

图 1
自由基+分子反应沿反应坐标势能曲线
Figure 1.
Energy profiles along the reaction coordinates for the radical+molecule reactions

烷基过氧化氢(ROOH, R=alkyl)及其自由基作为最简单的有机氢过氧化物, 不仅是十分重要的大气微量物种[1~7], 也是碳氢燃料燃烧过程中的重要中间体[8~10], 对碳氢燃料燃烧点火起着至关重要的作用[10].烷基过氧化氢与羟基自由基(OH•)的氢提取反应, 是碳氢化合物低温燃烧机理构建中最关键的反应类之一, 该反应类属于自由基与分子的反应.大量研究表明[11~24], 自由基与分子的反应不是基元反应, 而是分两步进行, 第一步是自由基与分子通过碰撞形成稳定的反应复合物(也称为范德华复合物、反应前驱体), 第二步是通过低能垒反应途径得到反应产物, 其过渡态能量比第一步反应的反应物能量还低, 因而其总包反应在低温下表现出反应速率常数随温度增加而减小, 即负活化能关系.为了解释自由基与分子反应的负活化能关系, 人们提出了直接机理模型[11]、平衡态模型[12]、稳态模型[11, 13, 14]和两态模型[15, 16].其中, 稳态模型是高温下可接受模型, 但也存在争议[17].本文研究的目的之一是通过量子化学计算, 阐明稳态模型成立的原因及条件.
碳氢化合物燃烧详细反应机理的自动构建, 通常由通用性好的核心机理和按反应类自动生成的反应两部分整合构成[25].由于不同大小的碳氢燃料燃烧涉及的基元反应的小分子部分基本一致, 因此常采用对单组分小分子体系如甲烷、乙烷、乙烯等燃烧实验数据广泛验证的C4以下反应机理, 经过点火延迟时间、火焰传播速度以及火焰结构等大量的实验验证和优化作为核心机理.对具体大分子碳氢燃料的详细燃烧反应机理的自动构建, 只要把由该大分子化合物出发变成小分子的反应添加到核心机理中, 即可作为该大分子碳氢化合物的燃烧详细机理, 这些添加的反应通常按反应类型由碳原子数大小逐级给出.由于这些按反应类型给出的反应数量比较多, 动力学参数通常按反应类型由反应规则给出, 以便于计算机编程, 即通过计算机机理自动生成程序产生.
羟基自由基提取烷基过氧化氢中氢的反应中, 其α位的氢提取反应是其主要通道, 该通道反应类中, 目前仅有个别反应的动力学数据有文献报道, 如Niki等[26]在温度为298 K时通过傅里叶红外光谱法(FTIR)间接得到OH•提取CH3OOH中α位氢的反应速率常数k=4.35×10-12 cm3•molecule-1•s-1; Vaghjiani等[27]在203~423 K温度范围内用激光诱导荧光法研究了OH•与CH3OOH的氢提取反应, 间接得到了OH•提取CH3OOH中α位氢的反应速率常数k(298 K)=(1.53±0.43)×10-12 cm3• molecule-1•s-1; 在由欧盟资助的欧洲专家小组(CEC)对燃烧模型动力学数据评估中[28], 基于Vaghjiani等的实验研究, 在250~1000 K温度范围内, 拟合得到OH•提取CH3OOH中α位氢的反应速率表达式k(T)=1.2× 10-12 exp (130/T); Luo等[29]在MC-QCISD计算水平下得到包含反应复合物的势能面, 采用正则变分过渡态理论计算得到OH•分别提取CH3OOH、C2H5OOH中α位氢的反应速率常数, 并在250~1500 K温度段拟合得反应速率表达式分别为k(T)=1.36×10-23T3.76exp (1012.57/ T)、k(T)=3.31×10-22T3.46exp (904.52/T).然而, 文献缺乏对该反应类的系统研究, 本文研究的目的之二是通过量子化学计算得到该类反应的动力学参数, 为碳氢化合物低温机理自动构建提供精确数据.
由于实际碳氢燃料涉及到的都是高碳数的大分子体系, 所以羟基自由基与ROOH的α位氢提取反应类也涉及大分子体系, 而大分子体系动力学参数的高精度计算是对量子化学计算方法的挑战.为此, Truong等[30~32]提出了反应类过渡态理论(RC-TST), 已广泛应用于有能垒的反应类体系动力学参数的精确计算. RC-TST理论认为同一类反应中的所有反应具有相同的活性中心.通常情况下将同一类反应中最小反应体系定义为主反应, 其余反应则为目标反应. Truong等研究发现, 主反应与目标反应的能垒差随从头算理论方法级别的变化很小, 即可通过低水平的从头算理论方法得到较高精度大分子体系的动力学参数.在我们的前期研究中[33, 34], 我们将主要用于热力学参数计算的等键反应推广到了含非典型化学键的过渡态, 对反应类过渡态理论提供了解释并用于低级别从头算能垒和动力学参数的修正, 从而达到反应类反应能垒及速率常数的精确计算.本文研究的目的之三是通过等键反应理论与过渡态理论相结合, 以羟基自由基与ROOH的α位氢提取反应为研究对象, 解决负活化能反应类大分子体系动力学参数的精确计算问题.
在自由基与分子反应的动力学研究中, 无论是平衡态模型[12], 还是稳态模型[11, 13, 14], 或是两态模型[15, 16], 都是基于反应前复合物机理, 认为反应分两步完成:第一步反应是通过碰撞可逆形成含有氢键的反应复合物, 该过程可以看成是无能垒的复合反应过程, 可用变分过渡态计算其正、逆反应速率常数, 而第二步反应是有能垒的不可逆反应.该反应复合物机理可表达如下:
|
|
其中, R表示反应物, RC表示反应复合物, P表示产物. k1、k-1分别为第一步的正、逆反应速率常数, k2为第二步反应速率常数.通过对RC采取稳态近似[11, 13, 14]得到总包反应速率常数表达式:
|
$ k = \frac{{{k_1}{k_2}}}{{{k_{ - 1}} + {k_2}}} $ |
对RC采取稳态近似的条件是RC难生成, 易消耗.而按图 1描述的沿反应坐标的势能面, 反映出的是反应复合物易生成(第一步正反应, 无能垒)、难消耗(第一步逆反应或第二步反应, 有能垒), 稳态近似的条件似乎不成立.但是, 按照过渡态理论或变分过渡态理论, 对理想气体反应的速率常数根据热力学函数可表示为:
|
$ k = \kappa (T)\frac{{{k_{\rm{B}}}T}}{h} \cdot {\left( {\frac{{RT}}{{{P^\theta }}}} \right)^{n - 1}}{\rm{exp}}\left[ { - \frac{{\Delta G_{\rm{m}}^ \ne }}{{RT}}} \right] $ |
式中, kB和h分别是Boltzmann常数和Planck常数, R是摩尔气体常数, T是热力学温度, pθ是标准态压力, n是反应物种数目, κ(T)为隧穿系数,
对于第一步反应, 由于是无能垒反应, 只能采用变分过渡态处理, 即沿反应物到复合物的反应坐标搜寻自由能G的极大值点作为变分过渡态G≠.从反应物A+B到变分过渡态G≠、再到复合物RC的过程, 有三个平动自由度和三个转动自由度逐步转变为分子内部受限运动, 其熵是明显减小的过程.根据定义G=H-TS, 则其自由能G除了受能量E或焓H影响外, 还受熵影响, 整个反应沿反应坐标的自由能变化在低温和高温条件下如图 2a、图 2b所示.
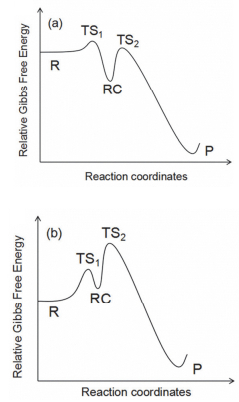
由于自由基与分子形成稳态复合物过程的反应焓变ΔH<0, 反应熵变ΔS<0, 所以在低温下k1>k-1, 随温度增加, 在某一转折温度Tc, k1=k-1.因此, 当温度远高于Tc时, k1<<k-1, 稳态近似成立.本文将通过量子化学计算得到具体反应的转折温度Tc, 从而得到稳态近似成立的条件.
自由基与分子反应的第一步反应的标准平衡常数
|
$ \Delta G_{1,{\rm{m}}}^0 = \Delta H_{1,{\rm{m}}}^0 - T\Delta S_{1,{\rm{m}}}^0 = - RT\;{\rm{ln}}\;K_1^0 $ |
浓度平衡常数
|
$ {K_{{\rm{c}},1}} = \frac{{{k_1}}}{{{k_{ - 1}}}} $ |
标准平衡常数
|
$ K_1^0 = {K_{{\rm{c}},1}}{(\frac{{RT}}{{{P^\theta }}})^{ - 1}} $ |
所以, 可以明确定义:当
自由基与分子反应的第二步反应是有能垒的反应, 在高温下通常k-1>>k2[13, 14], 稳态近似结果(1)式可进一步简化得到:
|
$ k = \frac{{{k_1}{k_2}}}{{{k_{ - 1}}}} = {K_{{\rm{c}},1}}{k_2} $ |
第二步反应是有能垒反应, 其速率常数计算可用传统过渡态理论计算[对应于(2) n=1情形]:
|
$ {k_2} = {\kappa _2}(T)\frac{{{k_{\rm{B}}}T}}{h} \cdot \exp \left[ { - \frac{{\Delta G_{2,{\mathop{\rm m}\nolimits} }^ \ne }}{{RT}}} \right] $ |
结合(3)~(7)式, 可得自由基与分子反应的总包反应速率常数:
|
$ \begin{array}{l} k = {K_{{\rm{c}},1}}{k_2} = \frac{{RT}}{{{P^\theta }}}{\rm{exp}}\left[ {\frac{{ - \Delta G_{1,m}^{^0}}}{{RT}}} \right]\kappa {}_2(T)\frac{{{k_{\rm{B}}}T}}{h} \cdot \\ {\rm{exp}}\left[ {\frac{{ - \Delta G_{2,{\rm{m}}}^ \ne }}{{RT}}} \right] = \kappa {}_2(T)\frac{{{k_{\rm{B}}}T}}{h}\frac{{RT}}{{{P^\theta }}}{\rm{exp}}\left[ {\frac{{ - \Delta G_{\rm{m}}^ \ne }}{{RT}}} \right]\\ \end{array} $ |
其中,
|
$ \Delta G_{\rm{m}}^ \ne = \Delta G_{1,{\rm{m}}}^0 + \Delta G_{2,{\rm{m}}}^ \ne = {G^ \ne } - {G_{\rm{R}}} $ |
因此, 在稳态近似成立的条件下, 自由基与分子反应总包反应速率常数的计算问题转化为反应物到过渡态Gibbs自由能变化的计算和第二步反应的隧穿系数的计算.
在我们前期的研究中, 将等键反应方法引入到反应类中大分子反应体系反应能垒的精确计算[33, 34].在反应类中, 选择最小分子体系为主反应P, 可用高水平从头算方法计算得到该反应的精确反应能垒
|
$ \Delta \Delta V_{\rm{P}}^ \ne = \Delta V_{\rm{P}}^{ \ne '} - \Delta V_{\rm{P}}^ \ne $ |
可作为对该反应类中其他反应(设为目标反应T)低水平从头计算能垒
|
$ \Delta V_{\rm{T}}^{ \ne '} = \Delta V_{\rm{T}}^ \ne + \Delta \Delta V_{\rm{P}}^ \ne $ |
其修正结果与直接用高精度从头算方法计算得到的精确能垒接近.将该修正方案引入到对低水平从头计算法计算的目标反应近似速率常数kT的修正:
|
$ k'_{\rm{T}} = {k_{\rm{T}}}{\rm{exp}}[( - \Delta \Delta V_{\rm{p}}^ \ne )/RT] $ |
本文选取了20个羟基自由基与ROOH的α位氢提取反应作为研究体系, R1作为主反应, 其余19个反应作为目标反应.所有反应列于表 1.
 下载:
导出CSV
下载:
导出CSV
| No. | Reaction |
| R1 | CH3OOH+OH•→CH2•OOH+H2O |
| R2 | CH3CH2OOH+OH•→CH3CH•OOH+H2O |
| R3 | C2H5CH2OOH+OH•→C2H5CH•OOH+H2O |
| R4 | C3H7CH2OOH+OH•→C3H7CH•OOH+H2O |
| R5 | CH3CH(CH3)CH2OOH+OH•→CH3CH(CH3)CH•OOH+H2O |
| R6 | C4H9CH2OOH+OH•→C4H9CH•OOH+H2O |
| R7 | C2H5CH(CH3)CH2OOH+OH•→C2H5CH(CH3)CH•OOH+H2O |
| R8 | C5H11CH2OOH+OH•→C5H11CH•OOH+H2O |
| R9 | CH3CH(CH3)C2H5CH2OOH+OH•→ CH3CH(CH3)C2H5CH•OOH+H2O |
| R10 | C3H7CH(CH3)CH2OOH+OH•→C3H7CH(CH3)CH•OOH+H2O |
| R11 | C6H13CH2OOH+OH•→C6H13CH•OOH+H2O |
| R12 | CH3CH(CH3)C3H6CH2OOH+OH•→ CH3CH(CH3)C3H6CH•OOH+H2O |
| R13 | C2H5CH(CH3)C2H4CH2OOH+OH•→ C2H5CH(CH3)C2H4CH•OOH+H2O |
| R14 | C4H9CH(CH3)CH2OOH+OH•→C4H9CH(CH3)CH•OOH+H2O |
| R15 | CH3CH(CH3)CH2CH(CH3)CH2OOH+OH•→ CH3CH(CH3)CH2CH(CH3)CH•OOH+H2O |
| R16 | C7H15CH2OOH+OH•→C7H15CH•OOH+H2O |
| R17 | CH3CH(CH3)C4H8CH2OOH+OH•→ CH3CH(CH3)C4H8CH•OOH+H2O |
| R18 | C5H11CH(CH3)CH2OOH+OH•→C5H11CH(CH3)CH•OOH+H2O |
| R19 | C2H5CH(CH3)CH2CH(CH3)CH2OOH+OH•→ C2H5CH(CH3)CH2CH(CH3)CH•OOH+H2O |
| R20 | C2H5CH(C2H5)C2H4CH2OOH+OH•→ C2H5CH(C2H5)C2H4CH•OOH+H2O |
所有量子化学计算均在Gaussian 09程序[35]下进行.用BHandHLYP/6-311G(d, p)方法对反应涉及到的所有物种进行几何结构优化、频率分析, 并作为低水平从头计算法用于单点能计算, 且用内禀反应坐标(IRC)对体系中反应通道进行了确认.频率校正因子为0.9335[36].在谐振分析中, 通过频率分析freq=hinderedrotor自动识别出对应于单键转动的低频振动.如果有模式被识别为受阻的或自由的内转动, 则自动校正热动力学函数.考虑到在BHandHLYP/6-311G(d, p)理论计算水平下自旋污染的影响, 我们检查了所有反应所涉及的各个物种湮灭前的<S2>期望值, 自旋多重度为2的物种都在0.752~0.765之间, 因此, 波函数的自旋污染可忽略.由于同一类反应具有相似的T1诊断值, 我们只对主反应所涉及的物种做了T1诊断, 结果表明, 可直接采用CCSD(T)/ CBS[37, 38]的单参考方法得到精确单点能, 不需考虑多参考效应.因此, 本文以CCSD(T)/CBS方法作为高水平从头计算法对R1~R5 5个反应的所有物种进行单点能计算.其外推方案采用CCSD(T)/cc-pVXZ(X=D, T, Q)外推到完全基组而得到:
|
$ {E^{{\rm{tot}}}} + {E^{{\rm{HF}}}} + {E^{{\rm{cor}}}} $ |
|
$ E_X^{{\rm{HF}}} = E_\infty ^{{\rm{HF}}} + B{e^{ - aX}} $ |
|
$ E_\infty ^{{\rm{cor}}} = E_X^{{\rm{cor}}} + A/{(X + 1/2)^3} $ |
其中, cc-pVDZ、cc-pVTZ和cc-pVQZ基组中X分别等于2、3和4, A、B、α都是常数, Hartree-Fock完全基组能量
焓的计算涉及到电子配分函数及平动、转动、振动配分函数的贡献; 熵只涉及到平动、转动、振动配分函数贡献, 其计算只依赖于几何结构优化和频率分析, 通常只需要较低级别从头算和较小基组.所以, 焓的计算因涉及电子配分函数的贡献, 其计算精度依赖于电子能量的精度, 需在高精度CCSD(T)/CBS方法下计算得到, 而熵变只需在BHandHLYP便可获得.本文选取R1~R5反应为代表反应, 计算了这些反应的转折温度Tc及相应的反应焓变
下载:
导出CSV
| R | ∆H1 a | ∆S1 a | Tc/K |
| R1 | -16.65 | -0.11 | 156.16 |
| R2 | -17.04 | -0.11 | 159.70 |
| R3 | -17.27 | -0.10 | 181.01 |
| R4 | -17.29 | -0.10 | 178.74 |
| R5 | -18.36 | -0.09 | 195.17 |
| a unit: kJ•mol-1. | |||
由表 2可知, R1~R5代表反应的转折温度分别为156.16、159.70、181.01、178.14、195.17 K, 反应的最高转折温度仅195.17 K, 远远低于通常的碳氢燃料燃烧模拟低温段(650~900 K)的最低温度.本文研究的是碳氢燃料燃烧模拟所需动力学参数的精确计算, 其温度范围远远高于转折温度Tc, 因而采用稳态近似是合理的.
在BHandHLYP/6-311G (d, p)水平下对所有反应所涉及的各个物种的结构进行优化和频率分析.过渡态反应中心几何结构所涉及的原子、键长及键角编号见图 3, 其中d1、d2为键长, A1、A2为键角, Ra为烷基取代基, Rb为氢取代基.本文研究的20个氢提取反应的过渡态反应中心几何结构参数列于表 3.从表 3可知, 该氢提取反应类过渡态的几何结构是相似的, 将断裂的键d1和新形成的键d2的最大绝对误差(MAE)只有0.001 nm和0.003 nm, 反应中心键角A1、A2的MAE也仅为1.8°和1.3°, 所以该氢提取反应类的过渡态几何结构是守恒的, 因此, 在该类氢提取反应中, 任意目标反应与主反应的差值所构建的反应都可作为等键反应[33].
 下载:
导出CSV
下载:
导出CSV
| TS | d1/nm | d2/nm | A1/(°) | A2/(°) |
| TS1 | 0.123 | 0.127 | 109.2 | 163.7 |
| TS2 | 0.122 | 0.129 | 107.8 | 164.7 |
| TS3 | 0.122 | 0.129 | 108.0 | 163.9 |
| TS4 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS5 | 0.122 | 0.130 | 107.7 | 163.4 |
| TS6 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS7 | 0.122 | 0.130 | 107.5 | 163.7 |
| TS8 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS9 | 0.122 | 0.129 | 107.9 | 164.0 |
| TS10 | 0.122 | 0.130 | 107.5 | 163.7 |
| TS11 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS12 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS13 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS14 | 0.122 | 0.130 | 107.5 | 163.8 |
| TS15 | 0.122 | 0.130 | 107.5 | 163.5 |
| TS16 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS17 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS18 | 0.122 | 0.130 | 107.5 | 163.7 |
| TS19 | 0.122 | 0.130 | 107.4 | 163.5 |
| TS20 | 0.122 | 0.129 | 107.9 | 163.9 |
| MAEa | 0.001 | 0.003 | 1.8 | 1.3 |
| a maximum absolute error. | ||||
本文采用等键反应方法对目标反应在BHandHLYP/ 6-311G (d, p)水平下的近似能垒进行了校正.为了验证该方法的可靠性, 选取R1~R5 5个代表反应作为研究对象, 分别采用低精度BHandHLYP/6-311G (d, p)方法和高精度CCSD(T)/CBS方法进行计算, 其计算结果与BHandHLYP方法修正前后反应能垒的比较列于表 4.
下载:
导出CSV
| Reaction | No. | CCSD(T)/CBS | DFTb | ∆(DFT)c | IRMd | ∆(IRM)e |
| R→TS | R1 | -2.87 | 15.65 | -18.52 | — | — |
| R2 | -9.68 | 9.47 | -19.15 | -9.05 | -0.63 | |
| R3 | -11.55 | 7.83 | -19.38 | -10.69 | -0.86 | |
| R4 | -12.02 | 7.53 | -19.55 | -10.99 | -1.03 | |
| R5 | -12.66 | 7.33 | -19.99 | -11.19 | -1.47 | |
| a Include zero point energy (ZPE); b Calculated at the BHandHLYP/6-311G (d, p) level of theory; c Difference between CCSD(T)/CBS barrier and BHandHLYP barrier; d BHandHLYP results after validation by isodesmic reaction method (IRM); e Difference between CCSD(T)/CBS value and IRM value. | ||||||
从表 4中可以看出, 低精度BHandHLYP/6-311G (d, p)计算方法与高精度CCSD(T)/CBS外推方法的反应能垒偏差约为19 kJ•mol-1, 修正后其最大反应能垒偏差降为1.47 kJ•mol-1, 偏差在化学精度以内, 表明用等键反应方法修正后的反应能垒与CCSD(T)的结果较为接近.因此, 对于大分子反应体系即可采用等键反应方法, 对低水平从头算的结果进行校正来得到高精度的计算结果.反应类中所有反应修正前后的反应能垒列于表 5.
下载:
导出CSV
| Target reaction | ΔVT*/(kJ•mol-1) | |
| DCMb | IRMc | |
| R2 | 9.47 | -9.05 |
| R3 | 7.83 | -10.69 |
| R4 | 7.53 | -10.99 |
| R5 | 7.33 | -11.19 |
| R6 | 7.47 | -11.05 |
| R7 | 7.08 | -11.44 |
| R8 | 7.46 | -11.06 |
| R9 | 7.3 | -11.22 |
| R10 | 6.86 | -11.66 |
| R11 | 7.57 | -10.95 |
| R12 | 7.15 | -11.37 |
| R13 | 7.49 | -11.03 |
| R14 | 6.78 | -11.74 |
| R15 | 6.93 | -11.59 |
| R16 | 7.64 | -10.88 |
| R17 | 7.31 | -11.21 |
| R18 | 6.78 | -11.74 |
| R19 | 6.83 | -11.69 |
| R20 | 7.31 | -11.21 |
| a Include zero point energy(ZPE); b Direct calculated at the BHandHLYP/ 6-311G (d, p) level of theory; c BHandHLYP results after validation by isodesmic reaction method (IRM). | ||
本文对反应类中20个反应的第二步反应在BHandHLYP/6-311G (d, p)水平、采用过渡态理论、隧穿系数经Wigner校正[39], 计算得到了该反应在298~3000 K温度范围内的近似速率常数, 并经等键反应方法校正后得到第二步反应精确的速率常数.再在稳态近似下得到总包反应速率常数, 按修正的Arrhenius方程k=ATnexp (-E/RT)进行拟合得到动力学参数(A, n, E), 结果列于表 6.
下载:
导出CSV
| Reaction | A/(cm3•molecule-1•s-1) | n | E/(kJ•mol-1) |
| R1 | 8.50×10-21 | 2.60 | -13.07 |
| R2 | 1.91×10-24 | 3.64 | -22.86 |
| R3 | 2.52×10-20 | 2.54 | -20.32 |
| R4 | 4.54×10-21 | 2.67 | -21.04 |
| R5 | 8.28×10-22 | 3.02 | -20.88 |
| R6 | 4.15×10-23 | 3.35 | -22.50 |
| R7 | 7.82×10-22 | 3.05 | -21.12 |
| R8 | 1.13×10-22 | 3.19 | -21.16 |
| R9 | 7.46×10-23 | 3.22 | -23.20 |
| R10 | 1.77×10-21 | 3.06 | -21.19 |
| R11 | 4.84×10-21 | 2.66 | -20.16 |
| R12 | 2.15×10-24 | 3.76 | -23.90 |
| R13 | 3.61×10-25 | 3.90 | -23.11 |
| R14 | 1.71×10-22 | 3.18 | -24.21 |
| R15 | 5.67×10-20 | 2.32 | -22.84 |
| R16 | 6.60×10-21 | 2.75 | -20.78 |
| R17 | 1.94×10-20 | 2.62 | -21.75 |
| R18 | 2.55×10-20 | 2.49 | -23.45 |
| R19 | 1.02×10-23 | 3.71 | -23.82 |
| R20 | 1.37×10-22 | 3.22 | -22.03 |
| a Temperature range in 298~3000 K. | |||
对于我们所研究的OH•提取ROOH中的α-H的反应类, 只有反应R1的速率常数有实验报道.对于R1反应, 本文计算的反应速率常数与文献值的对比列于表 7.由表 7可看出, 当温度为298 K时, 本文计算得到的总包反应速率常数为4.50×10-12 cm3•molecule-1•s-1, 略大于Vaghjiani等[27]的实验值1.53×10-12 cm3•mol-1•s-1, 接近于Niki等[26]的实验值4.35×10-12 cm3•mol-1•s-1.因此可看出本文计算的反应速率常数与文献值较为接近, 可为羟基自由基提取烷基过氧化氢中α位氢的反应类提供可靠的动力学数据.
下载:
导出CSV
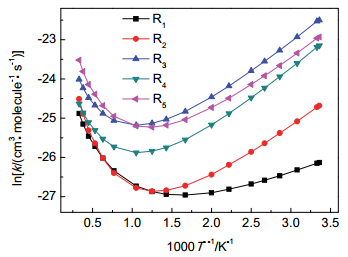
为了揭示该反应类的速率常数随温度的变化关系, 我们选取了R1~R5 5个反应为研究对象, 其总包反应速率常数随温度的变化趋势呈现于图 4.由图 4可知, 主反应R1在298~600 K温度范围内, 总包反应速率常数k随温度增加而减小, 即呈负活化能关系, 在600~3000 K温度范围内, 总包反应速率常数随温度增加而增大; 而R2~R5 4个反应则是在298~900 K温度段, 随温度增加而减小, 在900~3000 K温度段, 随温度增加而增大. R1主反应在298~600 K温度范围内表现出的负活化能关系, 与Vaghjiani的实验研究的反应趋势一致.因此本文揭示了所研究反应类只在低温段呈现负温度效应.
从以上的讨论中, 可得出以下几个结论:
OH•提取ROOH中α-H的这一氢提取反应是一类存在反应复合物的非基元反应.通过对反应类中R1~R5 5个代表反应的量子化学计算表明, 其第一步反应的最高转折温度仅有195.17 K, 远远低于碳氢燃料燃烧模拟通常关注的最低温度650 K, 因此采用稳态模型计算该反应体系的总包反应速率常数是合理的.
该类氢提取反应体系的过渡态几何结构守恒, 因此可采用等键反应法计算反应动力学参数.
采用等键反应方法校正反应能垒, 最大绝对偏差从低水平DFT的19.99 kJ•mol-1降到校正后的1.47 kJ• mol-1, 偏差在化学精度以内, 因此可得到该反应体系的精确反应能垒.
该氢提取反应体系只在低温段呈现负温度效应.
通过采用等键方法进行校正即可得到精确的动力学参数, 从而可解决碳氢燃料燃烧模拟中负活化能反应类大分子体系动力学参数的精确计算问题, 对碳氢化合物的低温燃烧机理研究具有重要意义.
Baasandorj, M.; Papanastasiou, D. K.; Talukdar, R. K.; Hasson, A. S.; Burkholder, J. B. Phys. Chem. Chem. Phys. 2010, 12, 12101. doi: 10.1039/c0cp00463d
Roehl, C. M.; Marka1, Z.; Fry, J. L.; Wennberg, P. O. Atmos. Chem. Phys. 2007, 7, 713. doi: 10.5194/acp-7-713-2007
Wang, C.; Chen, Z. Atmos. Environ. 2008, 42, 6614. doi: 10.1016/j.atmosenv.2008.04.033
Wang, C.; Chen, Z. Prog. Nat. Sci. 2006, 16, 1141. doi: 10.1080/10020070612330121
Lee, M.; Heikes, B. G.; O'Sullivan, D. W. Atmos. Environ. 2000, 34, 3475. doi: 10.1016/S1352-2310(99)00432-X
Frey, M. M.; Stewart, R. W.; McConnell, J. R.; Bales, R. C. J. Geophys. Res. 2005, 110(D23), D23301. doi: 10.1029/2005JD006110
Butkovskaya, N. I.; Kukui, A.; Pouvesle, N.; Bras, G. L. J. Phys. Chem. A 2004, 108, 7021.
Gross, A.; Mikkelsen, K. V.; Stockwell, W. R. Int. J. Quantum Chem. 2001, 84, 493. doi: 10.1002/(ISSN)1097-461X
Ranzi, E.; Cavallotti, C.; Cuoci, A.; Frassoldati, A.; Pelucchi, M.; Faravelli, T. Combust. Flame. 2015, 162, 1679. doi: 10.1016/j.combustflame.2014.11.030
Chen, D. N.; Jin, H. F.; Wang, Z. D.; Zhang, L. D.; Qi, F. J. Phys. Chem. A 2011, 115, 602. doi: 10.1021/jp1099305
Alvarez-Idaboy, J. R.; Mora-Diez, N.; Boyd, R. J.; Vivier-Bunge, A. J. Am. Chem. Soc. 2001, 123, 2018. doi: 10.1021/ja003372g
Bänsch, C.; Kiecherer, J.; Szöri, M.; Olzmann, M. J. Phys. Chem. A 2013, 117, 8343. doi: 10.1021/jp405724a
Alvarez-Idaboy, J. R.; Mora-Diez, N.; Vivier-Bunge, A. J. Am. Chem. Soc. 2000, 122, 3715. doi: 10.1021/ja993693w
Galano, A.; Alvarez-Idaboy, J. R.; Francisco-Márquez, M. J. Phys. Chem. A 2010, 114, 7525. doi: 10.1021/jp103575f
Greenwald, E. E.; North, S. W.; Georgievskii, Y.; Klippenstein, S. J. J. Phys. Chem. A 2005, 109, 6031.
Greenwald, E. E.; North, S. W.; Georgievskii, Y.; Klippenstein, S. J. J. Phys. Chem. A 2007, 111, 5582. doi: 10.1021/jp071412y
Shannon, R. J.; Taylor, S.; Goddard, A.; Blitz, M. A.; Heard, D. E. Phys. Chem. Chem. Phys. 2010, 12, 13511. doi: 10.1039/c0cp00918k
Uc, V. H.; Alvarez-Idaboy, J. R.; Galano, A.; Garcia-Cruz, I.; Vivier-Bunge, A. J. Phys. Chem. A 2006, 110, 10155. doi: 10.1021/jp062775l
Iuga, C.; Galano, A.; Vivier-Bunge, A. Chem. Phys. Chem. 2008, 9, 1453. doi: 10.1002/cphc.v9:10
Galano, A.; Alvarez-Idaboy, J. R.; Ruiz-Santoyo, M. E.; Vivier-Bunge, A. J. Phys. Chem. A 2002, 106, 9520. doi: 10.1021/jp020297i
Olivella, S.; Sole, A. J. Chem. Theory. Comput. 2008, 4, 941. doi: 10.1021/ct8000798
Vega-Rodriguez, A.; Alvarez-Idaboy, J. R. Phys. Chem. Chem. Phys. 2009, 11, 7649. doi: 10.1039/b906692f
Uc, V. H.; García-Cruz, I.; Hernandez-Laguna, A.; Vivier-Bunge, A. J. Phys. Chem. A 2000, 104, 7847. doi: 10.1021/jp993678d
Singleton, D. L.; Cvetanovic, R. J. J. Am. Chem. Soc. 1976, 98, 6812. doi: 10.1021/ja00438a006
Dente, M.; Bozzano, G.; Faravelli, T.; Marongiu, A.; Pierucci, S.; Ranzi, E. Adv. Chem. Eng. 2007, 32, 51. doi: 10.1016/S0065-2377(07)32002-4
Niki, H.; Maker, P. D.; Savage, C. M.; Breltenbach, L. P. J. Phys. Chem. 1983, 87, 2190. doi: 10.1021/j100235a030
Vaghjiani, G. L.; Ravishankara, A. R. J. Phys. Chem. 1989, 93, 1948. doi: 10.1021/j100342a050
Baulch, D. L.; Bowman, C. T.; Cobos, C. J.; Cox, R. A.; Just, T.; Kerr, J. A.; Pilling, M. J.; Stocker, D.; Troe, J.; Tsang, W.; Walker, R. W.; Warnatz, J. J. Phys. Chem. Ref. Data. 2005, 34, 757. doi: 10.1063/1.1748524
Luo, J.; Jia, X.; Gao, Y.; Song, G.; Yu, Y.; Wang, R.; Pan, X. J. Comput. Chem. 2011, 32, 987. doi: 10.1002/jcc.21684
Truong, T. N. J. Chem. Phys. 2000, 113, 4957. doi: 10.1063/1.1287839
Muszyńska, M.; Ratkiewicz, A.; Huynh, L. K.; Truong, T. N. J. Phys. Chem. A 2009, 113, 8327. doi: 10.1021/jp903762x
Huynh, L. K.; Ratkiewicz, A.; Truong, T. N. J. Phys. Chem. A 2006, 110, 473. doi: 10.1021/jp051280d
Wang, B. Y.; Li, Z. R.; Tan, N. X.; Yao, Q.; Li, X. Y. J. Phys. Chem. A 2013, 117, 3279. doi: 10.1021/jp400924w
Sun, X. H.; Yao, Q.; Li, Z. R.; Wang, J. B.; Li, X. Y. Theor. Chem. Acc. 2017, 136, 64. doi: 10.1007/s00214-017-2086-y
Frisch, M. J. ; Trucks, G. W. ; Schlegel, H. B. ; Scuseria, G. E. ; Robb, M. A. ; Cheeseman, J. R. ; Scalmani, G. ; Barone, V. ; Mennucci, B. ; Petersson, G. A. ; Nakatsuji, H. ; Caricato, M. ; Li, X. ; Hratchian, H. P. ; Izmaylov, A. F. ; Bloino, J. ; Zheng, G. ; Sonnenberg, J. L. ; Hada, M. ; Ehara, M. ; Toyota, K. ; Fukuda, R. ; Hasegawa, Jr. ; Ishida, M. ; Nakajima, T. ; Honda, Y. ; Kitao, O. ; Nakai, H. ; Vreven, T. ; Montgomery, J. A. ; J. ; Peralta, J. E. ; Ogliaro, F. ; Bearpark, M. ; Heyd, J. J. ; Brothers, E. ; Kudin, K. N. ; Staroverov, V. N. ; Kobayashi, R. ; Normand, J. ; Raghavachari, K. ; Rendell, A. ; Burant, J. C. ; Iyengar, S. S. ; Tomasi, J. ; Cossi, M. ; Rega, N. ; Millam, J. M. ; Klene, M. ; Knox, J. E. ; Cross, J. B. ; Bakken, V. ; Adamo, C. ; Jaramillo, J. ; Gomperts, R. ; Stratmann, R. E. ; Yazyev, O. ; Austin, A. J. ; Cammi, R. ; Pomelli, C. ; Ochterski, J. W. ; Martin, R. L. ; Morokuma, K. ; Zakrzewski, V. G. ; Voth, G. A. ; Salvador, P. ; Dannenberg, J. J. ; Dapprich, S. ; Daniels, A. D. ; Farkas, Ö. ; Foresman, J. B. ; Ortiz, J. V. ; Cioslowski, J. ; Fox, D. J. Gaussian 09, Revision A. 1, Gaussian Inc., Wallingford, CT, 2009.
Merrick, J. P.; Moran, D.; Radom, L. J. Phys. Chem. A 2007, 111, 11683. doi: 10.1021/jp073974n
Truhlar, D. G. Chem. Phys. Lett. 1998, 294, 45. doi: 10.1016/S0009-2614(98)00866-5
Huh, S. B.; Lee, J. S. J. Chem. Phys. 2003, 118, 3035. doi: 10.1063/1.1534091
Wigner, E. J. Chem. Phys. 1937, 5, 720. doi: 10.1063/1.1750107

图 1 自由基+分子反应沿反应坐标势能曲线
Figure 1 Energy profiles along the reaction coordinates for the radical+molecule reactions
R, RC, TS, P represent reactant, reactant complex, transition state, product, respectively

图 2 (a) 在低温下和(b)在高温下的自由基+分子反应沿反应坐标Gibbs自由能曲线,
Figure 2 Gibbs Free Energy profiles along the reaction coordinates for the radical+molecule reactions (a) at low temperature, and (b) at high temperature
R, TS1, RC, TS2 and P represent reactant, transition state of the first step reaction, reactant complex, transition state of the second step reaction and product, respectively

图 3 过渡态反应中心几何结构
Figure 3 Geometry structures for the reaction center of the transition states

图 4 5个代表反应的总包反应速率常数k随温度的变化趋势
Figure 4 Plot of the temperature dependent overall reaction constant k for five representative reactions
表 1 氢提取反应列表
Table 1. List of hydrogen abstraction reactions
| No. | Reaction |
| R1 | CH3OOH+OH•→CH2•OOH+H2O |
| R2 | CH3CH2OOH+OH•→CH3CH•OOH+H2O |
| R3 | C2H5CH2OOH+OH•→C2H5CH•OOH+H2O |
| R4 | C3H7CH2OOH+OH•→C3H7CH•OOH+H2O |
| R5 | CH3CH(CH3)CH2OOH+OH•→CH3CH(CH3)CH•OOH+H2O |
| R6 | C4H9CH2OOH+OH•→C4H9CH•OOH+H2O |
| R7 | C2H5CH(CH3)CH2OOH+OH•→C2H5CH(CH3)CH•OOH+H2O |
| R8 | C5H11CH2OOH+OH•→C5H11CH•OOH+H2O |
| R9 | CH3CH(CH3)C2H5CH2OOH+OH•→ CH3CH(CH3)C2H5CH•OOH+H2O |
| R10 | C3H7CH(CH3)CH2OOH+OH•→C3H7CH(CH3)CH•OOH+H2O |
| R11 | C6H13CH2OOH+OH•→C6H13CH•OOH+H2O |
| R12 | CH3CH(CH3)C3H6CH2OOH+OH•→ CH3CH(CH3)C3H6CH•OOH+H2O |
| R13 | C2H5CH(CH3)C2H4CH2OOH+OH•→ C2H5CH(CH3)C2H4CH•OOH+H2O |
| R14 | C4H9CH(CH3)CH2OOH+OH•→C4H9CH(CH3)CH•OOH+H2O |
| R15 | CH3CH(CH3)CH2CH(CH3)CH2OOH+OH•→ CH3CH(CH3)CH2CH(CH3)CH•OOH+H2O |
| R16 | C7H15CH2OOH+OH•→C7H15CH•OOH+H2O |
| R17 | CH3CH(CH3)C4H8CH2OOH+OH•→ CH3CH(CH3)C4H8CH•OOH+H2O |
| R18 | C5H11CH(CH3)CH2OOH+OH•→C5H11CH(CH3)CH•OOH+H2O |
| R19 | C2H5CH(CH3)CH2CH(CH3)CH2OOH+OH•→ C2H5CH(CH3)CH2CH(CH3)CH•OOH+H2O |
| R20 | C2H5CH(C2H5)C2H4CH2OOH+OH•→ C2H5CH(C2H5)C2H4CH•OOH+H2O |
 下载: 导出CSV
下载: 导出CSV
表 2 R1~R5 5个反应的反应焓变、熵变及转折温度
Table 2. The reaction enthalpy change, entropy change and the conversion temperature of R1~R5 reactions
| R | ∆H1 a | ∆S1 a | Tc/K |
| R1 | -16.65 | -0.11 | 156.16 |
| R2 | -17.04 | -0.11 | 159.70 |
| R3 | -17.27 | -0.10 | 181.01 |
| R4 | -17.29 | -0.10 | 178.74 |
| R5 | -18.36 | -0.09 | 195.17 |
| a unit: kJ•mol-1. | |||
下载: 导出CSV
表 3 过渡态反应中心几何参数
Table 3. Geometric parameters of reaction center of transition states
| TS | d1/nm | d2/nm | A1/(°) | A2/(°) |
| TS1 | 0.123 | 0.127 | 109.2 | 163.7 |
| TS2 | 0.122 | 0.129 | 107.8 | 164.7 |
| TS3 | 0.122 | 0.129 | 108.0 | 163.9 |
| TS4 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS5 | 0.122 | 0.130 | 107.7 | 163.4 |
| TS6 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS7 | 0.122 | 0.130 | 107.5 | 163.7 |
| TS8 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS9 | 0.122 | 0.129 | 107.9 | 164.0 |
| TS10 | 0.122 | 0.130 | 107.5 | 163.7 |
| TS11 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS12 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS13 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS14 | 0.122 | 0.130 | 107.5 | 163.8 |
| TS15 | 0.122 | 0.130 | 107.5 | 163.5 |
| TS16 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS17 | 0.122 | 0.129 | 107.9 | 163.9 |
| TS18 | 0.122 | 0.130 | 107.5 | 163.7 |
| TS19 | 0.122 | 0.130 | 107.4 | 163.5 |
| TS20 | 0.122 | 0.129 | 107.9 | 163.9 |
| MAEa | 0.001 | 0.003 | 1.8 | 1.3 |
| a maximum absolute error. | ||||
下载: 导出CSV
表 4 BHandHLYP方法修正前后势垒与CCSD(T)/CBS结果比较
Table 4. Comparison of BHandHLYP energy barriers (unit: kJ/mol) before and after validation with the CCSD(T)/CBS resultsa
| Reaction | No. | CCSD(T)/CBS | DFTb | ∆(DFT)c | IRMd | ∆(IRM)e |
| R→TS | R1 | -2.87 | 15.65 | -18.52 | — | — |
| R2 | -9.68 | 9.47 | -19.15 | -9.05 | -0.63 | |
| R3 | -11.55 | 7.83 | -19.38 | -10.69 | -0.86 | |
| R4 | -12.02 | 7.53 | -19.55 | -10.99 | -1.03 | |
| R5 | -12.66 | 7.33 | -19.99 | -11.19 | -1.47 | |
| a Include zero point energy (ZPE); b Calculated at the BHandHLYP/6-311G (d, p) level of theory; c Difference between CCSD(T)/CBS barrier and BHandHLYP barrier; d BHandHLYP results after validation by isodesmic reaction method (IRM); e Difference between CCSD(T)/CBS value and IRM value. | ||||||
下载: 导出CSV
表 5 目标反应的反应势垒
Table 5. Energy barriers for the target reactionsa
| Target reaction | ΔVT*/(kJ•mol-1) | |
| DCMb | IRMc | |
| R2 | 9.47 | -9.05 |
| R3 | 7.83 | -10.69 |
| R4 | 7.53 | -10.99 |
| R5 | 7.33 | -11.19 |
| R6 | 7.47 | -11.05 |
| R7 | 7.08 | -11.44 |
| R8 | 7.46 | -11.06 |
| R9 | 7.3 | -11.22 |
| R10 | 6.86 | -11.66 |
| R11 | 7.57 | -10.95 |
| R12 | 7.15 | -11.37 |
| R13 | 7.49 | -11.03 |
| R14 | 6.78 | -11.74 |
| R15 | 6.93 | -11.59 |
| R16 | 7.64 | -10.88 |
| R17 | 7.31 | -11.21 |
| R18 | 6.78 | -11.74 |
| R19 | 6.83 | -11.69 |
| R20 | 7.31 | -11.21 |
| a Include zero point energy(ZPE); b Direct calculated at the BHandHLYP/ 6-311G (d, p) level of theory; c BHandHLYP results after validation by isodesmic reaction method (IRM). | ||
下载: 导出CSV
表 6 反应动力学参数(A, n, E)
Table 6. Reaction kinetic parameters (A, n, E)a
| Reaction | A/(cm3•molecule-1•s-1) | n | E/(kJ•mol-1) |
| R1 | 8.50×10-21 | 2.60 | -13.07 |
| R2 | 1.91×10-24 | 3.64 | -22.86 |
| R3 | 2.52×10-20 | 2.54 | -20.32 |
| R4 | 4.54×10-21 | 2.67 | -21.04 |
| R5 | 8.28×10-22 | 3.02 | -20.88 |
| R6 | 4.15×10-23 | 3.35 | -22.50 |
| R7 | 7.82×10-22 | 3.05 | -21.12 |
| R8 | 1.13×10-22 | 3.19 | -21.16 |
| R9 | 7.46×10-23 | 3.22 | -23.20 |
| R10 | 1.77×10-21 | 3.06 | -21.19 |
| R11 | 4.84×10-21 | 2.66 | -20.16 |
| R12 | 2.15×10-24 | 3.76 | -23.90 |
| R13 | 3.61×10-25 | 3.90 | -23.11 |
| R14 | 1.71×10-22 | 3.18 | -24.21 |
| R15 | 5.67×10-20 | 2.32 | -22.84 |
| R16 | 6.60×10-21 | 2.75 | -20.78 |
| R17 | 1.94×10-20 | 2.62 | -21.75 |
| R18 | 2.55×10-20 | 2.49 | -23.45 |
| R19 | 1.02×10-23 | 3.71 | -23.82 |
| R20 | 1.37×10-22 | 3.22 | -22.03 |
| a Temperature range in 298~3000 K. | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们