
图 1.
酰胺的两种共振极限式
Figure 1.
Two resonance structures of an amide

有机化学教科书是以官能团为主线书写的.因此, 从某种意义上说, 有机化学是官能团的化学.酰胺官能团(N-单酰基胺)(1)广泛存在于自然界[1], 例如, α-氨基酸通过酰胺键连接而成蛋白质, 构成生命的基石.由于p-π共轭, 酰胺的C—N键具有部分双键性质(图 1), 是一类高度稳定的化合物.
酰胺在有机合成中被广泛用作胺的保护形式[2], 酰胺基是C—H键官能化的重要导向基[3].酰胺扮演的这些角色都是中间体, 因而需要后续反应把酰胺转化为目标化合物.此外, 由于酰胺处于高氧化态, 可转化为多种低氧化态化合物, 这既使之成为有机合成中多用途的中间体, 也对反应控制, 特别是反应的化学选择性提出挑战.因此, 尽管酰胺是人类最早认识和研究的官能团之一, 且酰胺的转化在有机和药物合成中有诸多需求, 然而由于酰胺是羧酸衍生物中羰基亲电性最低的化合物, 其直接转化需要剧烈的反应条件, 难以达到现代有机合成对化学选择性的要求.因而对于普通酰胺需求广泛的转化(例如, 酰胺还原烷基化和aza-Knoevenagel反应)需要采用多步骤过程, 这从以下几个代表性例子可以看出.
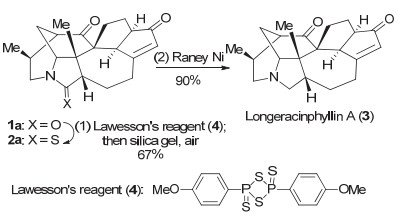
酰胺还原为胺[4]传统上用四氢铝锂或硼烷还原[5].由于四氢铝锂是强还原剂, 不但试剂本身具有危险性, 由于反应条件强烈, 还原反应往往缺乏化学选择性.而硼烷尽管温和, 有时可达到好的化学选择性, 但用作还原剂的甲硼烷和乙硼烷在常温下为气体, 需用价格不菲的硼烷-四氢呋喃或硼烷-二甲硫醚络合物, 且硼烷易与产物形成胺络合物, 增加了分离纯化的难度.因而二步法遂成为把酰胺还原为胺的常用方法.代表性的二步法首先把酰胺(如1a)转化为硫代酰胺(如2a), 然后用兰尼镍还原.由于该法表现出优良的化学选择性, 被广泛应用于生物碱的全合成.一个最新应用见诸李昂课题组[6]报道的复杂生物碱longeracinphyllin A (3)的全合成.其最后步骤为在酮与α, β-不饱和酮存在下, 对化合物1a进行酰胺的化学选择性还原(图式 1).
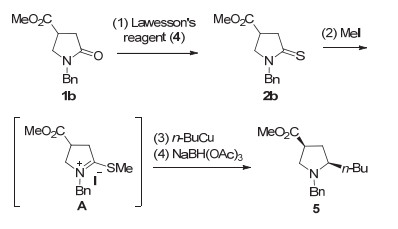
酰胺的还原烷基化是另一具有广泛需求的转化.传统的直接还原烷基化方法缺乏普适性(见下, 图式 3), 因而, 通常采用多步骤方法.常用的两种多步骤还原烷基化方法示于图式 2和图式 3.方法一先将酰胺1b转化为硫代酰胺2b, 然后进行S-甲基化, 接着有机金属试剂对中间体A 加成, 进一步用三醋酸硼氢化钠还原可得到烷基化产物5 (图式 2)[7].
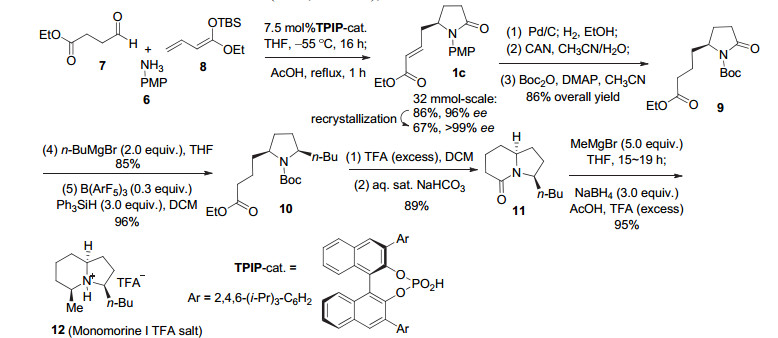
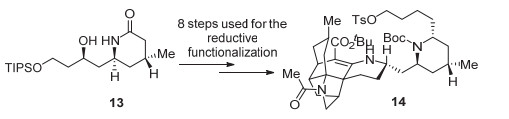
方法二以Schneider课题组[8]近期有关吲哚里西啶生物碱monomorine I (12)的合成展示(图式 3, 1c向10转化中的步骤2~5).该法需要首先进行N-去保护, 然后与Boc2O反应形成酰亚胺类化合物9 (N, N-二酰基胺类化合物, 此时原酰胺羰基的反应性与酮羰基类似), 再经两步完成还原烷基化得10. Schneider等的这一工作表明, 尽管通过有机催化可以极高的对映选择性(99% ee)从简单原料一步构建内酰胺1c, 且后续的吲哚里西啶酮11的还原烷基化可一步完成, 但γ-内酰胺1c的还原烷基化却需要四步(图式 3, 1c向10转化中的步骤2~5), 形成N-Boc保护的产物10.另一个典型实例见诸哈佛大学Shair小组[9]石松类生物碱的一体化全合成.在由内酰胺13转化为14的过程中, 涉及酰胺还原官能化的步骤多达8步(图式 4)!
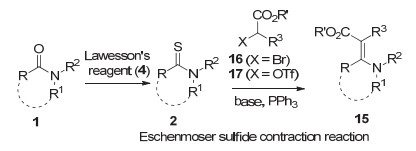
酰胺转化为烯胺酯(酮)化合物15是生物碱合成中又一个常用的转化[10, 11].现行的方法也需要把酰胺1首先转化为硫代酰胺2, 然后通过Eschenmoser缩硫反应[11], 即在三苯基膦作用下硫代酰胺与α-溴代酯16或α-三氟甲磺酰氧基酯17反应(图式 5).由于后者需要从相应的α-羟基酸合成, 因而, 当α-溴代酯为非商品化试剂时, 该法需要三步.
发展高效高选择性的有机合成方法是当前有机合成的主要目标.鉴于已知的具有普适性的酰胺转化基本为多步骤方法, 酰胺直接转化遂成为有机合成工作者的追求[12].由于酰胺的高稳定性和酰胺羰基的低亲电性, 发展化学选择性和具有普适性的酰胺转化方法的两种基本策略是进行酰胺活化和直接运用化学选择性试剂.此外, N-烷氧基酰胺这类特种酰胺, 既可自身活化也可类似于Weinreb酰胺, 形成稳定的亲核加成中间体, 因而可被直接转化为其他类型的化合物.
酰胺活化通常采用亲氧性Lewis酸型试剂和强亲电试剂.经典的Bischler-Napieralski环化反应, Vilsmeier试剂, 以及Vilsmeier-Haack反应均以POCl3或P2O5为活化剂和脱水剂.此外, SOCl2, CH2N2, PCl3, PCl5, Me2SO4, (COCl)2, (CF3CO)2O等, 以及高活性乙基化试剂(Et3O•BF4, Meerwein试剂)也被用于酰胺活化.现代版的酰胺活化试剂是三氟甲磺酸酐(Tf2O)[13].该试剂不但可有效地活化酰胺, 所形成的活化中间体比由传统活化剂形成的中间体活性更高, 因而近年来获得广泛应用.
1981年, 在把叔酰胺1d转化为烯酮亚胺盐B进而进行[2+2]环加成的方法中, Ghosez小组首次展示了Tf2O作为酰胺活化试剂的优点[14a], 以及Tf2O/DTBMP (2, 6-二叔丁基-4-甲基吡啶)组合(图式 6a)[14b]. 1990年, Martinez与Hanack及其合作者[15]报道, Tf2O与N, N-二甲基酰胺(DMF)形成的活化中间体可与非富电子的芳烃反应(图式 6b). 1991年, Fowler与Grierson等[16]合作报道了α, β-不饱和仲酰胺1e经Tf2O/Hűnig碱(DIPEA)活化直接还原氰化, 转化为双烯D (图式 6c)及其串联分子内Diels-Alder反应. 1994年, Banwell小组[17]通过Tf2O/DMAP (4-N, N-二甲氨基吡啶)活化, 发展了氨基甲酸酯19为底物的改良的Bischler-Napieralski环化反应(图式 6d). 1997年, Myers小组[18]在烯二炔类天然产物(+)-dynemicin A的全合成中, 研究了1f向21的转化, 发现2, 6-二叔丁基吡啶(DTBP)和2-氯吡啶(2-ClPyr.)均可与Tf2O匹配, 有效地促进反应(图式 6e). 1999年, Magnus小组把改良的Bischler-Napieralski环化[17]拓展为双Bischler-Napieralski环化, 实现了22向五环化合物23的一步转化(图式 6f)[19a], 从而发展了吲哚生物碱的快捷合成方法[19b].自2005年起, Bélanger课题组[20]着手探索基于叔酰胺与烯醇硅醚及烯胺等π-亲核体的分子内串联双环化策略, 以构建生物碱的各种复杂环系(图式 6g).

2006年, Movassaghi课题组报道了仲-N-苯基-苯甲酰胺经Tf2O/2-氯吡啶活化后与三甲基硅基乙炔铜合成α-三甲基硅基乙炔亚胺25a, 进而合成吡啶衍生物26的二步法(图式 6h)[21a], 并于次年优化为吡啶衍生物的一步合成法[21b].同年, 姚祝军小组以原位生成的Hendrickson试剂(Tf2O-2Ph3PO)[22]为活化试剂[23], 建立了基于串联反应进行抗癌中草药有效成分喜树碱27的简洁全合成(图式 6i)[24a].值得一提的是, 该策略最近被Haider小组[24b]用于Luotonin A衍生物的合成. 2008年, 姚祝军小组[24c]发展了Hendrickson试剂介入的α-三甲基硅基乙炔亚胺25b合成法(图式 6j).同年, Movassaghi小组[25]发展了以Tf2O/2-氯吡啶为活化试剂的改良的Bischler-Napieralski环化反应(图式 6k); 王彦广小组[26]展示了N-芳基仲酰胺1l经Tf2O/2-氯吡啶/2, 6二-氯吡啶联合活化, 与重氮乙酸乙酯29的反应, 建立了取代吲哚30的新合成法(图式 6l).值得一提的是, 这是2, 6二-氯吡啶首次用于酰胺活化, 而Movassaghi小组[27]在研究酰胺1m转化为吡啶衍生物32的过程中, 首次引入2-氟吡啶作为Tf2O的配套碱(图式 6m).
随后, Movassaghi小组[28]系统地研究、发展了基于酰胺活化合成结构复杂吲哚生物碱的方法学.
格氏试剂(RMgX)[29]和有机锂试剂(RLi)[29c, 29d, 29f, 30]是来源最为广泛的活泼有机金属试剂, 因而其对普通酰胺亲核加成的研究早已有之, 但无法形成具有普适性的合成方法(参见图式 3).特种酰胺如Weinreb酰胺是为把羧酸和羧酸酯转化为酮、醛而设计的, 无法解决普通酰胺的转化问题.黄培强课题组于2010年分别报道了普通叔酰胺1n经Tf2O原位活化, 用活泼有机金属试剂直接还原双烷基化(图式 7a)[31]和还原烷基化(图式 7b)[32]方法, 揭开了这一方法学的序幕.所发展的两个方法均具普适性, 分别被Carreira [33a]和Schneider小组[33b]采用, 并分别被用于(-)-FR901483[34a], 三尖杉碱[31b], 和(-)-morusimic acid[32b], (+)-preussin, (+)-preussin B[34b]等天然产物的全合成或形式全合成.
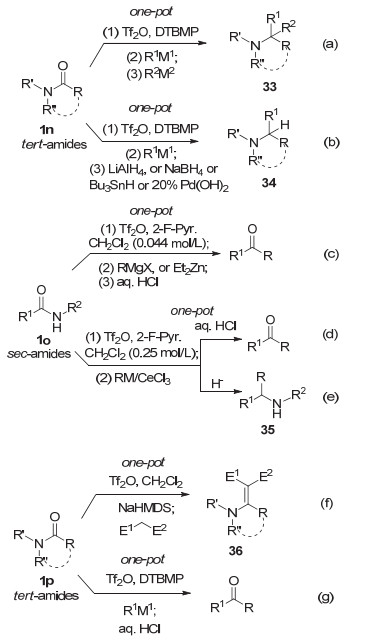
此后, Charette课题组[35]和黄培强课题组[36]于2012年分别独立发展了仲酰胺1o经Tf2O/2-氟吡啶活化, 与有机镁(锌)试剂和有机铈试剂反应的酮合成方法.此法具有很好的普适性, 表现出高度化学选择性(图式 7c, 7d).同年, 黄培强课题组[37]发展了仲酰胺经Tf2O原位活化, 进而用活泼有机金属试剂加成的直接还原烷基化方法(图式 7e).
接着, 黄培强课题组[38]分别于2014年和2015年发展了普通叔酰胺1p经Tf2O活化直接合成β, β-二官能化烯胺酯(酮)36的aza-Knoevenagel型反应(图式 7f), 和直接合成酮(图式 7g) [39]的普适性方法.
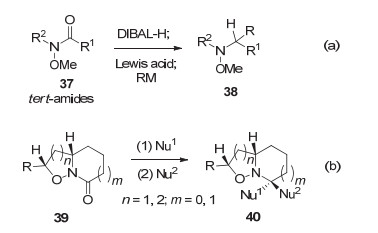
与普通酰胺相比, Weinreb酰胺属于特种酰胺, 不但表现出比普通酰胺更高的亲电性, 同时, 有机金属试剂(RMgX, RLi)与之加成可形成稳定的螯合中间体[40], 因而可用于酮和醛的合成而不发生过度加成.利用这些特点, 几乎与我们的工作[31a]同时, Chida小组于2010年报道了N-烷氧基酰胺37的直接还原烷基化[41a]和还原双烷基化[41b]方法(图式 8a).次年, Kouklovsky小组报道了亚硝基Diels-Alder环加成产物N-烷氧基酰胺39可被直接还原双烷基化[42a]和还原烷基化(图式 8b)[42b], 后者被用于哌啶生物碱的合成.
1993年, Ganem小组[43]开创性地展示了Schwartz试剂[Cp2Zr(H)Cl]试剂对仲酰胺1q的控制还原.该法被用于α-海人草酸43的简短合成, 表现出对酯基的兼容性(图式 9a)[43c].
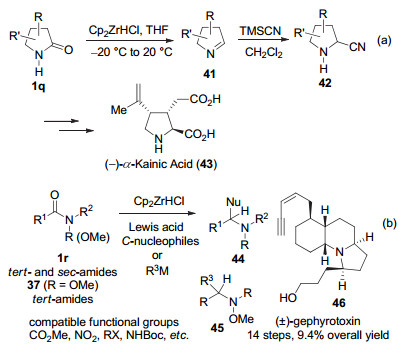
2012年, Chida小组[44a]发展了基于Schwartz试剂的N-甲氧基叔酰胺37的还原烯丙基化方法, 该法表现出很好的化学选择性.随后, 该法被扩展于叔酰胺和仲酰胺1r的还原官能化(图式 9b)[44b~44e].应用所建立的方法, 该小组完成了蟾蜍毒素生物碱gephyrotoxin (46)外消旋体的快捷全合成[44c, 44e].更全面的基于Schwartz试剂的合成方法学新近已有综述[12d, 45].
酰胺的催化转化有两种方式, 一是酰胺催化控制还原-C—C键形成, 二是酰胺的催化偶联反应.
酰胺催化转化的第一类方法依赖于酰胺催化控制还原方法的发展. 2009年, Nishikawa等[46]报道了含α-H的叔酰胺在Vaska’s络合物催化下的还原硅化.以此为基础, 2015年Dixon小组[47a]从硝基内酰胺1s出发, 发展了分子内硝基-Mannich型反应(图式 10a), 并用于吲哚生物碱的合成[47b].同年, Chida/Sato小组[48a]发展了N-甲氧基叔酰胺37b的催化还原-分子间官能化的普适性方法(图式 10b).该小组随后把底物拓展到N-羟基内酰胺37c, 由此建立了合成硝酮49的独特方法(图式 10c), 以及后者与烯烃的[2+3]环加成反应[48b, 48c].

黄培强课题组[49]于2017年发展了Ir和Cu双金属串联催化的叔酰胺1t的还原炔基化方法, 由此建立了炔丙胺型化合物50的新合成法(图式 10d).由于无需使用有机金属试剂, 这一方法表现出极好的化学选择性, 兼容醛等比酰胺活泼的官能团.
在上述方法中, Nishikawa等[46]提出烯胺为催化还原的产物.这先后得到Dixon等[47a]和Chida/Sato等[48a]的证实.按照这一机理, 叔酰胺需要含α-H.然而, 我们课题组的结果表明不含α-H的叔酰胺同样可进行催化还原官能化(图式 10d)[49].这随后得到Dixon小组[50]的证实.值得一提的是, Dixon的方法[47]仅第一步为催化反应, 第二步仍需使用化学计量的碱, 而Chida/Sato方法的第二步所用的有机金属试剂或烯醇负离子需要由化学计量的金属或碱生成[48].因此, 本课题组发展的方法[49]是首个全催化的酰胺还原官能化方法.
2016年, Adolfsson小组[51a]发展了Mo-催化的叔酰胺控制还原方法.该法表现出极好的化学选择性, 可兼容包括醛基在内的官能团.以此为基础, 他们发展了构建杂环化合物的新方法(图式 10e)[51b, 51c].
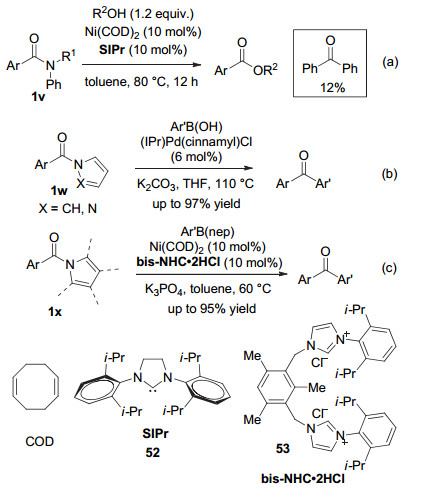
酰胺催化转化的第二类方法始于2015年, Garg和Houk小组合作, 率先实现了温和条件下酰胺向酯的镍催化转化(图式 11a)[52].尽管该反应只局限于N-芳基苯甲酰胺类化合物, 但仍然是酰胺催化转化的重要突破, 因而与Ni(价格较低廉且地球丰产金属)催化的其它转化一起, 被美国化学会《美国化学与工程新闻》(C & EN News)评为2015年化学的突破之一[53, 54].
然而, 当Garg等[55]试图把Ni-催化酰胺→酯转化拓展为酰胺→酮转化时, 产率只有20%.为此, 他们只得转而研究仲酰胺, 并采取分步策略:首先把仲酰胺转化为酰亚胺化合物(N-Boc酰胺), 然后进行Ni-催化的偶联反应[55].值得注意的是, 这一方法并未解决酰胺(无论是叔或仲酰胺)的催化偶联问题, 只是发展了酰亚胺的镍催化偶联方法.
2017年, Szostak小组[56]报道了Pd-催化下N‑酰基吡咯和N‑酰基吡唑1w与芳基硼酸的Suzuki-Miyaura交叉偶联反应(图式 11b).同期, 通过发展一个新的氮杂卡宾双齿配体53, 黄培强课题组[57]实现了镍-催化的N-酰基吡咯类叔酰胺1x与芳基硼酸新戊二醇酯的Suzuki交叉偶联反应(图式 11c).
近年酰胺直接转化的另一重要进展是温和条件下基于酰胺活化的酰胺转化和α-官能团化反应, Maulide[12e, 58]是这一方向的引领者.由于这些工作大多不涉及羰碳上的C—C键形成, 限于篇幅, 在此不作一一介绍, 将另文作全面回顾和解读.令人欣喜的是, 除了前述国内课题组的贡献, 近年来, 酰胺转化的多个方面吸引了国内愈来愈多课题组的关注, 并取得可喜的成绩[59].
综上, 酰胺直接转化以及相关的催化转化在近年获得了多方位的突破和应用, 尤其是温和条件下的酰胺化学选择性转化, 成为有机合成设计中多用途合成砌块的重要产生方式.随着酰胺直接转化化学逐步成为一个新的前沿研究领域, 这一“经典”官能团将在有机化学, 天然产物全合成和药物化学焕发出新的活力, 进一步丰富有机合成文献和传统教科书知识.
Brown, R. S. In The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, Eds. : Greenberg, A. ; Breneman, C. M. ; Liebman, J. F., John Wiley & Sons, Hoboken, 2000, pp. 85~114.
(a) Ma, X. Y. ; An, X. T. ; Zhao, X. H. ; Du, J. Y. ; Deng, Y. H. ; Zhang, X. Z. ; Fan, C. A. Org. Lett. 2017, 19, 2965. (b) Shu, C. ; Li, L. ; Tan, T. D. ; Yuan, D. Q. ; Ye, L. W. Sci. Bull. 2017, 62, 352. (c) Kong, D. Y. ; Li, M. N. ; Wang, R. ; Zi, G. F. ; Hou, G. H. Org. Biomol. Chem. 2016, 14, 1216. (d) Li, Y. ; Li, J. ; Ding, H. F. ; Li, A. Natl. Sci. Rev. 2017, 4, 397. (e) Chen, W. ; Zhang, H. B. Sci. China: Chem. 2016, 59, 1065. (f) Yu, K. ; Gao, B. L. ; Ding, H. F. Acta Chim. Sinica 2016, 74, 410. (余宽, 高北岭, 丁寒锋, 化学学报, 2016, 74, 410) (g) Yu, X. Y. ; Zhou, F. ; Chen, J. R. ; Xiao, W. J. Acta Chim. Sinica 2017, 75, 86. (余晓叶, 周帆, 陈加荣, 肖文精, 化学学报, 2017, 75, 86. )
(a) Wu, Q. F. ; Shen, P. X. ; He, J. ; Wang, X. B. ; Zhang, F. ; Shao, Q. ; Zhu, R. Y. ; Mapelli, C. ; Qiao, J. X. ; Poss, M. A. ; Yu, J. Q. Science 2017, 355, 499. (b) Kainz, Q. M. ; Matier, C. D. ; Bartoszewicz, A. ; Zultanski, S. L. ; Peters, J. C. ; Fu, G. C. Science 2016, 351, 681. (c) Luo, F. H. ; Long, Y. ; Li, Z. K. ; Zhou, X. G. Acta Chim. Sinica 2016, 74, 805. (罗飞华, 龙洋, 李正凯, 周向葛, 化学学报, 2016, 74, 805. )
For recent reviews, see: (a) Chardon, A. ; Morisset, E. ; Rouden, J. ; Blanchet, J. Synthesis 2018, 50, 984. (b) Volkov, A. ; Tinnis, F. ; Slagbrand, T. ; Trillo, P. ; Adolfsson, H. Chem. Soc. Rev. 2016, 45, 6685. (c) Zhang, L. L. ; Han, Z. B. ; Zhang, L. Li, M. X. ; Ding, K. L. Chin. J. Org. Chem. 2016, 36, 1824. (张琳莉, 韩召斌, 张磊, 李明星, 丁奎岭, 有机化学, 2016, 36, 1824. ) (d) Smith, A. M. ; Whyman, R. Chem. Rev. 2014, 114, 5477. (e) Werkmeister, S. ; Junge, K. ; Beller, M. Org. Process Res. Dev. 2014, 18, 289. (f) Addis, D. ; Das, S. ; Junge, K. ; Beller, M. Angew. Chem., Int. Ed. 2011, 50, 6004.
Seyden-Penne, J. Reductions by the Alumino-and Borohydrides in Organic Synthesis, 2nd ed., Wiley-VCH, New York, 1997.
Li, J. ; Zhang, W. H. ; Zhang, F. ; Chen, Y. ; Li, A. J. Am. Chem. Soc. 2017, 139, 14893. correction: J. Am. Chem. Soc. 2018, 140, 2384.
(a) Mateo, P. ; Cinqualbre, J. ; Mojzes, M. M. ; Schenk, K. ; Renaud, P. J. Org. Chem. 2017, 82, 12318. See also: (b) Murai, T. ; Mutoh, Y. ; Ohta, Y. ; Murakami, M. J. Am. Chem. Soc. 2004, 126, 5968. For a review, see: (c) Murai, T. ; Mutoh, Y. Chem. Lett. 2012, 41, 2.
(a) Abels, F. ; Lindemann, C. ; Koch, E. ; Schneider, C. Org. Lett. 2012, 14, 5972. (b) Abels, F. ; Lindemann, C. ; Schneider, C. Chem. -Eur. J. 2014, 20, 1964.
Lee, A. S.; Liau, B. B.; Shair, M. D. J. Am. Chem. Soc. 2014, 136, 13442. doi: 10.1021/ja507740u
Michael, J. P.; de Koning, C. B.; Gravestock, D.; Hosken, G. D.; Howard, A. S.; Jungmann, C. M.; Krause, R. W. M.; Parsons, A. S.; Pelly, S. C.; Stanbury, T. V. Pure Appl. Chem. 1999, 71, 979. doi: 10.1351/pac199971060979
Hussaini, S. R.; Chamala, R. R.; Wang, Z. Tetrahedron 2015, 71, 6017. doi: 10.1016/j.tet.2015.06.026
For reviews, see: (a) Seebach, D. Angew. Chem., Int. Ed. 2011, 50, 96. (b) Pace, V. ; Holzer, W. Aust. J. Chem. 2013, 66, 507. (c) Pace, V. ; Holzer, W. ; Olofsson, B. Adv. Synth. Catal. 2014, 356, 3697. (d) Sato, T. ; Chida, N. Org. Biomol. Chem. 2014, 12, 3147. (e) Kaiser, D. ; Maulide, N. J. Org. Chem. 2016, 81, 4421. (f) Sato, T. ; Chida, N. J. Synth. Org. Chem. Jpn. 2016, 74, 599. (g) Li, X. J. ; Sun, Y. ; Zhang L. ; Peng, B. Chin. J. Org. Chem. 2016, 36, 2530(李晓锦, 孙艳, 张磊, 彭勃, 有机化学, 2016, 36, 2530). (h) Evano, G. ; Lecomte, M. ; Thilmany, P. ; Theunissen, C. Synthesis 2017, 49, 3183. (i) Adachi, S. ; Kumagai, N. ; Shibasaki, M. Tetrahedron Lett. 2018, 59, 1147.
(a) Stang, P. J. ; White, M. R. Aldrichim. Acta 1983, 16, 15. (b) Baraznenok, I. L. ; Nenajdenko, V. G. ; Balenkova, E. S. Tetrahedron 2000, 56, 3077. For an excellent mechanistic investigation on the role of base additive in conjuction with Tf2O, see: (c) Mátravö lgyi, B. ; Hergert, T. ; Bálint, E. ; Bagi, P. ; Faigl, F. J. Org. Chem. 2018, 83, 2282.
(a) Falmagne, J. B. ; Escudero, J. ; Talebsahraoui, S. ; Ghosez, L. Angew. Chem. Int. Ed. Engl. 1981, 20, 879. (b) Chen, L. Y. ; Ghosez, L. Tetrahedron Lett. 1990, 31, 4467.
Martinez, A. G.; Alvarez, R. M.; Barcina, J. O.; Cerero, S. M.; Vilar, E. T.; Fraile, A. G.; Hanack, M.; Subramanian, L. R. J. Chem. Soc., Chem. Commun. 1990, 1571. https://www.researchgate.net/publication/272787896_Article_title_Between_Institutional_Political_and_Policy_Agenda_An_Analysis_of_Issue_Congruence_in_the_2004-2008_Election_Cycle_in_Slovenia_Reference_JSSC615_Journal_title_Communist_and_Post-Communist
Sisti, N. J.; Fowler, F. W.; Grierson, D. S. Synlett 1991, 816. https://www.researchgate.net/publication/272787896_Article_title_Between_Institutional_Political_and_Policy_Agenda_An_Analysis_of_Issue_Congruence_in_the_2004-2008_Election_Cycle_in_Slovenia_Reference_JSSC615_Journal_title_Communist_and_Post-Communist
Banwell, M. G.; Cowden, C. J.; Gable, R. W. J. Chem. Soc., Perkin Trans. 1 1994, 3515. https://www.researchgate.net/publication/272787896_Article_title_Between_Institutional_Political_and_Policy_Agenda_An_Analysis_of_Issue_Congruence_in_the_2004-2008_Election_Cycle_in_Slovenia_Reference_JSSC615_Journal_title_Communist_and_Post-Communist
Myers, A. G.; Tom, N. J.; Fraley, M. E.; Cohen, S. B.; Madar, D. J. J. Am. Chem. Soc. 1997, 119, 6072. doi: 10.1021/ja9703741
(a) Magnus, P. ; Gazzard, L. ; Hobson, L. ; Payne, A. H. ; Lynch, V. Tetrahedron Lett. 1999, 40, 5135. (b) Magnus, P. ; Gazzard, L. ; Hobson, L. ; Payne, A. H. ; Rainey, T. J. ; Westlund, N. ; Lynch, V. Tetrahedron 2002, 58, 3423.
(a) Bélanger, G. ; Larouche-Gauthier, R. ; Ménard, F. ; Nantel, M. ; Barabé, F. Org. Lett. 2005, 7, 4431. (b) Bélanger, G. ; Larouche-Gauthier, R. ; Ménard, F. ; Nantel, M. ; Barabé, F. J. Org. Chem. 2006, 71, 704.
(a) Movassaghi, M. ; Hill, M. D. J. Am. Chem. Soc. 2006, 128, 4592. (b) Movassaghi, M. ; Hill, M. D. ; Ahmad, O. K. J. Am. Chem. Soc. 2007, 129, 10096.
Hendrickson, J. B.; Hussoin, M. D. J. Org. Chem. 1987, 52, 4137. doi: 10.1021/jo00227a041
(a) You, S. L. ; Razavi, H. ; Kelly, J. W. Angew. Chem., Int. Ed. 2003, 42, 83; (b) You, S. -L. ; Kelly, J. W. Org. Lett. 2004, 6, 1681.
(a) Zhou, H. B. ; Liu, G. S. ; Yao, Z. J. Org. Lett. 2007, 9, 2003. (b) Atia, M. ; Bogdán, D. ; Brügger, M. ; Haider, N. ; Mátyus, P. Tetrahedron 2017, 73, 3231. (c) Dong, Q. L. ; Liu, G. S. ; Zhou, H. B. ; Chen, L. ; Yao, Z. J. Tetrahedron Lett. 2008, 49, 1636.
Movassaghi, M.; Hill, M. D. Org. Lett. 2008, 10, 3485. doi: 10.1021/ol801264u
Cui, S. L.; Wang, J.; Wang, Y. G. J. Am. Chem. Soc. 2008, 130, 13526. doi: 10.1021/ja805706r
Medley, J. W.; Movassaghi, M. J. Org. Chem. 2009, 74, 1341. doi: 10.1021/jo802355d
(a) White, K. L. ; Mewald, M. ; Movassaghi, M. J. Org. Chem. 2015, 80, 7403. (b) Mewald, M. ; Medley, J. W. ; Movassaghi, M. Angew. Chem., Int. Ed. 2014, 53, 11634.
(a) Handbook of Grignard Reagents, Eds. : Silverman, G. S. ; Rakita, P. E., Marcel Dekker, New York, 1996; (b) Grignard Reagent: New Developments, Ed. : Richey, H. G. Jr., Wiley, Chichester, 2000; (c) Main Group Metals in Organic Synthesis, Eds. : Yamamoto, H. ; Oshima, K., Wiley-VCH, Weinheim, 2004; (d) Handbook of Functionalized Organometallics Application in Synthesis, Ed. : Knochel, P., Wiley-VCH, Weinheim, 2005; (e) Klatt, T. ; Markiewicz, J. T. ; Sä mann, C. ; Knochel, P. J. Org. Chem. 2014, 79, 4253; (f) Bao, R. L. -Y. ; Zhao, R. ; Shi, L. Chem. Commun. 2015, 51, 6884, correction: Chem. Commun. 2015, 51, 9744.
(a) Foubelo, F. ; Yus, M. Chem. Soc. Rev. 2008, 37, 2620; (b) Chinchilla, R. ; Nájera, C. ; Yus, M. Tetrahedron 2005, 61, 3139; (c) The Chemistry of Organolithium Compounds, Eds. : Rappoport, Z. ; Marek, I., Wiley-VCH, Weinheim, 2004.
(a) Xiao, K. -J. ; Luo, J. -M. ; Ye, K. -Y. ; Wang, Y. ; Huang, P. -Q. Angew. Chem., Int. Ed. 2010, 49, 3037. (b) Huo, H. -H. ; Luo, J. -M. ; Xia, X. -E. ; Zhang, H. -K. ; Wang, Y. ; Huang, P. -Q. Chem. Eur. J. 2013, 19, 13075.
(a) Xiao, K. -J. ; Wang, Y. ; Ye, K. -Y. ; Huang, P. -Q. Chem. Eur. J. 2010, 16, 12792. (b) Xiao, K. -J. ; Wang, Y. ; Huang, Y. -H. ; Wang, X. -G. ; Huang, P. -Q. J. Org. Chem. 2013, 78, 8305.
(a) Guérot, C. ; Tchitchanov, B. H. ; Knust, H. ; Carreira, E. M. Org. Lett. 2011, 13, 780. (b) Lindemann, C. ; Schneider, C. Synthesis 2016, 48, 828.
(a) Huo, H. -H. ; Xia, X. -E. ; Zhang, H. -K. ; Huang, P. -Q. J. Org. Chem. 2013, 78, 455. (b) Huang, P. -Q. ; Geng, H. ; Tian, Y. -S. ; Peng, Q. -R. ; Xiao, K. -J. Sci. China: Chem. 2015, 58, 478.
Bechara, W. S.; Pelletier, G.; Charette, A. B. Nat. Chem. 2012, 4, 228. doi: 10.1038/nchem.1268
(a) Xiao, K. -J. ; Wang, A. -E; Huang, Y. -H. ; Huang, P. -Q. Asian J. Org. Chem. 2012, 1, 130. (b) Huang, P. -Q. ; Huang, Y. -H. ; Geng, H. ; Ye, J. -L. Sci. Rep. 2016, 6, 28801. (c) Huang, P. -Q. ; Huang, Y. -H. Chin. J. Chem. 2017, 35, 613.
Xiao, K.-J.; Wang, A.-E; Huang, P.-Q. Angew. Chem. Int. Ed. 2012, 51, 8314. doi: 10.1002/anie.v51.33
Huang, P.-Q.; Ou, W.; Xiao, K.-J.; Wang, A.-E Chem. Commun. 2014, 50, 8761. doi: 10.1039/C4CC03826F
Huang, P.-Q.; Wang, Y.; Xiao, K.-J.; Huang, Y.-H. Tetrahedron 2015, 71, 4248. doi: 10.1016/j.tet.2015.04.074
(a) Castoldi, L. ; Holzer, W. ; Langer, T. ; Pace, V. Chem. Commun. 2017, 53, 9498. (b) Pace, V. ; Murgia, I. ; Westermayer, S. ; Langer, T. ; Holzer, W. Chem. Commun. 2016, 52, 7584.
(a) Shirokane, K. ; Kurosaki, Y. ; Sato, T. ; Chida, N. Angew. Chem. Int. Ed. 2010, 49, 6369. (b) Yoritate, M. ; Meguro, T. ; Matsuo, N. ; Shirokane, K. ; Kurosaki, Y. ; Sato, T. ; Chida, N. Chem. Eur. J. 2014, 20, 8210. See also: (c) Jaekel, M. ; Qu, J. ; Schnitzer, T. ; Helmchen, G. Chem. Eur. J. 2013, 19, 16746.
(a) Vincent, G. ; Guillot, R. ; Kouklovsky, C. Angew. Chem. Int. Ed. 2011, 50, 1350. (b) Vincent, G. ; Karila, D. ; Khalil, G. ; Sancibrao, P. ; Gori, D. ; Kouklovsky, C. Chem. -Eur. J. 2013, 19, 9358.
(a) Schedler, D. J. A. ; Godfrey, A. G. ; Ganem, B. Tetrahedron Lett. 1993, 34, 5035. (b) Schedler, D. J. A. ; Li, J. ; Ganem, B. J. Org. Chem. 1996, 61, 4115. (c) Xia, Q. ; Ganem, B. Org. Lett. 2001, 3, 485.
(a) Nakajima, M. ; Oda, Y. ; Wada, T. ; Minamikawa, R. ; Shirokane, K. ; Sato, T. ; Chida, N. Chem. Eur. J. 2014, 20, 17565. (b) Oda, Y. ; Sato, T. ; Chida, N. Org. Lett. 2012, 14, 950. (c) Shirokane, K. ; Wada, T. ; Yoritate, M. ; Minamikawa, R. ; Takayama, N. ; Sato, T. ; Chida, N. Angew. Chem. Int. Ed. 2014, 53, 512. (d) Fukami, Y. ; Wada, T. ; Meguro, T. ; Chida, N. ; Sato, T. Org. Biomol. Chem. 2016, 14, 5486. (e) Shirokane, K. ; Tanaka, Y. ; Yoritate, M. ; Takayama, N. Sato, T. ; Chida, N. Bull. Chem. Soc. Jpn. 2015, 88, 522. See also: (f) Pace, V. ; Vega-Hernández, K. de la; Urban, E. ; Langer, T. Org. Lett. 2016, 18, 2750.
Więcław, M. M.; Stecko, S. Eur. J. Org. Chem. 2018, DOI: 10.1002/ejoc.201701537
Motoyama, Y.; Aoki, M.; Takaoka, N.; Aoto, R.; Nagashima, H. Chem. Commun. 2009, 1574. https://www.researchgate.net/publication/272787896_Article_title_Between_Institutional_Political_and_Policy_Agenda_An_Analysis_of_Issue_Congruence_in_the_2004-2008_Election_Cycle_in_Slovenia_Reference_JSSC615_Journal_title_Communist_and_Post-Communist
(a) Gregory, A. W. ; Chambers, A. ; Hawkins, P. ; Jakubec, A. ; Dixon, D. J. Chem. -Eur. J. 2015, 21, 111. (b) Tan, P. W. ; Seayad, J. ; Dixon, D. J. Angew. Chem. Int. Ed. 2016, 55, 13436.
(a) Nakajima, M. ; Sato, T. ; Chida, N. Org. Lett. 2015, 17, 1696. (b) Katahara, S. ; Kobayashi, S. ; Fujita, K. ; Matsumoto, T. ; Sato, T. ; Chida, N. J. Am. Chem. Soc. 2016, 138, 5246. (c) Katahara, S. ; Kobayashi, S. ; Fujita, K. ; Matsumoto, T. ; Sato, T. ; Chida, N. Bull. Chem. Soc. Jpn. 2017, 90, 893.
Huang, P.-Q.; Ou, W.; Han, F. Chem. Commun. 2016, 52, 11967. doi: 10.1039/C6CC05318A
(a) Fuentes de Arriba, A. L. ; Lenci, E. ; Sonawane, M. ; Formery, O. ; Dixon, D. J. Angew. Chem. Int. Ed. 2017, 56, 3655. (b) Xie, L. G. ; Dixon, D. J. Chem. Sci. 2017, 8, 7492.
(a) Tinnis, F. ; Volkov, A. ; Slagbrand, T. ; Adolfsson, H. Angew. Chem., Int. Ed. 2016, 55, 4562. (b) Slagbrand, T. ; Kervefors, G. ; Tinnis. F. ; Adolfsson, H. Adv. Synth. Catal. 2017, 359, 1990. (c) Trillo, P. ; Slagbrand, T. ; Tinnis F. ; Adolfsson, H. Chem. Commun. 2017, 53, 9159.
Hie, L.; Fine Nathel, N. F.; Shah, T.; Baker, E. L.; Hong, X.; Yang, Y. F.; Liu, P.; Houk, K. N.; Garg, N. K. Nature 2015, 524, 79. doi: 10.1038/nature14615
Ritter, S. K. Chem. Eng. News 2015, 93(49), 23. https://www.researchgate.net/publication/272787896_Article_title_Between_Institutional_Political_and_Policy_Agenda_An_Analysis_of_Issue_Congruence_in_the_2004-2008_Election_Cycle_in_Slovenia_Reference_JSSC615_Journal_title_Communist_and_Post-Communist
(a) Ritter, S. K. Chem. Eng. News Archive 2015, 93(30), 9. (b) Ruider, S. A. ; Maulide, N. Angew. Chem. Int. Ed. 2015, 54, 13856.
Weires, N. A.; Baker, E. L.; Garg, N. K. Nat. Chem. 2016, 8, 75. doi: 10.1038/nchem.2388
Meng, G.; Szostak, R.; Szostak, M. Org. Lett. 2017, 19, 3596. doi: 10.1021/acs.orglett.7b01575
Huang, P.-Q.; Chen, H. Chem. Commun. 2017, 53, 12584. doi: 10.1039/C7CC07457C
(a) Kaiser, D. ; Teskey, C. J. ; Adler, P. ; Maulide, N. J. Am. Chem. Soc. 2017, 139, 16040. (b) Shaaban, S. ; Tona, V. ; Peng, B. ; Maulide, N. Angew. Chem. Int. Ed. 2017, 56, 10938. (c) Torre, A. ; Kaiser, D. ; Maulide, N. J. Am. Chem. Soc. 2017, 139, 6578. (d) Kaiser, D. ; de la Torre, A. ; Shaaban, S. ; Maulide, N. Angew. Chem., Int. Ed. 2017, 56, 5921. (e) Mauro, G. D. ; Maryasin, B. ; Kaiser, D. ; Shaaban, S. ; González, L. ; Maulide, N. Org. Lett. 2017, 19, 3815. (f) Tona, V. ; Maryasin, B. ; de la Torre, A. ; Sprachmann, J. ; González, L. ; Maulide, N. Org. Lett. 2017, 19, 2662. (g) Gawali, V. S. ; Simeonov, S. ; Drescher, M. ; Knott, T. ; Scheel, O. ; Kudolo, J. ; Kä hlig, H. ; Hochenegg, K. ; Hochenegg, U. ; Roller, A. ; Todt, H. ; Maulide, N. ChemMedChem 2017, 12, 1819. (h) Peng, B. ; Geerdink, D. ; Fares, C. ; Maulide, N. Angew. Chem. Int. Ed. 2014, 53, 5462.
(a) Xiao, P. H. ; Tang, Z. X. ; Wang, K. ; Chen, H. ; Guo, Q. Y. ; Chu, Y. ; Gao, L. ; Song, Z. L. J. Org. Chem. 2018, 83, 1687. (b) Li, L. H. ; Niu, Z. J. ; Liang, Y. M. Chem. Eur. J. 2017, 23, 15300. (c) Li, X. W. ; Lin, F. G. R. ; Huang, K. M. ; Wei, J. L. ; Li, X. Y. ; Wang, X. Y. ; Geng, X. Y. ; Jiao, N. Angew. Chem. Int. Ed. 2017, 56, 12307. (d) Xie, C. M. ; Luo, J. S. ; Zhang, Y. ; Zhu, L. L. ; Hong, R. Org. Lett. 2017, 19, 3592. (e) Chen, J. J. ; Long, W. H. ; Fang, S. W. ; Yang, Y. G. ; Wan, X. B. Chem. Commun. 2017, 53, 13256. (f) Ding, G. N. ; Wu, X. Y. ; Jiang, L. L. ; Zhang, Z. G. ; Xie, X. M. Org. Lett. 2017, 19, 6048. (g) Zhang, Q. ; Yuan, J. W. ; Yu, M. F. ; Zhang, R. ; Liang, Y. J. ; Huang, P. ; Dong, D. W. Synthesis 2017, 49, 4996. (h) Jiang Meng, J. ; Jia, R. ; Leng, J. ; Wen, M. ; Yu, X. ; Deng, W. -P. Org. Lett. 2017, 19, 4520. (i) Shi, L. ; Tan, X. ; Long, J. ; Xiong, X. ; Yang, S. ; Xue, P. ; Lv, H. ; Zhang, X. Chem. -Eur. J. 2017, 23, 546. (j) Yuan, M. L. ; Xie, J. H. ; Zhou, Q. L. ChemCatChem 2016, 8, 3036. (k) Yuan, M. L. ; Xie, J. H. ; Zhu, S. F. ; Zhou, Q. L. ACS Catal. 2016, 6, 3665. (l) Xing, S. Y. ; Ren, J. ; Wang, K. ; Cui, H. ; Xia, T. ; Zhang, M. ; Wang, D. D. Adv. Synth. Catal. 2016, 358, 3093. (m) Lang, Q. -W. ; Hu, X. -N. ; Huang, P. -Q. Sci. China: Chem. 2016, 59, 1638. (n) Mou, X. Q. ; Xu, L. ; Wang, S. H. ; Yang, C. Tetrahedron Lett. 2015, 56, 2820. (o) Zhang, T. X. ; Zhang, Y. ; Zhang, W. X. ; Luo, M. -M. Adv. Synth. Catal. 2013, 355, 2775. (p) Xie, W. ; Zhao, M. ; Cui, C. Organometallics 2013, 32, 7440. (q) Zhao, M. N. ; Ren, Z. H. ; Wang, Y. Y. ; Guan, Z. H. Chem. Commun. 2012, 48, 8105.

图式 1 酰胺还原二步法在longeracinphyllin A全合成中的应用
Scheme 1 Stepwise reduction of amide 1a in the total synthesis of longeracinphyllin A

图式 2 酰胺还原烷基化的多步骤方法一
Scheme 2 Multistep method 1 for the reductive alkylation of amides

图式 3 酰胺还原烷基化的多步骤方法2与内酰胺11的直接转化
Scheme 3 Multistep method 2 and direct reductive alkylation of lactam 11

图式 4 内酰胺13还原官能化所需的步骤数
Scheme 4 Steps required for the reductive functionalization of lactam 13

图式 5 酰胺转化为烯胺酮(酯)化合物的经典方法
Scheme 5 The classical method for the transformation of amides into enaminones and β-enaminoesters

图式 6 三氟甲磺酸酐原位活化后π-和σ-亲核试剂与酰胺的反应
Scheme 6 Reactions of π- and σ-nucleophiles with amides via in situ activated with Tf2O

图式 7 三氟甲磺酸酐原位活化后活泼有机金属试剂与酰胺的反应
Scheme 7 Reactions of reactive organometallic reagents with amides via in situ activated with Tf2O

图式 9 基于Schwartz试剂的酰胺直接还原官能化
Scheme 9 Schwartz reagent-based reductive functionalizations of amides

图式 10 基于催化控制还原的酰胺直接还原官能化
Scheme 10 Catalytic partial reduction-based reductive functionalizations of amides

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:

