催化剂作用生成π-烯丙基钯中间体, 同时释放出一分子的叔丁氧基阴离子和CO2.叔丁氧基阴离子作为碱拔掉氧化吲哚底物76的苄位质子, 形成的阴离子中间体被手性的氢键催化剂稳定.然后, 该物种在烯醇阴离子的Re面与烯丙基钯反应, 得到手性的目标产物.作者通过磷谱和氢谱实验证明了氢键催化剂并未和钯配位, 只是和76的阴离子通过氢键形成络合物, 从而控制产物的手性.
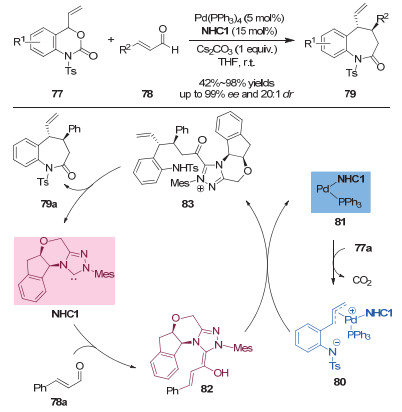
2016年, Glorius课题组[35]通过钯和手性氮杂卡宾协同催化策略, 实现了烯基苯并噁嗪酮和α, β-不饱和醛形式上的不对称[4+3]环加成产物(图 25, 上).随后, 该小组运用动力学实验对此反应的机理进行了研究[36].如图 25所示, 作者认为手性氮杂环卡宾在反应中具有双重作用, 既作为亲核性催化剂活化不饱和醛, 也作为配体与钯络合促使氢键导向的支链选择性烯丙基化反应.
在产生多个手性中心的不对称烯丙基化反应中, 如果将手性的有机催化剂和手性的过渡金属结合起来, 就有可能实现单一手性源难以实现的立体控制. 2013年, Carreira课题组[37]通过联合使用手性的铱催化剂和金鸡纳碱衍生的伯胺催化剂, 实现了支链醛的立体多样性烯丙基化反应.通过简单的改变配体以及伯胺催化剂的构型, 作者以较好的收率、完美的非对映选择性和对映选择性得到四个立体异构体(图 26).
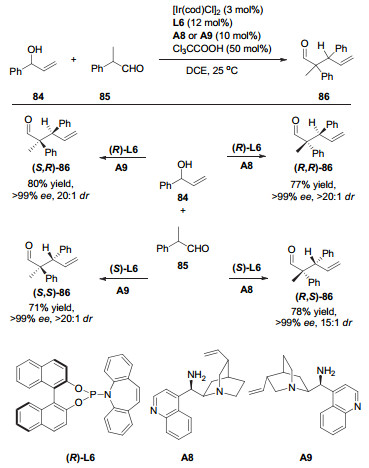
在此基础上, 他们通过改变手性胺催化剂, 成功地将该反应拓展到直链烷基醛.作者发现, 脯氨酸衍生的手性二级胺催化剂可以高效地活化直链烷基醛, 实现铱和有机胺协同催化的不对称烯丙基化反应.同样, 通过改变有机胺催化剂以及配体的立体构型, 反应以很好的对映以及非对映选择性得到4种立体产物(图 27)[38]. 2014年, 他们将此策略成功应用于天然产物Δ9-tetrahydrocannabinols所有异构体的不对称全合成[39].这种策略在2015年还被他们自己用于α-氨基乙醛和α-羟基乙醛的不对称烯丙基化反应[40], 成功得到了4种立体构型的产物.
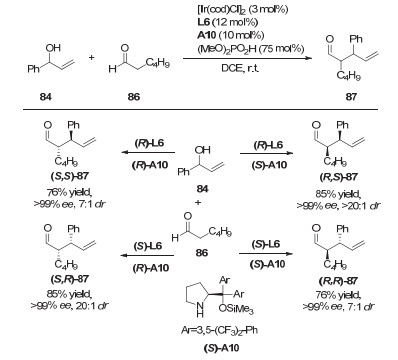
2017年, Hartwig等[41]利用手性铱络合物和手性Lewis碱协同催化的策略, 实现了2-芳基乙酸酯的不对称烯丙基化反应(图 28).当使用手性苯并四咪唑(BTM)作为Lewis碱和铱的亚磷酰胺络合物时, 反应可以得到高达>99%的ee, 以及>20:1的dr值.需要指出的是, 五氟苯基酯作为亲核试剂前体是该反应的关键, 其它芳香酯衍生物作为底物只能得到非常低的产率和立体选择性.而且, 通过改变BTM以及铱配合物的立体构型, 还可以得到所有立体构型的产物.
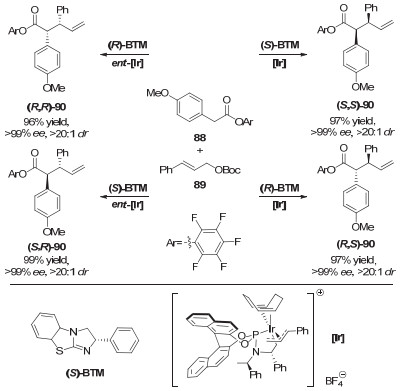
事实上, 2016年以来张万斌课题组[42]、王春江课题组[43]以及Hartwig课题组[44]通过协同的Lewis酸与手性铱络合物催化, 还先后实现了α-羟基苯乙酮、氨基酸衍生物和茚酮衍生物等化合物的不对称烯丙基化反应.通过改变两种手性源的手性, 同样可以给出4种立体构型.
2015年, J rgensen等[45]实现了α, β-不饱和醛γ位的不对称烯丙基化反应(图 29).在手性二级胺和手性亚磷酰胺-铱配合物的协同催化下, 以消旋的烯丙醇为烯丙基源, 反应以较好的对映和非对映选择性得到了支链型取代产物.随后, 作者将金属铱催化剂换成金属钯催化剂, 以烯丙基酯为烯丙基源以及使用非手性的配体和手性的二级胺催化剂可以以高对映选择性得到直链型取代产物.
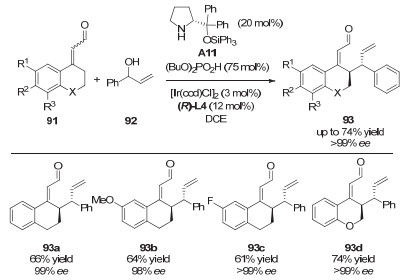
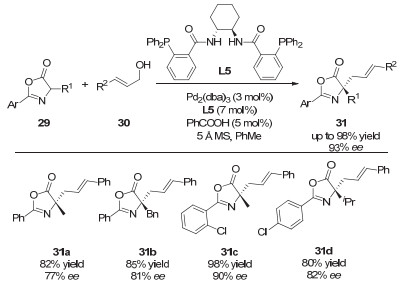
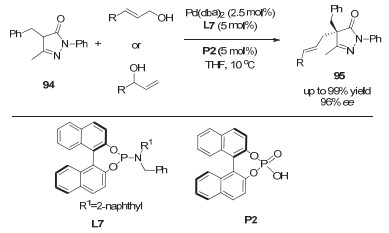
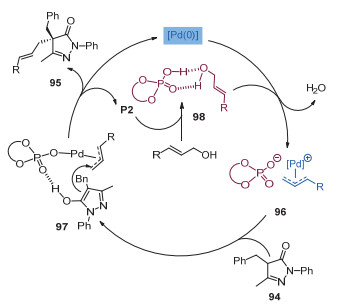
2013年, 龚流柱课题组[46]通过使用手性的钯催化剂和手性磷酸催化剂, 首次实现了5-吡唑啉酮的不对称烯丙基化反应, 得到了多取代的、含有季碳手性中心的吡唑啉酮衍生物(图 30).文中指出, 手性亚磷酰胺配体和手性磷酸的共同使用对反应对映选择性的提升具有非常重要的作用.如图 31所示, 手性磷酸一方面通过氢键作用生成络合物98, 辅助C—O键断裂来形成π-烯丙基钯配合物, 另一方面手性磷酸根作为抗衡阴离子, 与手性钯络合物之间的协同效应, 明显地提升了反应的ee值.
之后, 该小组利用类似的催化体系, 直接以端烯或1, 4-戊二烯为烯丙基源, 进一步完善了5-吡唑啉酮的不对称烯丙基化反应, 生成了具有手性季碳中心的5-吡唑啉酮衍生物(图 32)[47].
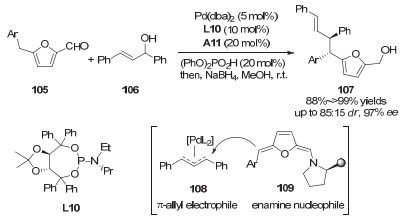
2017年, 龚流柱课题组[48]还实现了糠醛衍生物的不对称烯丙基化反应(图 33).通过联合使用手性二级胺催化剂和手性钯催化剂, 作者以很高的产率和对映选择性得到手性的呋喃衍生物107.文中指出, 糠醛衍生物不对称转化的难点在烯胺中间体反应位点与催化剂手性中心相距较远, 通过两种手性催化剂的组合很好地解决了这个问题.单独使用手性胺催化剂虽然可以得到较高的产率, 但反应的对映选择性和非对映选择性不甚理想, 在反应体系中加入手性亚磷酰胺配体之后, 目标产物的dr值和ee值显著提高.
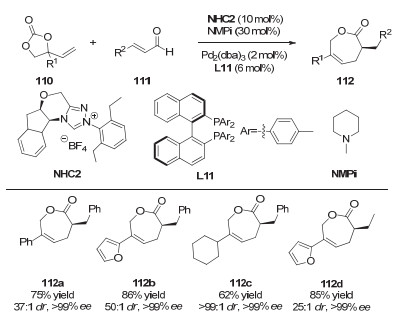
2018年, Glorius等[49]将手性氮杂环卡宾催化和不对称钯催化相结合, 发展了一例不对称的[5+2]环加成反应(图 34).该反应可以以高收率、高对映选择性给出手性ε-己内酯产物.值得一提的是, 与该小组之前的工作不同[35, 36], 手性氮杂环卡宾在此反应中不再作为金属钯的配体, 而仅仅是用于形成Breslow中间体的有机小分子催化剂.
综上所述, 近十几年来过渡金属和有机小分子协同催化的不对称烯丙基化反应取得了长足的发展, 已经成为传统单一过渡金属催化模式的一个有力补充.这种协同催化策略的应用不仅极大地拓宽了烯丙基化反应中亲核试剂的类型, 而且还实现了很多单一催化剂难以实现的反应以及立体选择性控制.
在今后的研究中, 这一领域仍有两个方面值得进一步研究: (1)原料简单化:尽管已有零星报道, 但如何使用简单易得的原料, 例如烯丙醇甚至烯烃作为烯丙基源, 实现步骤经济性、原子经济性的不对称烯丙基化仍是一个重要的研究内容; (2)催化体系绿色化:目前大多数不对称烯丙基化反应需要使用钯、铑、铱等贵金属催化剂, 如何通过协同催化策略来实现铁、钴、镍等第一过渡金属催化的不对称烯丙基化反应[50], 同样也是一项值得探索的研究内容.相信在不久的将来, 通过合成化学家们持续不断的努力, 一定会实现更加绿色、更加高效和选择性更高的不对称烯丙基化反应.
For selected reviews, see: (a) Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921. (b) Mohr, J. T.; Stoltz, B. M. Chem.-Asian J. 2007, 2, 1476. (c) Lu, Z.; Ma, S.-M. Angew. Chem., Int. Ed. 2008, 47, 258. (d) Butt, N. A.; Zhang, W. Chem. Soc. Rev. 2015, 44, 7929. (e) Butt, N. A.; Yang, G.; Zhang, W. Chem. Rec. 2016, 16, 2687. (f) Deng, Y.; Yang, W.; Yang, X.; Yang, D. Chin. J. Org. Chem. 2017, 37, 3039 (in Chinese). (邓颖颍, 杨文, 杨新, 杨定乔, 有机化学, 2017, 37, 3039.)
(a) Tsuji, J.; Takahashi, H.; Morikawa, M. Tetrahedron Lett. 1965, 6, 4387. (b) Tsuji, J. Acc. Chem. Res. 1969, 2, 144.
(a) Atkins, K. E.; Walker, W. E.; Manyik, R. M. Tetrahedron. Lett. 1970, 11, 3821. (b) Hata, G.; Takahashi, K.; Miyake, A. J. Chem. Soc., Chem. Commun. 1970, 1392.
Trost, B. M.; Strege, P. E. J. Am. Chem. Soc. 1977, 99, 1649. doi: 10.1021/ja00447a064
(a) Trost, B. M.; Machacek, M. R.; Aponick, A. Acc. Chem. Res. 2006, 39, 747. (b) Hartwig, J. F.; Stanley, L. M. Acc. Chem. Res. 2010, 43, 1461. (c) Zhuo, C.-X.; Zheng, C.; You, S.-L. Acc. Chem. Res. 2014, 47, 2558. (d) Qu, J.; Helmchen, G. Acc. Chem. Res. 2017, 50, 2539. (e) Yu, Y.-N.; Xu, M.-H. Acta Chim. Sinica 2017, 75, 655 (in Chinese). (于月娜, 徐明华, 化学学报, 2017, 75, 655.)
Weaver, J. D.; Recio, A.; Grenning, A. J.; Tunge, J. A. Chem. Rev. 2011, 111, 1846. doi: 10.1021/cr1002744
(a) Yan, X. X.; Liang, C. G.; Zhang, Y.; Hong, W.; Cao, B. X.; Dai, L. X.; Hou, X. L. Angew. Chem., Int. Ed. 2005, 44, 6544. (b) Zheng, W.-H.; Zheng, B.-H.; Zhang, Y.; Hou, X.-L. J. Am. Chem. Soc. 2007, 129, 771. (c) Zhang, K.; Peng, Q.; Hou, X.-L. Angew. Chem., Int. Ed. 2008, 47, 1741. (d) Liu, W.; Chen, D.; Zhu, X.-Z.; Wan, X.-L.; Hou, X.-L. J. Am. Chem. Soc. 2009, 131, 8734. (e) Lei, B.-L.; Ding, C.-H.; Yang, X.-F.; Wan, X.-L.; Hou, X.-L. J. Am. Chem. Soc. 2009, 131, 8734. (f) Li, X.-H.; Zheng, B.-H.; Ding, C.-H.; Hou, X.-L. Org. Lett. 2013, 15, 6086.
(a) Shao, Z.; Zhang, H. Chem. Soc. Rev. 2009, 38, 2745. (b) Zhong, C.; Shi, X. Eur. J. Org. Chem. 2010, 2999. (c) Allen, A. E.; MacMillan, D. W. C. Chem. Sci. 2012, 3, 633. (d) Du, Z.; Shao, Z. Chem. Soc. Rev. 2013, 42, 1337. (e) Deng, Y.; Kumar, S.; Wang, H. Chem. Commun. 2014, 50, 4272. (f) Inamdar, S. M.; Shinde, V. S.; Patil, N. T. Org. Biomol. Chem. 2015, 13, 8116. (g) Meazza, M.; Rios, R. Synthesis 2016, 48, 960. (h) Afewerki, S.; Córdova, A. Chem. Rev. 2016, 116, 13512. (i) Fu, J.; Huo, X.; Li, B.; Zhang, W. Org. Biomol. Chem. 2017, 15, 9747. (j) Sun, Z.; He, J.; Qu, M.; Li, K. Chin. J. Org. Chem. 2015, 35, 1250 (in Chinese). (孙哲, 何金梅, 屈孟男, 李侃社, 有机化学, 2015, 35, 1250.)
(a) Chen, G.; Deng, Y.; Gong, L.; Mi, A.; Cui, X.; Jiang, Y.; Choi, M. C. K.; Chan, A. S. C. Tetrahedron: Asymmetry 2001, 12, 1567. (b) Nakoji, M.; Kanayama, T.; Okino, T.; Takemoto, Y. Org. Lett. 2001, 3, 3329.
(a) Mukherjee, S.; Yang, J. W.; Hoffmann, S.; List, B. Chem. Rev. 2007, 107, 5471. (b) Chen, Y.-C. Synlett 2008, 13, 1919. (c) Xu, L.-W.; Lu, Y.-X. Org. Biomol. Chem. 2008, 6, 2047.
Ibrahem, I.; Córdova, A. Angew. Chem., Int. Ed. 2006, 45, 1952. doi: 10.1002/(ISSN)1521-3773
Bihelovic, F.; Matovic, R.; Vulovic, B.; Saicic, R. N. Org. Lett. 2007, 9, 5063. doi: 10.1021/ol7023554
Vulovic, B.; Bihelovic, F.; Matovic, R.; Saicic, R. N. Tetrahedron 2009, 65, 10485. doi: 10.1016/j.tet.2009.10.006
(a) Zhao, X.; Liu, D.; Guo, H.; Liu, Y.; Zhang, W. J. Am. Chem. Soc. 2011, 133, 19354. (b) Zhao, X.; Liu, D.; Xie, F.; Liu, Y.; Zhang, W. Org. Biomol. Chem. 2011, 9, 1871.
Huo, X.; Yang, G.; Liu, D.; Liu, Y.; Gridnev, I. D.; Zhang, W. Angew. Chem., Int. Ed. 2014, 53, 6776. doi: 10.1002/anie.201403410
Huo, X.; Quan, M.; Yang, G.; Zhao, X.; Liu, D.; Liu, Y.; Zhang, W. Org. Lett. 2014, 16, 1570. doi: 10.1021/ol5000988
Trost, B. M.; Quancard, J. J. Am. Chem. Soc. 2006, 128, 6314 doi: 10.1021/ja0608139
Zhou, H.; Yang, H.; Liu, M.; Xia, C.; Jiang, G. Org. Lett. 2014, 16, 5350. doi: 10.1021/ol502535z
Afewerki, S.; Ibrahem, I.; Rydfjord, J.; Breistein, P.; Córdova, A. Chem. Eur. J. 2012, 18, 2972. doi: 10.1002/chem.201103366
Ma, G.; Afewerki, S.; Deiana, L.; Palo-Nieto, C.; Liu, L.; Sun, J.; Ibrahem, I.; Córdova, A. Angew. Chem., Int. Ed. 2013, 52, 6050. doi: 10.1002/anie.201300559
Afewerki, S.; Ma, G.; Ibrahem, I.; Liu, L.; Sun, J.; Córdova, A. ACS Catal. 2015, 5, 1266. doi: 10.1021/cs501975u
Halskov, K. S.; N sborg, L.; Tur, F.; J rgensen, K. A. Org. Lett. 2016, 18, 2220. doi: 10.1021/acs.orglett.6b00852
Laugeois, M.; Ponra, S.; Ratovelomanana-Vidal, V.; Michelet, V.; Vitale, M. R. Chem. Commun. 2016, 52, 5332. doi: 10.1039/C6CC01775D
Meazza, M.; Rios, R. Chem. Eur. J. 2016, 22, 9923. doi: 10.1002/chem.201601893
Leth, L. A.; Glaus, F.; Meazza, M.; Fu, L.; Th gersen, M.-K.; Bitsch, E.-A.; J rgensen, K. A. Angew. Chem., Int. Ed. 2016, 55, 15272. doi: 10.1002/anie.v55.49
(a) Yoshida, M.; Terumine, T.; Masaki, E.; Hara, S. J. Org. Chem. 2013, 78, 10853. (b) Yoshida, M.; Masaki, E.; Terumine, T.; Hara, S. Synthesis 2014, 46, 1367.
(a) Zhou, H.; Zhang, L.; Xu, C.; Luo, S. Angew. Chem., Int. Ed. 2015, 54, 12645. (b) Li, B.; Liu, R.; Liang, R.; Jia, Y. Acta Chim. Sinica 2017, 75, 448 (in Chinese). (李保乐, 刘人荣, 梁仁校, 贾义霞, 化学学报, 2017, 75, 448.) (c) Li, J.; Tan, C.; Mu, X.; Gong, J.; Yang, Z. Chin. J. Chem. 2017, 35, 562.
(a) Muzart, J.; Le Bras, J. Chem. Soc. Rev. 2014, 43, 3003. (b) Koschker, P.; Breit, B. Acc. Chem. Res. 2016, 49, 1524. (c) Zimmer, R.; Dinesh, C. U.; Nandanan, E.; Khan, F. A. Chem. Rev. 2000, 100, 3067.
Zhou, H.; Wang, Y.; Zhang, L.; Cai, M.; Luo, S. J. Am. Chem. Soc. 2017, 139, 3631. doi: 10.1021/jacs.7b00437
(a) Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Angew. Chem., Int. Ed. 2004, 43, 1566. (b) Uraguchi, D.; Terada, M. J. Am. Chem. Soc. 2004, 126, 5356. (c) Wu, X.; Li, M.; Gong, L. Acta Chim. Sinica 2013, 71, 1091 (in Chinese). (吴祥, 李明丽, 龚流柱, 化学学报, 2013, 71, 1091.)
Mukherjee, S.; List, B. J. Am. Chem. Soc. 2007, 129, 11336. doi: 10.1021/ja074678r
Jiang, G.; List, B. Angew. Chem., Int. Ed. 2011, 50, 9471. doi: 10.1002/anie.v50.40
(a) Wang, P.-S.; Lin, H.-C.; Zhai, Y.-J.; Han, Z.-Y.; Gong, L.-Z. Angew. Chem., Int. Ed. 2014, 53, 12218. (b) Zhang, Z.-J.; Tao, Z, -L.; Arafate, A.; Gong, L.-Z. Acta Chim. Sinica 2017, 75, 1196 (in Chinese). (张子競, 陶忠林, 阿拉法特·阿地力, 龚流柱, 化学学报, 2017, 75, 1196.) (c) Tang, H.; Huo, X.; Meng, Q.; Zhang, W. Acta Chim. Sinica 2016, 74, 219 (in Chinese). (汤淏溟, 霍小红, 孟庆华, 张万斌, 化学学报, 2016, 74, 219.)
Boucherif, A.; Duan S.-W.; Yuan, Z.-G.; Lu, L.-Q.; Xiao, W.-J. Adv. Synth. Catal. 2016, 358, 2594. doi: 10.1002/adsc.v358.16
Guo, C.; Fleige, M.; Janssen-Müller, D.; Daniliuc, C. G.; Glorius, F. J. Am. Chem. Soc. 2016, 138, 7840. doi: 10.1021/jacs.6b04364
Guo, C.; Janssen-Müller, D.; Fleige, M.; Lerchen, A.; Daniliuc, C. G.; Glorius, F. J. Am. Chem. Soc. 2017, 139, 4443. doi: 10.1021/jacs.7b00462
Krautwald, S.; Sarlah, D.; Schafroth, M. A.; Carreira, E. M. Science 2013, 340, 1065. doi: 10.1126/science.1237068
Krautwald, S.; Schafroth, M. A.; Sarlah, D.; Carreira, E. M. J. Am. Chem. Soc. 2014, 136, 3020. doi: 10.1021/ja5003247
Schafroth, M. A.; Zuccarello, G.; Krautwald, S.; Sarlah, D.; Carreira, E. M. Angew. Chem., Int. Ed. 2014, 53, 13898. doi: 10.1002/anie.201408380
Sandmeier, T.; Krautwald, S.; Zipfel, H. F.; Carreira, E. M. Angew. Chem., Int. Ed. 2015, 54, 14363. doi: 10.1002/anie.201506933
Jiang, X.-Y.; Beiger, J. J.; Hartwig, J. F. J. Am. Chem. Soc. 2017, 139, 87. doi: 10.1021/jacs.6b11692
(a) Huo, X.; He, R.; Zhang, X.; Zhang, W. J. Am. Chem. Soc. 2016, 138, 11093. (b) Huo, X.; He, R.; Fu, J.; Zhang, J.; Yang, G.; Zhang, W. J. Am. Chem. Soc. 2017, 139, 9819. (c) He, R.; Liu, P.; Huo, X.; Zhang, W. Org. Lett. 2017, 19, 5513. (d) Huo, X.; Zhang, J.; Fu, J.; He, R.; Zhang, W. J. Am. Chem. Soc. 2018, 140, 2080. (e) Huo, X.; Fu, J.; He, X.; Chen, J.; Xie, F.; Zhang, W. Chem. Commun. 2018, 54, 599.
Wei, L.; Zhu, Q.; Xu, S.-M.; Chang, X.; Wang, C.-J. J. Am. Chem. Soc. 2018, 140, 1508. doi: 10.1021/jacs.7b12174
Jiang, X.; Boehm, P.; Hartwig, J. F. J. Am. Chem. Soc. 2018, 140, 1239. doi: 10.1021/jacs.7b12824
Næsborg, L.; Halskov, K. S.; Tur, F.; Mønsted, S. M. N.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2015, 54, 10193. doi: 10.1002/anie.201504749
Tao, Z.-L.; Zhang, W.-Q.; Chen, D.-F.; Adele, A.; Gong, L.-Z. J. Am. Chem. Soc. 2013, 135, 9255. doi: 10.1021/ja402740q
Lin, H.-C.; Wang, P.-S.; Tao, Z.-L.; Chen, Y.-G.; Han, Z.-Y.; Gong, L.-Z. J. Am. Chem. Soc. 2016, 138, 14354. doi: 10.1021/jacs.6b08236
Su, Y.-L.; Han, Z.-Y.; Li, Y.-H.; Gong, L.-Z. ACS Catal. 2017, 7, 7917. doi: 10.1021/acscatal.7b02667
Singha, S.; Patra, T.; Daniliuc, C. G.; Glorius, F. J. Am. Chem. Soc. 2018, 140, 3551. doi: 10.1021/jacs.8b00868
Cong, X.; Zhai, S.; Zeng, X. Org. Chem. Front. 2016, 3, 673. doi: 10.1039/C6QO00011H

图 2 协同的钯/吡咯烷催化:醛、酮α位的分子间烯丙基化
Figure 2 Synergetic Pd/pyrrolidine catalysis: intermolecular α-allylations of aldehydes and ketones

图 3 协同的钯/二级胺催化:醛α位的分子内烯丙基化反应
Figure 3 Synergetic Pd/secondary amine catalysis: intramolecular allylation reactions of aldehydes

图 4 钯和吡咯烷协同催化酮α位直接烯丙基化的机理
Figure 4 A proposed mechanism of direct α-allylations of ketones via synergetic Pd/pyrrolidine catalysis

图 5 协同的钯/吡咯烷催化:醛酮与烯丙基胺的烯丙基化反应
Figure 5 Synergetic Pd/pyrrolidine catalysis: allylation reactions of aldehydes and ketones with allylic amines

图 6 协同的钯/吡咯烷催化:醛酮与烯丙醇的烯丙基化反应
Figure 6 Synergetic Pd/pyrrolidine catalysis: allylation reactions of aldehydes and ketones with allylic alcohols

图 7 钯催化:吲哚3位的不对称烯丙基化反应
Figure 7 Palladium-catalyzed enantioselective C-3 allylation of indoles

图 8 协同的钯/酸催化:吖内脂与烯丙醇的烯丙基化反应
Figure 8 Synergetic Pd/acid catalysis: allylation reactions of azlactone with allylic alcohol

图 9 协同的钯/二级胺催化:醛α位对映选择性的烯丙基化反应
Figure 9 Synergetic Pd/secondary amine catalysis: enantioselective α-allylic alkylations of aldehydes

图 10 协同的钯/二级胺催化:不对称的分子间环化反应
Figure 10 Synergetic Pd/secondary amine catalysis: asymmetric intermolecular cyclizations

图 11 钯和二级胺协同催化分子间不对称环化反应的机理
Figure 11 A proposed mechanism of asymmetric cyclizations through synergetic Pd/secondary amine catalysis

图 12 协同的钯/二级胺催化:合成多取代的手性螺环氧化吲哚
Figure 12 Synergetic Pd/secondary amine catalysis: asymmetric construction of highly substituted spirocyclic oxindoles

图 13 协同的钯/二级胺催化:其它不对称的[3+2]环加成
Figure 13 Synergetic Pd/secondary amine catalysis: other asymmetric [3+2] cycloadditions

图 14 协同的钯/二级胺催化:烯基苯并噁嗪酮的不对称脱羧[4+2]环加成反应
Figure 14 Synergetic Pd/secondary amine catalysis: asymmetric decarboxylative [4+2] cycloadditions of vinyl benzoxazinones

图 15 协同的钯/一级胺催化:支链醛的不对称烯丙基化
Figure 15 Synergetic Pd/primary amine catalysis: asymmetric allylic alkylation of branched aldehydes

图 16 协同的钯/双胺催化剂催化: β-酮酸酯的不对称烯丙基化
Figure 16 Synergetic Pd/diamine catalysis: enantioselective allylic alkylation of β-carbonyl esters

图 17 协同的钯/双胺催化: β-酮酸酯和联烯的不对称烯丙基化反应
Figure 17 Synergetic Pd/diamine catalysis: enantioselective allylations of β-carbonyl esters with allenes

图 18 钯和一级胺协同催化β-酮酸酯和联烯不对称烯丙基化反应机理
Figure 18 A proposed mechanism of asymmetric allylic alkylations of β-carbonyl esters with allenes through synergetic Pd/primary amine catalysis

图 19 协同的钯/相转移催化:席夫碱甘氨酸酯不对称烯丙基化反应
Figure 19 Synergetic Pd/Phase-transfer catalysis: enantioselective allylations of t-butyl(diphenylmethylene)-glycinate

图 20 协同的钯/磷酸催化:支链醛的不对称α-烯丙基化
Figure 20 Synergetic Pd/phosphoric acid catalysis: asymmetric α-allylations of branched aldehydes

图 21 钯和磷酸协同催化支链醛的不对称α-烯丙基化机理
Figure 21 A proposed mechanism of asymmetric α-allylations of branched aldehydes through synergetic Pd/phosphoric acid catalysis

图 22 协同的钯/磷酸/伯胺催化:支链醛和端烯的氧化不对称烯丙基化
Figure 22 Synergetic Pd/phosphoric acid/primary amine catalysis: asymmetric oxidative allylations of branched aldehydes with terminal alkenes

图 23 协同的钯/硫脲催化: 3-芳基氧化吲哚的不对称烯丙基化
Figure 23 Synergetic Pd/thiourea catalysis: asymmetric allylations of 3-aryl oxindoles

图 24 钯/硫脲协同催化3-芳基氧化吲哚的不对称烯丙基化
Figure 24 A proposed mechanism of asymmetric allylations of 3-aryl oxindoles through synergetic Pd/thiourea catalysis

图 25 协同的钯/氮杂卡宾催化:不对称的[4+3]环加成及其反应机理
Figure 25 Synergetic Pd/N-heterocyclic carbene catalysis: asymmetric [4+3] cycloadditions and a proposed mechanism

图 26 协同的铱/一级胺催化:支链醛的不对称α-烯丙基化
Figure 26 Synergetic Ir/primary amine catalysis: asymmetric α-allylations of branched aldehydes

图 27 协同的铱/二级胺催化:直链醛的不对称α-烯丙基化
Figure 27 Synergetic Ir/secondary amine catalysis: asymmetric α-allylations of linear aldehydes

图 28 协同的铱/Lewis碱催化:芳基乙酸酯的不对称烯丙基化
Figure 28 Synergetic Ir/Lewis base catalysis: enantioselective allylations of aryl acetic acid esters

图 29 协同的铱/二级胺催化: α, β-不饱和醛的γ位不对称烯丙基化
Figure 29 Synergetic Ir/secondary amine catalysis: enantioselective γ-allylations of α, β-unsaturated aldehydes

图 30 协同的钯/磷酸催化: 5-吡唑啉酮与烯丙醇的不对称烯丙基化
Figure 30 Synergetic Pd/phosphoric acid catalysis: enantioselective allylations of pyrazol-5-ones with allylic alcohols

图 31 钯和磷酸协同催化5-吡唑啉酮不对称烯丙基化反应的机理
Figure 31 A proposed mechanism of enantioselective allylations of pyrazol-5-ones via synergetic asymmetric Pd/phosphoric acid catalysis

图 32 协同的钯/质子酸催化: 5-吡唑啉酮与端烯或1, 4-戊二烯的不对称烯丙基化
Figure 32 Synergetic Pd/protonic acid catalysis: enantioselective allylations of pyrazol-5-ones with terminal alkenes or 1, 4-pentadiene

图 33 协同的钯/二级胺催化:糠醛衍生物的不对称烯丙基化
Figure 33 Synergetic Pd/secondary amine catalysis: enantioselective allylations of furfural derivatives

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
