Received Date:
27 December 2017 Available Online:
15 June 2018
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21773257, 21373233 and 91441108)
Abstract:
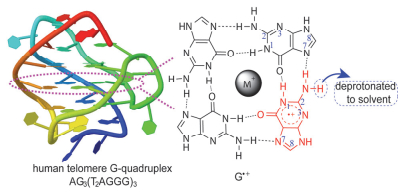
G-Quadruplex can be a promising candidate as molecular electronic device due to the ability of transferring hole. Extensive studies have reported that fast deprotonation of guanine radical cation (G·+) to form a neutral radical G(-H)· is the most important reaction in competition with hole transfer in DNA, hindering potential applications of DNA in molecular electronics. We thus carry out joint experimental and theoretical studies on deprotonation of G·+ in human telomere G-quadruplex AG3(T2AG3)3by using nanosecond laser flash photolysis and quantum chemical calculations. Upon 355 nm laser photolysis of Na2S2O8, instantaneously generated SO4·- radical oxidizes G base in the G-quadruplex to G·+. In the time-resolved absorption spectra that record the reaction of G-quadruplex with SO4·- at different temperatures, the transient absorptions of G(N(2)-H)· featured by absorption band at 640 nm are observed. It turns out that the G-quadruplex deprotonation product is G(N(2)-H)· and the deprotonation site is thereby validated to be amino proton. To obtain the activation energy of the G·+ deprotonation in G-quadruplex, the N(2)-H deprotonation rate constants at different temperatures varying from 280 to 300 K in steps 5 K are measured at a high G-quadruplex concentration, where the deprotonation has been proved to be the rate-limiting step in our previous work. Based upon Arrhenius equation, the deprotonation activation energy of G·+ in G-quadruplex is determined to be 20.0±1.0 kJ/mol. Further, the potential energy profile for the G·+ deprotonation in G-quadruplex is calculated at M062X/6-31G(d) level by carefully taking into account hydration environment of G·+ in G-quadruplex. The calculated energy barrier of 26.4 kJ/mol matches with the measured activation energy value, indicating the calculated potential energy profile can describe the deprotonation process of G·+ in the G-quadruplex. These theoretical and experimental results provide valuable dynamics information and mechanistic insights for potential applications of DNA structures in electronic device.
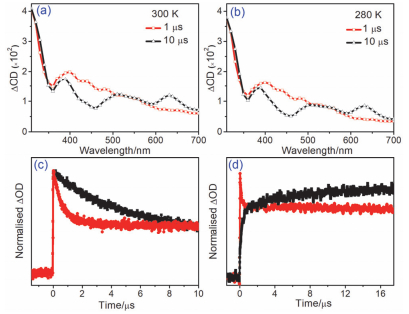
Figure 2.
Transient UV-vis absorption spectra for human telomere G-quadruplex AG3(T2AG3)3 ([G]=4 mmol/L)+Na2S2O8 (400 mmol/L) in buffer solution (pH=7.5) after 355 nm laser photolysis at (a) 300 K; (b) 280 K; (c) kinetics traces at 440 nm after excitation of Na2S2O8 (400 mmol/L) solution in the absence or presence of human telomere G-quadruplex ([G]=4 mmol/L) at 300 K and (d) kinetics traces for G•+ decay at 480 nm and G(N(2)-H)• growth at 640 nm at 300 K
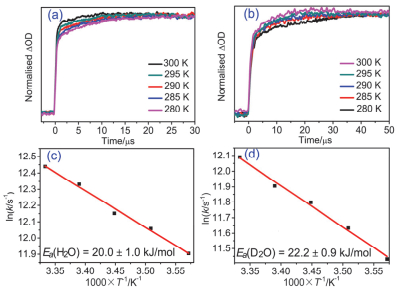
Figure 3.
Normalized kinetic traces at 640 nm at different temperatures in (a) H2O buffer and (b) D2O buffer, arrhenius plots of the deprotonation rate constants at temperatures in the range of 280~300 K in steps of 5 K, with the activation energy indicated in (c) H2O buffer and (d) D2O buffer. Solid line is the fitting line
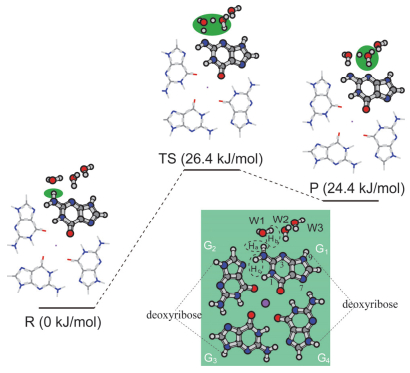
Figure 4.
Optimized structures and relative energies obtained at the M062X/6-31G(d) level for G•+deprotonation in a G-quartet under the 3H2O-PCM model (all energies given are relative to the reactant complex. Carbon, oxygen, nitrogen, hydrogen atoms and sodium ion are denoted with gray, red, blue, white and pink balls, respectively)
Faraggi, M.; Broitman, F.; Trent, J. B.; Klapper, M. H. J. Phys. Chem. 1996, 100, 14751. doi: 10.1021/jp960590g
[11]
Cleveland, C. L.; Barnett, R. N.; Bongiorno, A.; Joseph, J.; Liu, C. S.; Schuster, G. B.; Landman, U. J. Am. Chem. Soc. 2007, 129, 8408. doi: 10.1021/ja071893z
Cerón-Carrasco, J. P.; Requena, A.; Perpète, E. A.; Michaux, C.; Jacquemin, D. J. Phys. Chem. B2010, 114, 13439. doi: 10.1021/jp101711z
[20]
Takada, T.; Kawai, K.; Fujitsuka, M.; Majima, T. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 14002. doi: 10.1073/pnas.0402756101
[21]
Choi, J.; Park, J.; Tanaka, A.; Park, M. J.; Jang, Y. J.; Fujitsuka, M.; Kim, S. K.; Majima, T. Angew. Chem., Int. Ed. 2013, 52, 1134. doi: 10.1002/anie.201208149
[22]
Delaney, S.; Barton, J. K. Biochemistry2003, 42, 14159. doi: 10.1021/bi0351965
[23]
Szalai, V. A.; Thorp, H. H. J. Am. Chem. Soc. 2000, 122, 4524. doi: 10.1021/ja0001355
[24]
Song, D.; Yang, W.; Qin, T.; Wu, L.; Liu, K.; Su, H. J. Phys. Chem. Lett. 2014, 5, 2259. doi: 10.1021/jz501040a
[25]
Wolter, M.; Elstner, M.; Kubař, T. J. Chem. Phys. 2013, 139, 125102. doi: 10.1063/1.4821594
[26]
Barnett, R. N.; Bongiorno, A.; Cleveland, C. L.; Joy, A.; Landman, U.; Schuster, G. B. J. Am. Chem. Soc. 2006, 128, 10795. doi: 10.1021/ja061795y
[27]
Rokhlenko, Y.; Geacintov, N. E.; Shafirovich, V. J. Am. Chem. Soc. 2012, 134, 4955. doi: 10.1021/ja212186w
[28]
Rokhlenko, Y.; Cadet, J.; Geacintov, N. E.; Shafirovich, V. J. Am. Chem. Soc. 2014, 136, 5956. doi: 10.1021/ja412471u
[29]
Saintome, C.; Amrane, S.; Mergny, J. L.; Alberti, P. Nucleic Acids Res. 2016, 44, 2926. doi: 10.1093/nar/gkw003
Li, X.; Cai, Z.; Sevilla, M. D. J. Phys. Chem. B2001, 105, 10115. doi: 10.1021/jp012364z
[36]
Kumar, A.; Sevilla, M. D. J. Phys. Chem. B2009, 113, 11359. doi: 10.1021/jp903403d
[37]
Horvath, M. P.; Schultz, S. C. J. Mol. Biol. 2001, 310, 367. doi: 10.1006/jmbi.2001.4766
[38]
Parkinson, G. N.; Lee, M. P. H.; Neidle, S. Nature2002, 417, 876. doi: 10.1038/nature755
[39]
Marx, D.; Tuckerman, M. E.; Hutter, J.; Parrinello, M. Nature1999, 397, 601. doi: 10.1038/17579
[40]
Berkelbach, T. C.; Lee, H. S.; Tuckerman, M. E. Phys. Rev. Lett. 2009, 238302.
[41]
Frisch, M. J. ; Trucks, G. W. ; Schlegel, H. B. ; Scuseria, G. E. ; Robb, M. A. ; Cheeseman, J. R. ; Montgomery, J. A. ; Vreven, T. ; Kudin, K. N. ; Burant, J. C. ; Millam, J. M. ; Iyengar, S. S. ; Tomasi, J. ; Barone, V. ; Mennucci, B. ; Cossi, M. ; Scalmani, G. ; Rega, N. ; Petersson, G. A. ; Nakatsuji, H. ; Hada, M. ; Ehara, M. ; Toyota, K. ; Fukuda, R. ; Ha-segawa, J. ; Ishida, M. ; Nakajima, T. ; Honda, Y. ; Kitao, O. ; Nakai, H. ; Klene, M. ; Li, X. ; Knox, J. E. ; Hratchian, H. P. ; Cross, J. B. ; Bakken, V. ; Adamo, C. ; Jaramillo, J. ; Gomperts, R. ; Stratmann, R. E. ; Yazyev, O. ; Austin, A. J. ; Cammi, R. ; Pomelli, C. ; Ochterski, J. W. ; Ayala, P. Y. ; Morokuma, K. ; Voth, G. A. ; Salvador, P. ; Dannen-berg, J. J. ; Zakrzewski, V. G. ; Dapprich, S. ; Daniels, A. D. ; Strain, M. C. ; Farkas, O. ; Malick, D. K. ; Rabuck, A. D. ; Raghavachari, K. ; Foresman, J. B. ; Ortiz, J. V. ; Cui, Q. ; Baboul, A. G. ; Clifford, S. ; Cioslowski, J. ; Stefanov, B. B. ; Liu, G. ; Liashenko, A. ; Piskorz, P. ; Komaromi, I. ; Martin, R. L. ; Fox, D. J. ; Keith, T. ; Al-Laham, M. A. ; Peng, C. Y. ; Nanayakkara, A. ; Challacombe, M. ; Gill, P. M. W. ; Johnson, B. ; Chen, W. ; Wong, M. W. ; Gonzalez, C. ; Pople, J. A. Gaussian 09, Revision A. 01, Gaussian, Inc., Wallingford, CT, 2009.
Figure 2
Transient UV-vis absorption spectra for human telomere G-quadruplex AG3(T2AG3)3 ([G]=4 mmol/L)+Na2S2O8 (400 mmol/L) in buffer solution (pH=7.5) after 355 nm laser photolysis at (a) 300 K; (b) 280 K; (c) kinetics traces at 440 nm after excitation of Na2S2O8 (400 mmol/L) solution in the absence or presence of human telomere G-quadruplex ([G]=4 mmol/L) at 300 K and (d) kinetics traces for G•+ decay at 480 nm and G(N(2)-H)• growth at 640 nm at 300 K
Figure 3
Normalized kinetic traces at 640 nm at different temperatures in (a) H2O buffer and (b) D2O buffer, arrhenius plots of the deprotonation rate constants at temperatures in the range of 280~300 K in steps of 5 K, with the activation energy indicated in (c) H2O buffer and (d) D2O buffer. Solid line is the fitting line
Figure 4
Optimized structures and relative energies obtained at the M062X/6-31G(d) level for G•+deprotonation in a G-quartet under the 3H2O-PCM model (all energies given are relative to the reactant complex. Carbon, oxygen, nitrogen, hydrogen atoms and sodium ion are denoted with gray, red, blue, white and pink balls, respectively)

 下载:
下载:

 下载:
下载:





