Vacuum Metallurgical National Engineering Laboratory, Kunming University of Science and Technology, Kunming 650093
b.
Yunnan Key Laboratory of Nonferrous Metals Vacuum Metallurgy, Kunming University of Science and Technology, Kunming 650093
c.
State Key Laboratory of Complex Nonferrous Metal Resources Clear Utilization in Yunnan Province, Kunming University of Science and Technology, Kunming 650093
Received Date:
30 March 2018 Available Online:
15 July 2018
Fund Project:
Project supported by the the National Natural Science Foundation of China (Nos. 51704136, 11765010), the Yunnan applied basic research project of China (No. 2016FB087) and the Yunnan Academy of Liberal Exploration Funds of China (No. 2017HA006)
Abstract:
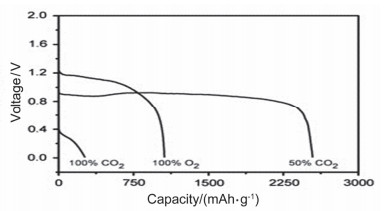
Due to the heavy use of fossil fuels, the emission of carbon dioxide has been steadily increased and the climate has been deteriorated severely. In order to solve these problems, various physical and chemical methods were used to reduce the amount of carbon dioxide in the atmosphere, but the result is not so effective. Metal carbon dioxide batteries not only can capture carbon dioxide, but also can be used as clean energy storage devices. At the same time, the development and research of metal carbon dioxide batteries also promote the development of the electric vehicle industry towards a more economical, environmentally friendly and sustainable direction. Based on these advantages, metal carbon dioxide battery has developed rapidly in recent years. Li-CO2 batteries exhibit an extremely high discharge capacity of 17625 mAh/g and a cut-off capacity of 1000 mAh/g at a current density of 100 mA/g, running for 100 cycles at low overpotentials. Quasi-solid state Na-CO2 batteries are non-flammable and have strong electrolyte-locking ability. It can run 400 cycles at 500 mA/g with a fixed capacity of 1000 mAh/g in pure CO2. Its electrochemical performance has the potential to be further improved. Al-CO2 battery has good application prospects and economic benefits due to the low cost of Al as well as great economic value of the sodium aluminate as discharge product. Mg-CO2 battery shows a discharge voltage plateau of 0.9 V when the volume ratio of CO2/O2 is 1:1, which is higher than that of pure O2. This paper mainly introduces the research progress of metal (lithium, sodium, aluminum, and magnesium) carbon dioxide battery, and compares the electrochemical performance of metal (lithium, sodium) carbon dioxide battery with metal (lithium, sodium) oxygen battery, puts forward the current problems of metal carbon dioxide batteries, and provides the solutions. Finally, the future development of metal carbon dioxide batteries is reviewed.
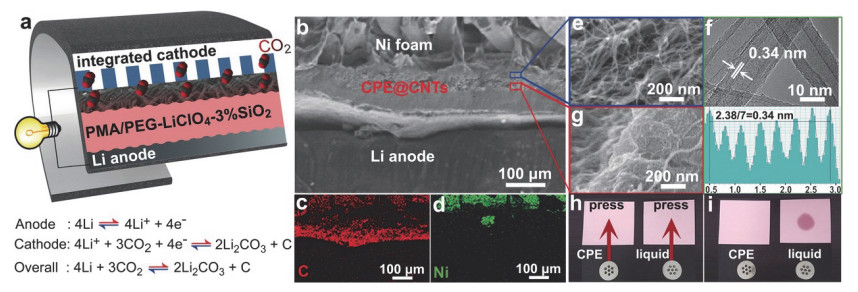
Figure 2.
The structure of all-solid-state Li-CO2 batteries. (a) Schematic diagram of Li-CO2batteries with metal Li foil anode and incorporating a CPE@CNTs cathode. (b) SEM image of the Li/CPE@CNTs. (c, d) EDX mapping of the integration structure in (b). (e) SEM image and (f) HRTEM image of CNTs with lattice distance of 0.34 nm. (g) SEM image of the CPE@CNTs interface. (h, i) Leakage test. Li-CO2 batteries with CPE and liquid electrolyte (1 mol/L LiClO4/TEGDME solution). (h) Before and (i) after pressing on a piece of dry paper[30]
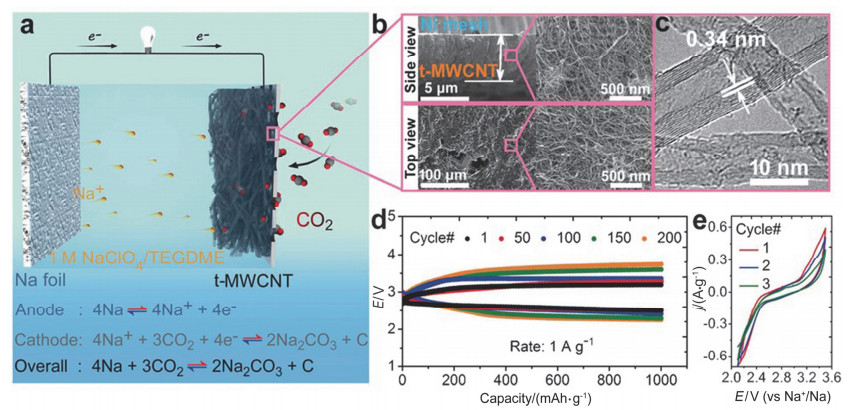
Figure 4.
The structure and recharge ability of room-temperature Na-CO2 batteries. (a) Structure of Na-CO2 batteries with metal Na foil anode, ether-based electrolyte, and t-MWCNT cathode. (b) SEM images of cathode from top and side views. (c) HRTEM image of t-MWCNT. (d) Discharge and charge profiles of Na-CO2 batteries at 1 A/g. (e) CV curves of Na–CO2 batteries with scan rate of 0.1 mV/s[21]
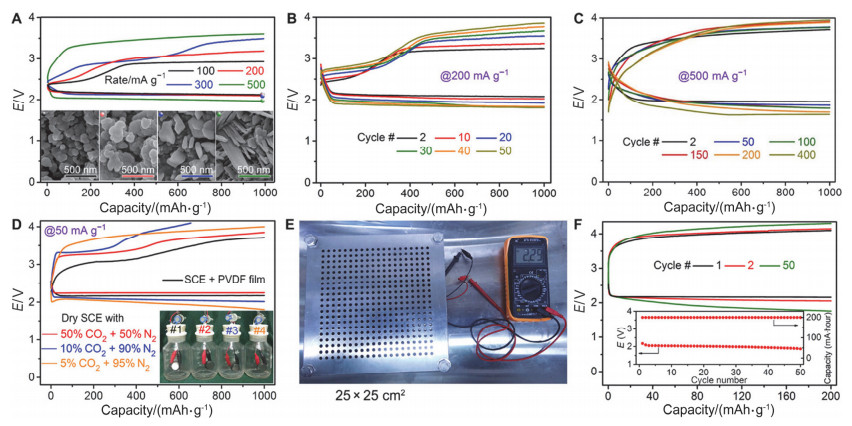
Figure 5.
The performances of quasi-solid state Na-CO2 batteries with rGO-Na anodes. (A) Rate capability. Insets: SEM images of discharged products at different rates. The black, red, blue, and green scale bars correspond to rates of 100, 200, 300, and 500 mA/g, respectively. (B) Discharge/charge profiles and (C) corresponding variation of the terminal discharge voltage with a cutoff capacity of 1000 mAh/g at 200 mA/g. (D) Initial discharge/charge profiles in SCE with different CO2 partial pressure. Rate, 50 mA/g. Four bottles use different test conditions. #1, SCE atmosphere with PVDF film protection; #2, dry SCE with 50%CO2 and 50% N2; #3, dry SCE with 10% CO2 and 90% N2; #4, dry SCE with 5% CO2 and 95%N2. (E) A photograph of pouch-type battery (20 cm×20 cm, 10 g) packed in a plastic bag with two stainless steel plates (25 cm×25 cm) to fix. (F) Discharge/charge profiles at 10 mA with a reversible capacity of 200 mAh. Inset: Corresponding variation of the middle discharge voltage [15]
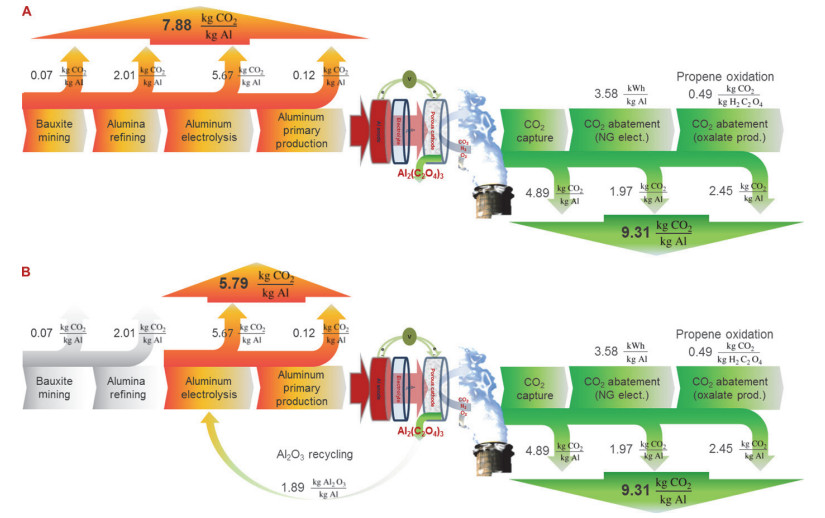
Figure 6.
Preliminary system analysis. (A) Overall balance of CO2 emissions captured/abated by the primary Al/80% CO2 electrochemical system contrasted with emissions of aluminum metal production. (B) Overall balance of CO2 emissions, allowing the recycling of Al2O3 for production of aluminum metal [16]
Friedlingstein, P.; Houghton, R. A.; Marland, G.; Hackler, J.; Boden, T. A.; Conway, T. J.; Canadell, J. G.; Raupach, M. R.; Ciais, P.; Quere, C. L. Nature Geosci. 2010, 3, 811. doi: 10.1038/ngeo1022
Gao, S.; Lin, Y.; Jiao, X. C.; Sun, Y. F.; Luo, Q. Q.; Zhang, W. H.; Li, D. Q.; Yang, J. L.; Xie, Y. Nature 2016, 529, 68. doi: 10.1038/nature16455
[6]
Rosen, B. A.; Salehi-Khojin, A.; Thorson, M. R.; Zhu, W.; Whipple, D. T.; Paul, J. A. Science 2011, 334, 643. doi: 10.1126/science.1209786
[7]
Zhang, S.; Kang P, Ubnoske, S.; Brennaman, M. K.; Song, N.; House, R. L.; Glass, J. T.; Meyer, T. J. J. Am. Chem. Soc. 2014, 136, 7845. doi: 10.1021/ja5031529
Zhang, Z.; Wang, X. G.; Zhang, X.; Xie, Z. J.; Chen, Y. N.; Ma, L. P.; Peng, Z. Q.; Zhou, Z. Adv. Sci. 2018, 5, 2198. doi: 10.1007/BF03246155
[14]
Yang, S. X.; Qiao, Y.; He, P.; Liu, Y. J.; Cheng, Z.; Zhu, J. J.; Zhou, H. S. Energy Environ. Sci. 2017, 10, 972. doi: 10.1039/C6EE03770D
[15]
Hu, X. F.; Li, Z. F.; Zhao, Y. R.; Sun, J. C.; Zhao, Q.; Wang, J. B.; Tao, Z. L.; Chen, J. Sci. Adv. 2017, 3, e1602396. http://www.oalib.com/references/15695966
McCloskey, B. D.; Valery, A, ; Luntz, A. C.; Gowda, S. R.; Wallraff, G. M.; Garcia, J. M.; Mori, T.; Krupp, L. E.; J. M. J. Phys. Chem. Lett. 2013, 4, 2989. doi: 10.1021/jz401659f
[35]
Lim, H. D.; Song, H.; Kim, J.; Gwon, H.; Bae, Y.; Park, K. Y.; Hong, J.; Kim, H.; Kim, T.; Kim, Y. H.; Lepro, X.; Robles, R. O.; Baughman, R. H.; Kang, K. Angew. Chem., Int. Ed. 2014, 53, 3926. doi: 10.1002/anie.201400711
[36]
Wang, R.; Yu, X.; Bai, J.; Li, H.; Huang, X. J.; Chen, L. Q.; Yang, X. Q. J. Power Sources 2012, 218, 113. doi: 10.1016/j.jpowsour.2012.06.082
[37]
Yin, W.; Grimaud, A.; Lepoivre, F.; Yang, C. Z.; Tarascon, J. M. J. Phys. Chem. Lett. 2016, 8, 214.
[38]
Xie, Z. J.; Zhang, X.; Zhang, Z.; Zhou, Z. Adv. Mater. 2017, 29, 1605891. doi: 10.1002/adma.201605891
Lim, H. K.; Lim, H. D.; Park, K. Y.; Seo, D. H.; Gwon, H.; Hong, J.; Goddard, W. A.; Kim, H.; Kang, K. J. Am. Chem. Soc. 2013, 135, 9733. doi: 10.1021/ja4016765
[42]
Fang, C.; Luo, J. M.; Jin, C. B.; Yuan, H. D.; Sheng, O. W.; Huang, H.; Gan, Y. P.; Xia, Y.; Liang, C.; Zhang, J.; Zhang, W. K.; Tao, X. Y. ACS Appl. Mater. Interfaces 2018, DOI: 10.1021/acsami.8b04034.
[43]
Xu, S. M.; Lu, Y. Y.; Wang, H. S.; Abruna, H. D.; Archer, L. A. J. Mater. Chem. A 2014, 2, 17723. doi: 10.1039/C4TA04130E
[44]
Gough, L. P.; Day, W. C. US Geological Survey 2010.
Figure 2
The structure of all-solid-state Li-CO2 batteries. (a) Schematic diagram of Li-CO2batteries with metal Li foil anode and incorporating a CPE@CNTs cathode. (b) SEM image of the Li/CPE@CNTs. (c, d) EDX mapping of the integration structure in (b). (e) SEM image and (f) HRTEM image of CNTs with lattice distance of 0.34 nm. (g) SEM image of the CPE@CNTs interface. (h, i) Leakage test. Li-CO2 batteries with CPE and liquid electrolyte (1 mol/L LiClO4/TEGDME solution). (h) Before and (i) after pressing on a piece of dry paper[30]
Figure 4
The structure and recharge ability of room-temperature Na-CO2 batteries. (a) Structure of Na-CO2 batteries with metal Na foil anode, ether-based electrolyte, and t-MWCNT cathode. (b) SEM images of cathode from top and side views. (c) HRTEM image of t-MWCNT. (d) Discharge and charge profiles of Na-CO2 batteries at 1 A/g. (e) CV curves of Na–CO2 batteries with scan rate of 0.1 mV/s[21]
Figure 5
The performances of quasi-solid state Na-CO2 batteries with rGO-Na anodes. (A) Rate capability. Insets: SEM images of discharged products at different rates. The black, red, blue, and green scale bars correspond to rates of 100, 200, 300, and 500 mA/g, respectively. (B) Discharge/charge profiles and (C) corresponding variation of the terminal discharge voltage with a cutoff capacity of 1000 mAh/g at 200 mA/g. (D) Initial discharge/charge profiles in SCE with different CO2 partial pressure. Rate, 50 mA/g. Four bottles use different test conditions. #1, SCE atmosphere with PVDF film protection; #2, dry SCE with 50%CO2 and 50% N2; #3, dry SCE with 10% CO2 and 90% N2; #4, dry SCE with 5% CO2 and 95%N2. (E) A photograph of pouch-type battery (20 cm×20 cm, 10 g) packed in a plastic bag with two stainless steel plates (25 cm×25 cm) to fix. (F) Discharge/charge profiles at 10 mA with a reversible capacity of 200 mAh. Inset: Corresponding variation of the middle discharge voltage [15]
Figure 6
Preliminary system analysis. (A) Overall balance of CO2 emissions captured/abated by the primary Al/80% CO2 electrochemical system contrasted with emissions of aluminum metal production. (B) Overall balance of CO2 emissions, allowing the recycling of Al2O3 for production of aluminum metal [16]

 下载:
下载:

 下载:
下载:








