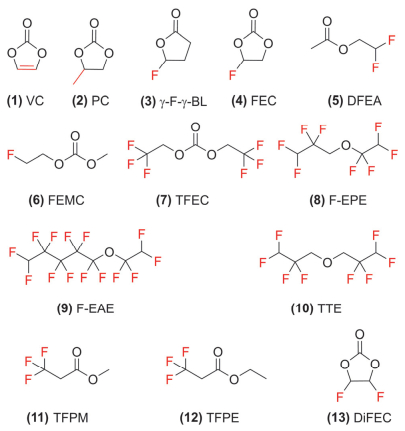
图 1
VC、PC及氟代溶剂或添加剂的分子结构
Figure 1.
Molecular structures of VC, PC, and fluoro-substituted solvent and additives

电解液被称作锂离子电池的“血液”, 其对锂离子电池(LIBs)性能的发挥有着至关重要的作用, 尤其是电极与电解液界面结构和性质.随着纯电动汽车(EVs)及混合电动汽车(HEVs)的快速发展, 对LIBs能量密度、循环寿命以及安全性要求不断提高, 然而, 在传统电解液体系中, 正极材料在高电压、高温下发生剧烈的结构变化和界面副反应, 给实际应用带来巨大挑战.开发适配的电解液添加剂改善电极与电解液界面结构, 是提高锂离子电池电化学性能最经济有效的方法之一, 受到各界的广泛关注[1~3].
三元材料{Li[NixCoyMz]O2 (0<x, y, z<1, M=Mn, 缩写NMC; M=Al, 缩写NCA)}如NMC111、NMC532等, 因其利用了Ni、Co、Mn等元素间的协同效应, 使得其综合性能优异, 是最具应用前景的一类正极材料.然而, 在传统电解液体系中, 其同样存在电极与电解液界面在高电压、高温下不稳定的缺点, 尤其给高镍三元材料的循环寿命和安全性带来严重挑战.目前, 解决方法有两种:一方面, 改进或开发新电极材料, 另一方面, 开发适配三元材料的电解液添加剂是最简单有效的方法[4~7].本文主要从物质结构出发, 综述了近5年来电解液添加剂在三元NMC及NCA锂离子电池中的应用及作用机理, 并进行总结和展望, 这些添加剂包括碳酸亚乙烯酯(VC)、氟代物、新型锂盐、含P、含B、含S、腈类、其它类型以及复合添加剂.
碳酸亚乙烯酯(VC, 1, 图 1), 最早被日本的Sanyo Electric Company公司发现, 其介电常数高、黏度低, 是目前最常用的一种电解液添加剂, 随后, Saft发现少量的VC能在电位大于1 V下被还原(C=C键的存在), 在石墨负极表面形成保护膜, 抑制溶剂碳酸丙烯酯(PC, 2, 图 1)对石墨的插层反应, 显著提高了PC基电解液的电化学性能.基于VC, 他们还提出一系列含C=C键的物质作为电解液添加剂.自此, 拉开了以VC为代表的电解液添加剂应用研究的序幕[8].
近年来, VC电解液添加剂得到了深入研究.长久以来, VC的作用机理被认为是, 在电池化成过程中, 其在负极表面优先于碳酸乙烯酯(EC, 电解液的组分之一)被还原, 形成固态电解质界面膜(SEI)[9].随后, 研究者在正极表面也发现了VC的分解产物, 而且Ouatani等[10]认为VC在正极表面分解的主要机理是自由基聚合反应.于是, 关于VC如何提高电池电化学性能的主要原因便出现了两种观点:一是认为VC在负极被还原形成更稳定的SEI膜, 二是认为其聚合反应形成正极电解质界面膜(CEI)降低了正极表面电解液氧化的速率.第一种观点在此不以赘述, 而关于第二种观点, 研究认为导致高镍NMC容量衰减的主要原因包括正极表面相变和副反应[7].当NMC811/G截止电压从4.1 V提高到4.3 V, 循环后NMC811表面岩盐相结构明显增加, 加入2% VC(后文如未说明, 均为质量比)可以明显抑制岩盐相生长, 此时阻抗减小, 但极化增加, 这说明极化不完全是表面相变造成的.作者认为循环过程容量衰减的原因不完全是岩盐相生长, 电解液氧化也是一个重要原因[11]. Burns等[12]将包含电解液氧化这一类的电流概括为以下四个方面: (a)负极SEI膜形成消耗Li+, 伴随的ILi; (b)正极表面穿梭反应或过渡金属溶出产生的Iox, a; (c)电解液氧化消耗电解液中的Li+, Iox, b; (d)正极结构破坏, Ip.
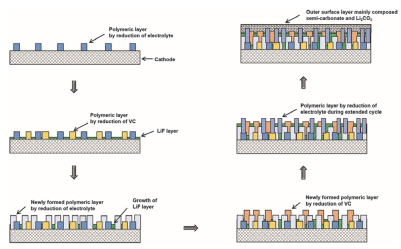
添加VC后电池的放电容量衰减速率没有明显变化, 但库伦效率较未添加有所提高, 同时充电容量变化减小, 这三个现象表明VC的主要作用在于减小Iox, b.同时, 为了表征电解液氧化反应的热效应, Downie等[13]用等温微量热法测定NMC111/G随电压变化的寄生反应热, 添加2% VC, 尤其在电压大于4.23 V时, 能够显著降低其寄生反应热. Lee等[14]进一步研究了脱锂态NMC532和NMC622的热稳定性, 发现随着Ni含量增加, VC越有利于材料的热稳定性, XPS深度刻蚀表明: VC与其它电解液溶剂参与了CEI膜的形成, 最终使得表面富含聚合物, 热稳定性提高(如图 2所示).
VC还可以降低正极界面阻抗, 但其含量超过2%会导致负极阻抗急剧增大, 而硫酸亚乙酯(ES, 65, 图 13)可以降低负极阻抗, 因此VC+ES的协同作用弥补了各自的不足[15]. Madec等[16]进一步分析二者的作用机理, 如表 1所示, 无论是否存在Li+, ES的还原能最低, 因而在电极反应过程中最有可能被还原.但存在Li+的情况下, VC的溶剂化能低于ES, 因而溶剂化VC的还原能要远低于未溶剂化的ES.当VC+ES混合时, VC会在首次化成过程中优先被还原, 而ES只是被缓慢和少量的消耗, 使得VC在CEI和SEI膜的形成中占主导地位.这种性质使得VC与其它添加剂混合使用时, 既能发挥其它添加剂的特性, 又能保留VC良好的成膜性能, 对于开发复合添加剂具有重要意义(详见第10节).
 下载:
导出CSV
下载:
导出CSV
| EC | VC | ES | EMC | |
| ΔEred, without Li*/(kcal"mol-1) | 26.9 | 26.4 | -0.7 | 34.9 |
| ΔEred, with Li**/(kcal"mol-1) | -89.5 | -88.1 | -131.8 | -91.5 |
| ΔEsolv/(kcal"mol-1) | -47.1 | -43.9 | -42.7 | -42.6 |
| The calculation of ∆Ered and ∆Esolv is based on the MP2/6-311+G(2d, p) level in gas phase. The geometry was optimized at the B3LYP/6-311+G(2d, p) level[16]. | ||||
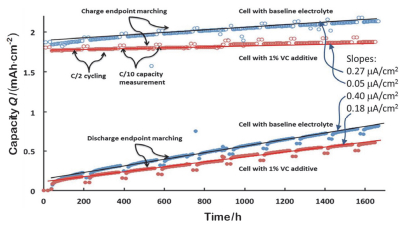
VC最显著的特点是可以提高库伦效率和充电容量保持率.因此, Burns等[17]认为VC可减少电池内部寄生反应, 提高循环寿命.然而, Deshpande等[18]研究发现, 1% VC降低了每次充放电容量的变化速率, 提高了循环库伦效率, 但放电容量衰减无明显变化(如图 3所示).这说明循环过程中存在两种副反应, 都导致了这个充放电容量的变化.一种是循环过程中发生的体积膨胀等导致负极SEI膜的破坏和再修补, 使正极中的Li被过度消耗, 造成极大的容量损失; 另外一种是正负极间的穿梭反应. VC仅降低了后者, 因为穿梭反应整体上没有消耗锂盐和溶剂, 这个过程可逆且不会导致容量损失.因此虽然VC提高了库伦效率, 但库伦效率并不是一个表征循环寿命可靠的指标.
VC对于NMC半电池和全电池的影响有何区别?Qian等[19]发现, 提高VC含量对NMC111/Li循环性能有利, 而对NMC111/G不利. XPS研究发现, CEI膜的成分和厚度是最主要的影响因素.对于NMC111/Li, 循环过程中Li负极与电解液持续发生副反应, 且NMC111表面生成大量PEO聚合物, 恶化了电池性能.而VC的加入可以抑制PEO的产生, 因此提高VC的含量有利于半电池的循环性能.也正因为半电池存在较多上述副反应, 所以并不适合表征电池的循环性能[20]. Qian等[21]研究表明, CEI膜的厚度在前100次循环变化不大, 而随后的50次循环, 厚度急剧增加, 循环性能随之降低. GC-MS表明, 只要VC存在, 电解液分解就较少, 随着VC持续被消耗, 后50圈电解液分解急剧增加, 同时CEI厚度增加, 导致电池的循环性能随之下降.相反, NMC111/G在首次化成过程中, SEI膜的形成较为完善, 石墨负极与电解液间的副反应大大减少, 因此电解液中残留的VC相对稳定.而VC过量, 电解液电导率下降, 同时CEI膜的厚度急剧增加, 内部阻抗增大, 不利于电池的电化学性能.因此, VC添加量最适合的范围为0~2%. Burns等[9]发现, 提高VC的含量到4%或6%, 在低倍率下的循环性能更佳, 这说明, 如果单纯追求长循环寿命, VC的添加量可以更高, 而如果要求倍率性能, 则应控制VC的添加量在1%~2%之间.
总之, VC作为一个普适性的电解液添加剂, 在较低电压和温度下, 1%或2% VC几乎对所有电池体系的电化学性能都有一定程度改善. Burns等[9]认为传统电解液体系中应该加入少量VC.而且, Wang等[22]认为, 大部分文献报道中, 在电解液中加入一种添加剂很容易获得比不添加更好的电化学性能, 但均不如添加2% VC的电解液.因此, 只有一种新型添加剂的电化学性能比2% VC更好或相似, 那么它才可能具有实际意义.然而, VC的缺点是高温或高电压下的电化学性能较差[23], 实际应用中还需复合其它添加剂使用.
氟代溶剂及其添加剂的研究, 基于F原子具有强电负性和弱极性, 因而其凝固点降低、闪点提高、氧化稳定性提高以及电解液与电极之间的兼容性增强, 相应地提高了电解液低温性能、抗氧化性能、阻燃性能及电极润湿性[8, 24].目前, F代溶剂包括氟代内酯、氟代环状碳酸酯、氟代羧酸、氟代线性碳酸酯及氟代醚等(3~7, 图 1)[8], 图 1依次列举了代表性物质.综合考虑黏度、介电常数、电导率以及溶解度等因素, 本文着重阐述被研究较多的存在EC溶剂的电解液体系如氟代碳酸乙烯酯(FEC, 4, 图 1)、氟代碳酸甲乙酯(FEMC, 6, 图 1), 以及无EC的氟代溶剂及添加剂.
FEC作为EC的单F取代产物, 其熔点为17 ℃, 低于EC (37 ℃).此外, 其最高占据轨道(HOMO)能级和最低未被占据轨道(LUMO)能级都低于EC, 因而其氧化稳定性提高, 而还原稳定性降低, 因此在低电位下, 容易在石墨负极、硅负极被还原, 具有良好的成膜性[8, 25].但其相对介电常数和黏度都高于EC, 因此LiPF6在FEC中的电导率比在EC中低.本小节主要阐述FEC作为添加剂, 在NMC/G和NMC/Si中的应用及机理分析.
首先, 对NMC111/Li半电池的研究认为, FEC形成了富含LiF的CEI膜, 使得其在循环过程中更加稳定, 厚度变化较小, 因而提高了电池的循环性能[21], 但部分观点认为过多的LiF会增加阻抗[26].
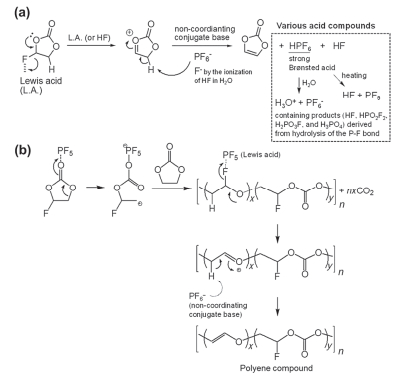
然而, FEC在负极还原性较强, 容易引起其他副反应, 而且对不同负极材料(石墨、硅)影响也有区别.在石墨负极中, Kim等[25]提出FEC在LiPF6基电解液中的两种可能反应机理.第一种机理如图 4(a)所示, 路易斯酸或HF的存在会促进FEC的脱HF反应, 生成VC、HF及其它酸, 如H3OPF6、HPO2F2、H2PO3F及H3PO4; 第二种机理如图 4(b), 路易斯酸PF5还会催化FEC开环生成部分氟代的聚合物.因此, 上述两个过程实际上促进了非配位共轭碱PF6-的分解, 使得电解液酸度升高, 加剧了电解液的分解, 而且高温下更加显著.另外, Streich等[27]通过在线电化学质谱(OEMS)研究发现, FEC增加了电极表面Li2CO3的溶解度, 因而促进了反应(1)、(2), 增加了体系中POF3和CO2的含量.而且, 由于化成过程中FEC在负极被还原, 覆盖了CO2的反应位点, 使得体系产生的CO2不能有效地被还原, 导致CO2气体累积. Xiong等[28]将充电态NMC111和石墨电极分别浸泡在电解液中, 并在高温下密封存储, 结果NMC111密封袋中产生大量CO2而石墨没有.同样条件下, 充电态的
|
$ {\rm{L}}{{\rm{i}}_{\rm{2}}}{\rm{C}}{{\rm{O}}_{\rm{3}}}{\rm{ + LiP}}{{\rm{F}}_{\rm{6}}} \to {\rm{3LiF + PO}}{{\rm{F}}_{\rm{3}}}{\rm{ + C}}{{\rm{O}}_{\rm{2}}} $ |
|
$ {\rm{L}}{{\rm{i}}_{\rm{2}}}{\rm{C}}{{\rm{O}}_{\rm{3}}}{\rm{ + P}}{{\rm{F}}_{\rm{5}}} \to {\rm{2LiF + PO}}{{\rm{F}}_{\rm{3}}}{\rm{ + C}}{{\rm{O}}_{\rm{2}}} $ |
NMC111/G软包电池也没有发现CO2, 这表明CO2可能在嵌锂态石墨负极被消耗了.因此作者推断, 在高温或高电压下, 如果NMC111/G中嵌锂态石墨负极没有类似地“清除”功能, 那么正极界面产气和阻抗增加将是电池恶化的主要原因.类似地, Xiong等[29]证明了FEC/TFEC (7, 图 1)电解液使得石墨负极表面SEI膜对CO2的“清除”能力降低.而且, 相比EC/EMC(碳酸甲乙酯)电解液, F代电解液使NMC/G产生了更多的H2.此外, Ma等[30]通过加速量热仪(ARC)研究嵌锂态石墨与电解液反应活性随温度的变化规律, 10% FEC使得放热反应温度降低到50 ℃, 且随着温度升高, 自加热速率(SHR)增大, 这说明FEC过量会导致上述分解反应更加剧烈, 因而, FEC在石墨负极中应用较少.
近年来, FEC在Si负极表面的成膜性受到极大关注[31]. FEC和VC一样, 可以提高NMC532/Si-G在4.2 V下的循环性能.分析循环后的Si-G负极发现, 虽然存在35%的容量损失, 但FEC降低了活性Li和Si的损失, 且循环后负极阻抗变化较小, 说明是正极阻抗增加导致了这部分容量损失[32, 33].而且, Kim等[25]研究发现少量FEC可以显著提高Si/Li的循环性能.一般地, FEC分解产生VC和HF.此外, FEC在Si负极表面发生化学还原生成聚乙炔和LiF, 以及电化学开环生成聚碳酸乙烯酯(CH2-CHOCO2Li)等物质.但过量的FEC在PF5等路易斯酸的催化下, 尤其高温时会产生更多酸性物质, 导致NMC532过渡金属的溶解, 并在Si-G负极表面沉积, 产生自放电, 使得负极电位升高, 引发更多的副反应.相比FEC, VC分解产物没有HF, 降低了对材料的腐蚀. Klett等[33]研究NMC532/Si-G发现, 随着循环次数增加, FEC的含量逐渐减少, 因而20% FEC在前100次循环没有导致较大容量衰减, 而后续循环过程中衰减明显.总之, 相比石墨负极, FEC在硅负极上成膜性更优异, 效果更佳, 这可能是由于石墨和硅负极表面物理化学性质不同导致的[34].
EMC为线性碳酸酯, 具有较低的熔点(-53 ℃)和黏度, 低温性能优异, 一般与介电常数高(黏度大)的环状碳酸酯如EC等混合使用. FEMC作为EMC的单氟取代物, F引入使得其极性增加、介电常数变大, FEMC (1.4 mPa•s)黏度大于EMC (0.65 mPa•s), 但其HOMO和LUMO能级都低于EMC, 因而其更抗氧化, 而更易被还原[8].
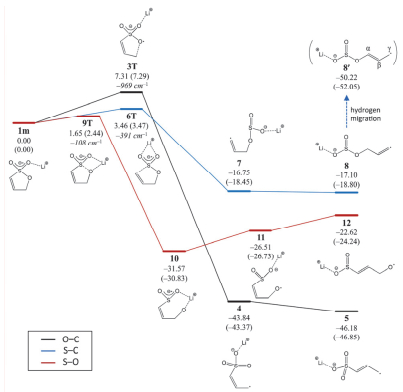
研究表明[35, 36], FEMC作为添加剂可在NMC532表面形成稳定的CEI膜, 抑制高电压循环中的结构变化, 提高NMC532/Li半电池的循环性.然而, FEMC对石墨负极兼容性较差, 容易导致电池首次效率降低, 一般配合SEI成膜添加剂如VC[36]来提高首效. Lee等[35]提出了FEMC在高电压下形成CEI膜的两种反应机理.机理1[见图 5(a)], FEMC的C-O首先发生单电子转移到金属阳离子(如Ni3+→Ni2+)上, 由于F的强吸电子效应, (CF3-O)失去电子后断裂, 形成的自由基进一步重排形成CO2等小分子.其中b与NMC532表面金属形成MF2 (M=Ni、Co及Mn), 同时也会通过亲核进攻形成M-CH2CF3. F-则易与Li+反应生成LiF, 或者直接进攻NMC532, 形成M-F.机理2[见图 5(b)], FEMC的C-O发生单电子转移反应, 生成的阳离子c, 进一步分解为d和气体(C2H4和CO2), d继续与EC或EMC, 以及Li+反应生成一系列有机物.总之, 上述分解产物MF2和C-F类物质优化并稳固了CEI膜结构.
除了FEMC, 其它含F物质一般作为EC等溶剂的共溶剂或者添加剂使用, 可提高电解液的抗氧化性和成膜性. Wang等[37, 38]报道了F-EPE (8, 图 1)和F-EAE (9, 图 1)两种部分氟代有机物作为添加剂, 它们具有高氧化稳定性和优良的负极成膜性, 使得NMC111/G在4.5 V下的循环性能得到提高.此外, Tornheim等[39]还对比了VC、TEP (31, 图 9)、TTE (10, 图 1)及TTFP (37, 图 9)等在4.6 V恒压控制下的漏电流大小, 结果显示:添加TTE和VC的电池恒压阶段的氧化漏电流均降低, 但二者机理不同, VC通过形成更稳定的CEI膜, 而TTE则由于本身具有强氧化稳定性.同时, VC和TTE容量保持率最低, 对应最小的氧化漏电流和最高的阻抗.这是因为实验中将N/P比提高到3, 使得正极发生电解液氧化时, 负极有足够的容量来维持电压平台, 而不产生极化, 因此容量的衰减主要是电极表面阻抗导致.这种实验方法有助于确定单一添加剂是否有效地钝化了正极界面.此外, Zheng等[40]研究发现, TFPM (11, 图 1)及TFPE (12, 图 1)在NMC111表面具有优良的成膜性能, 可以提高其4.6 V下的循环性能.
EC作为溶剂在石墨负极成膜性优于PC, 一直以来, 在低电压体系(<4.3 V)中有着广泛的应用.但其也有明显的缺点:一是熔点高(36.4 ℃)、黏度大, 因而低温性能较差; 二是与VC和FEC相比, 其氧化稳定性差, 容易产气及增加阻抗[41~43].随着对电池能量密度的需求的提高, 提高充电截止电压是一种有效方法, 但电极与电解液之间的副反应会严重降低电池的电化学性能.尤其电压大于4.5 V时, EC等碳酸酯的分解更剧烈, 因此亟待开发电位窗口更宽的新型溶剂[44].目前, 如砜类、腈类、离子液体及氟代物已经被广泛用作高电压溶剂, 它们含有强吸电子基团, 使得电解液氧化稳定性增强.但在无EC存在的情况下, 它们大多对石墨负极的兼容较差, 因而需要加入“助剂”优化SEI膜.文献报道中常见的有VC、FEC, PES (63, 图 13)、SA (89, 图 18)等[41], 本小节主要介绍含F有机物作为“助剂”的研究现状.
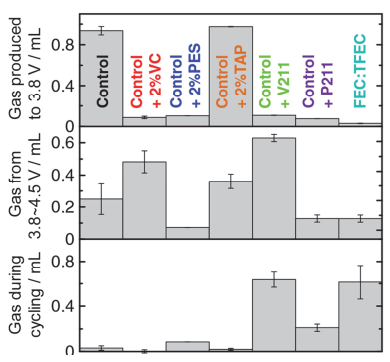
FEC是报道较多一种“助剂”, 相比VC, 其形成的SEI膜阻抗低. Liu等[45]认为, VC形成高阻抗的SEI膜, 引发锂沉积, 且在低温和高倍率下更明显.沉积的Li又与电解液反应, 消耗活性锂导致容量损失, 而FEC可以有效地抑制这个问题. Downie等[44]通过等温微热量法研究NMC442/G在不同电压下的寄生反应热, 相比EC/EMC电解液, FEC/TFEC体系低电压下严重产气, 而只有在电压>4.4 V时, 寄生反应热降低.而且, 如图 6所示, 含FEC电解液在高电压循环和高温存储中大量产气, 而传统电解液产气较少, 这表明FEC抑制了负极表面“清除”气体的功能(详见第3.1节).为此, Xia等[46]在FEC/TFEC中添加PES来抑制电池化成及存储过程中的产气.对称电池研究发现, 4.5 V下, 虽然NMC442正极在FEC/TFEC/PES电解液中能够保持稳定, 但负极的阻抗和产气量依然很大.因此如何降低产气和负极阻抗还需进一步研究[47].
除了FEC/TFEC, FEC/EMC体系也得到广泛研究. Xia等[41]在EMC基电解液中加入少量VC、FEC及DiFEC (13, 图 1)等作为“助剂”, 提高EMC基电解液对负极的兼容性. Ma等[42]在优化这些“助剂”的最佳添加量时发现, EMC在化成阶段有一个明显的还原峰, 其还原产物醇盐随后会发生酯交换反应, 通过这个反应可以检测“助剂”的含量是否可以有效地钝化负极. “助剂”的量过多会让其部分被氧化, 导致循环和存储中产气和阻抗增大; 过少又不能很好地钝化负极.研究认为, 其最佳含量范围是, VC (2%)、FEC和DiFEC (2%~5%).此外, 还有FEC/FEMC溶剂体系, 它在EC/EMC基础上全替换为氟代物. Im等[48]研究发现, FEMC作为主溶剂, 少量FEC作为添加剂, 在电压低于4.5 V时, NMC532/G的循环性能与EC/EMC相当或者更差; 而电压达到4.7 V, FEC/FEMC电解液可以显著提高电池的循环性能, 其优点主要体现在优化正极界面上(详见第3.2节), 但其负极成膜性较差, 因此FEMC基电解液还有待进一步研究.
总之, 含F添加剂得益于F原子的强电负性, 使得其抗氧化性能增强, 同时电解液在高电压下稳定性增加.然而, F代添加剂在不同温度、电压及正负极界面上的作用机理不同.而且, 在全氟代体系中, 正负极/电解液间的界面膜不稳定, 导致后续循环过程中阻抗、产气及电压降增加, 虽然加入一些“助剂”如VC、PES可以得到适当抑制, 但还需进一步优化.关于新型添加剂的开发, 一方面, 可以将F取代扩展到更多结构中, 进行多功能化, 如含Si、P、S、B、N等物质; 另一方面, 深入研究含F添加剂与其它添加剂的协同作用机理, 从而获得综合性能更优的电解液.
目前, LiPF6依然占据商业化锂离子电池锂盐的主导地位.这是因为其具有较高的溶解度和氧化稳定性, 以及适中的价格, 在所有锂盐中最具应用性, 但缺点是化学及热稳定性较差.随着对安全性和能量密度需求的不断提高, LiPF6基电解液已经不能满足高温和高电压的要求. Chrétien等[49]研究了LiPF6基电解液中锂盐的存在形式, 包括LiF、Li2CO3、LiOH、Li2O、LiOCH3及LiOC2H5等.发现NMC111对锂盐更加敏感, Li2CO3、LiOCH3及LiOC2H5对NMC111形成CEI膜有利, 而LiOH、Li2O对NMC111和石墨负极均有害. Ren等[50]还发现商业化LiPF6中的杂质LiCl会使石墨表面SEI膜阻抗增加, 恶化其循环稳定性, 而LiF杂质对SEI膜形成的影响较小.
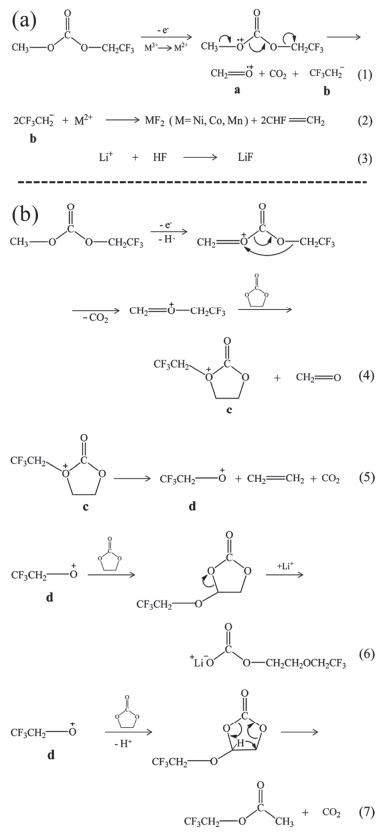
目前, 新型锂盐的研究主要是改变阴离子种类[51], 但或多或少都存在一些严重缺点, 并不能完全替代LiPF6, 因此大多数研究者只是将它们作为添加剂使用.本小节综述了近年来在三元材料中应用的锂盐添加剂, 包括B基盐LiBOB (14, 图 7)、LiODFB (15, 图 7)等, F代物LiTFSI (16, 图 7)、LiFSI (17, 图 7)等, 以及其它新型锂盐.
LiBOB最早由Xu等[52]在2001年报道, 是研究最广泛的一种锂盐.此前, 与其类似的B基锂盐被大量研究, 如LiBBB (18, 图 7)、LiFSB (19, 图 7)、LiBPB (20, 图 7)等, 但这些锂盐都因为溶解度或氧化稳定性较差等原因没有得到重视[8]. LiBOB在EC/DMC中只能溶解0.8 mol/L, 但在腈类、醚类及环状碳酸酯溶剂可溶解大于1 mol/L.其热稳定性较高, 对正负极均有较好的兼容性, 缺点是氧化稳定性较差(>4.2 V)[3, 8]. ARC表明[53, 54], LiBOB在石墨负极和充电态NMC111上热稳定性影响较小, 但对LCO影响较大, 这是由于LiBOB与含Co正极材料兼容性不好[55].此外, LiBOB对NMC和NCA材料也表现出不同的性质, 提高LiBOB的浓度, 脱锂态NMC的热稳定性降低, 而NCA正好相反. VC作为添加剂时也存在这个现象, 相反地是, 提高VC含量到10%, NMC111热稳定性基本不变, 但NCA却显著降低[55, 56].这可能与二者Co的含量以及Mn和Al不同有关, 具体原因有待继续研究.而另外一些研究通过排除Li负极的影响, 并认为LiBOB可在NMC111/Li中正极表面形成钝化层, 抑制电解液分解, 提高电池高电压下的循环性能[57, 58].相反, Xiang等[59]通过对比NCA/G与NCA/Li, 认为导致高倍率下NCA/Li容量衰减的原因在于Li负极表面恶化.因此提出用导电性更高的LiTFSI-LiBOB混合锂盐, 形成富含硫的SEI膜, 提高了Li+迁移速率.综上, LiBOB对LCO、NMC和NCA材料的影响有明显区别, Co和Mn的存在可能与LiBOB不兼容, 相比LCO和NMC, LiBOB可明显提高NCA材料热稳定性, 具体机理还需深入研究.
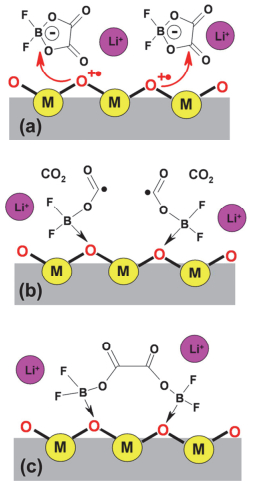
LiODFB是继LiBOB后, 另外一种具有前景的锂盐, 最早是由Central Glass公司报道.其兼具了LiBOB的成膜性和LiBF4的低温性能, 相比LiBOB, 在线性碳酸酯中溶解度更高, 因而电导率更高, 但仍然低于LiPF6.此外, 其钝化Al箔的能力更强、热稳定性更好[8]. LiODFB的成膜性与LiBOB类似, 在化成过程中, 分别在1.97 V和1.81 V左右被还原, 并参与形成SEI膜[60].而EIS表明, 全电池的阻抗大小顺序为: LiBOB>LiBF4>LiODFB>LiPF6. LiODFB阻抗更小, 一个原因是: LiBOB中一个草酸根被还原后, 另一个草酸根开环与其它BOB-发生电化学聚合, 引发交联反应, 使得阻抗增大; 而LiODFB只有一个草酸根, 不会发生类似交联反应[61, 62].深入研究发现, LiBOB和LiODFB在被还原过程中分别释放草酸根和F-, 促使形成了SEI膜内部无机层[63]. Lee[64]和Cha[65]等通过XPS确定LiODFB形成的SEI膜富含LiF.此外, ODFB-会与EC直接反应生成碳酸盐和聚合物, 形成SEI膜外部有机层.另外, LiODFB与LCO、NMC及LNMO[64~66]等多种正极材料兼容性较好. Shkrob等[63]提出LiODFB在金属氧化物表面的一种反应机理, 如图 8所示, 首先是M-O表面O空穴中心与ODFB-反应, 生成路易斯酸F2BOC·O自由基(图 8b), 随后与M-O表面的O配位, 同时相互之间通过两个自由电子结合成键, 并稳定地存在于M-O表面.此外, 由于LiODFB与PF5的络合作用大于EC, 因此可抑制PF5与溶剂之间发生副反应, 产生HF、CO2及C2H4等有害物质[67].而且, LiODFB还可抑制LiFSI对Al集流体的腐蚀(高电位下LiFSI与Al反应生成Al(FSI)3, 溶解到电解液中, 造成Al的损失), 其化成时优先在正极表面形成钝化膜(富含Al-F、B-O/B-F和Al2O3)[68, 69].综上, 这些优点使得LiODFB极具替代LiPF6的潜力.
除LiBOB、LiODFB之外, 文献报道的还有LiB(CN)4[70]、Li2B12F12-xHx[71, 72]等, 但由于它们溶解度较低、对电极兼容性差等原因没有得到广泛应用.最近报道[72]以Li2B12F12-xHx为锂盐, LiODFB为添加剂, 在富锂材料中表现出优异的抗过充和热稳定性, 但较高的成本限制了其进一步应用.
氟代酰亚胺锂盐最初由氟代亚胺锂盐发展而来, 如HN(COCF3)2 (21, 图 7), 最早在1973年被报道, 其Li+的解离度依赖于N原子上取代基的吸电子能力[8].而X-CO-N-CO-X (X=C或F)结构中, C=O对N的电荷离域能力较差, 导致锂盐溶解度较低, 用SO2取代C=O形成酰亚胺结构, X-SO2-N-SO2-X, 当X为氟代烷基和F时, 便形成了现在广泛研究的LiTFSI和LiFSI.其中LiFSI的溶解度更高, 而热稳定性稍低(C-F键比S-F键稳定).虽然它们对石墨负极兼容性较好, 但也存在严重缺点, 即电压大于3.5 V时, 它们会腐蚀Al集流体, 因而不适用于三元等高电压正极材料[3, 8], 而且LiTFSI和LiFSI均不能单独作为锂盐使用.
目前, 大量研究关注于如何消除其对Al集流体的腐蚀.一方面改变本征结构, 开发新型锂盐; 另一方面加入其它添加剂来抑制这个过程.一些新型锂盐包括LiFNFSI (22, 图 7)[3]、LiBETI (23, 图 7)[8]、LiN(SO2C3F6SO2) (24, 图 7)[8]等, 它们对Al集流体的腐蚀大大减小, 而且电导率较高, 但还没有得到充分研究.
另外, LiODFB[69, 73]、LiPF6[17, 74]等作为添加剂也能抑制腐蚀过程, 且抑制能力大小如下: LiDFOB>LiBF4 ≈LiPF6>LiBOB[73].此外, 酰亚胺锂盐本身热稳定性好、离子电导率高, 也可作为添加剂使用. LiTFSI和VC一起添加到LiPF6电解液中, 可减少产气, 提高高温存储性能[75].另外, LiFSI离子电导率较高, 可抑制电池在高倍率下循环时Li枝晶的形成[76].
除此之外, 还有一部分研究适配酰亚胺锂盐的溶剂体系. Petibon等[77]研究不含EC的LiPF6/LiFSI/EA(乙酸乙酯)电解液, 发现少量的LiPF6不但可以抑制LiFSI对Al箔的腐蚀, 而且LiFSI还可以避免EC在石墨负极被过度还原, 提高了石墨负极兼容性.进一步, Pohl等[78]提出LiTFSI/TMS-OPN (85, 图 17)电解液, NMC111/G在2.8~4.2 V下循环性能较好, 且对Al箔的腐蚀较小.
目前研究的新型锂盐添加剂种类繁多, 其中一些是LixPFyOz类似物, 另外一些是具有不同阴阳离子的锂盐.一方面, LixPFyOz实际上是LiPF6的分解产物, 也是SEI膜的组分之一.研究认为, PS (62, 图 13)和VC复合使用时, 电池循环性能提高, 这得益于SEI膜中LixPFyOz的含量提高, 且其在CEI膜和SEI膜中的含量有差别, 说明LiPF6在正负极的反应活性不同[79].基于此, Yang等[80]证明添加剂LiPO2F2参与了SEI和CEI膜的形成, 减少了界面膜中LiF的含量, 同时界面阻抗降低, 提高了电化学性能.类似地, PES (63, 图 13)[81]、LiDFP (25, 图 7)[82]、LiDMP (26, 图 7)[83]等均被证明具有类似的作用.
另一方面, 改变阴阳离子种类.首先是改变阴离子, 如LiDMSI (27, 图 7)[84]在4.3 V左右优先被氧化, NMC111/G高电压性能提高.此外, LiTFSM (28, 图 7)和LiPFSM (29, 图 7)本身在60 ℃以上热稳定性较好, 但其结构与LiTFSI类似, 可能在高电压下导致Al腐蚀, 不适用于高电压正极材料[85]. Xu等[86]提出LiTDI (30, 图 7)可以发生LiTDI-H2O配位, 消除电解液中残留水分.结果显示, 即使加入2000 mg/kg的水, 也没有观察到LiPF6的分解, 相应地, NMC/G在高电压及高温下的电化学性能提高.此外, 改变阳离子如CsPF6, 可以抑制PC对石墨负极的插层反应, 提高NCA/G在-40~60 ℃时的电化学性能[87].值得一提的是, Wagner等[88]将Mg(TFSI)2加入到LiPF6-EC/EMC电解液中, NMC111/G在4.6 V下循环性能明显提高, XPS和ToF-SIMS表明, Mg(TFSI)2参与形成了CEI膜, 且Mg2+的存在促进了PF6-的水解, 产生了更多的PFxOy-, 使得CEI膜更加稳定.这与传统认为金属离子会恶化电池性能有所相悖.
总之, LiPF6的地位在未来一段时间可能仍然不会改变, 但其对水敏感和热稳定性差的问题始终没有得到有效解决.要克服这个难题, 一方面, 现阶段, 大力开发新型电解液添加剂, 抑制LiPF6的分解, 不失为一种经济有效的手段; 另一方面, 从长远看, 需与溶剂、隔膜等结合, 开发适配电极材料的新型锂盐, 从而获得离子电导率更高、热稳定性更好及价格更低廉的电解液.
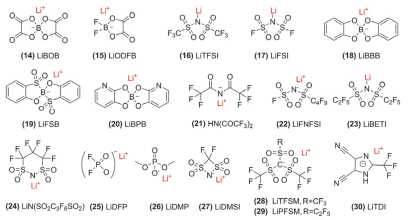
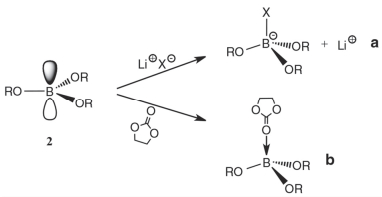
最早, 含P有机物如TEP (31, 图 9)等主要被用作锂离子电池电解液的阻燃共溶剂.随后发现, 少量不饱和含P有机物[如TAP (32, 图 9)]可以稳定电极/电解液界面, 提高电化学性能[3, 8].早期专利涉及到的含P添加剂还包括EDP (33, 图 9)、EPO (34, 图 9)等[8], 但并没有相关的机理研究.本节主要介绍近年来含P有机物作为电解液添加剂在三元材料中的应用及机理.
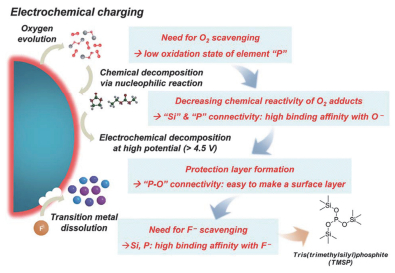
受TMSP (35, 图 9)可在Mg合金表面形成P-O-Mg键的启发, Yan等[89]将TMSP应用到NMC532正极材料中, 其在NMC表面发生P-O-M (M=Ni, Co, Mn)络合作用, 优化了CEI膜结构, 提高了电化学性能[90].因此TMSP的主要作用被认为是优化CEI膜[91], 而在全电池中机理则不同.通过DFT计算HOMO/LUMO能级可以预测物质的氧化还原难易程度, 但电解液组分间的H质子转移反应会改变物质的固有氧化还原电位[92, 93]. GC计算表明[93], Li+(TMSP)与Li+(EC)的溶剂化能相近(见表 2), 因此TMSP可能会取代EC, 形成由其主导的Li+溶剂化层, 而TMSP还原电位较低, 因而使得电解液的还原电位降低, 改变了SEI膜的成分和形成机制. TMSP被还原后, P-O断裂产生的自由基扩散至正极表面被氧化, 形成CEI膜.同时, 由于镍锰酸锂(LiNMO)正极表面的去质子化能垒较大, TMSP被氧化后, H质子更倾向转移到另外一个TMSP分子上引发聚合反应, 而不会继续在正极表面发生氧化, 使得表面酸性增加.
下载:
导出CSV
| Complex | G4MP2 | M05-2X/6-31+G** |
| Li+(EC) | 0.48~0.61 | 0.45~0.61 |
| Li+(DMC)(cis-cis) | 0.22~0.4 | 0.33 |
| Li+(DMC)(cis-trans) | 0.6 | |
| Li+(FEC) | 0.9 | 0.92 |
| Li+(FEC)(Li-F formed) | 2.25 | 1.7 |
| Li+(VC) | 0.80 | 0.23 |
| Li+(PS) | 0.46 | 0.04 |
| Li+(TMSP) | 0.64 | 0 |
| LiDFOB | 1.57 | 1.71 |
| (LiDFOB)2 | 2.12 | 2.18 |
| LiTDi | 0.75 | 0.72 |
| Li2TDi | 1.10 | 1.11 |
| TFSI- | 1.4 | |
| (Li+)2 TFSI-(LiF formed) | 2.1~2.9 | |
| (LiPF6)2(LiF formed) | 1.61 | 1.7 |
| (PF6-)(Li+)3(PF6-) | 2.1 | 2.2 |
| (LiBF4)2 | 0.0 | -0.4 |
| (BF4-) (Li+)3(BF4-) | 0.2~0.3 | -0.1 |
另外, TMSPi (36, 图 9)相比于TMSP, 具有不饱和P结构, 更容易被氧化, 具有更好的成膜性能[94]. Han等[95]通过EELS检测Mn的价态, 证明TMSPi参与了CEI膜的形成, 且抑制了富锂正极在循环中的不可逆相转变, 同时HF含量减少, 降低了其对正极的腐蚀[97]. Yim等[96]总结了TMSPi的4个主要作用机理(如图 10):第一, 三价P易被氧化, 从而消耗正极界面产生的氧气; 第二, TMSPi中亲电性的P和Si易与LixOy反应, 减少其引起的电解液分解; 第三, TMSPi在3.8 V左右氧化分解生成烷基磷(RO3P)保护层; 第四, TMSPi结构中的硅基醚(O-Si-C)可以与HF反应, 抑制其对过渡金属的腐蚀. Song等[98]研究了四种含磷添加剂与HF的反应活性, 结果如下: TMSPi>TFEP (37, 图 9)>TMP (38, 图 9)>TPP (39, 图 9). Kim等[99]进一步发现, TMSPi中的Si-O与HF的反应占主导地位, Si-O键断裂后生成的PO3类物质, 氧化稳定性较低, 它们会优先氧化形成致密的CEI膜.而且, 将TMSPi中-CH3替换为供电子能力更强的基团, 可更有效地利用Si-O键.随后, Han等[100]提出DFT筛选高电压含磷添加剂的条件: (1)氧化电位低于VC; (2)还原电位低于EC; (3)跟HF和LiF反应活性与TMSPi相当或更佳, 基于以上条件最终筛选出TTBP、TOP、T-adamantyl-P及DPDP (40~43, 图 9)四种成膜添加剂.此外, 虽然TEPi (44, 图 9)与TMSPi结构类似, 但其结构不含三甲基硅(TMS)官能团, 不能形成富含P和O的CEI膜, 也不能抑制金属氧自由基的形成以及电解液的H转移反应[101].
另外, TMSPi+VC复合添加剂具有协同作用. TMSPi可抑制正极界面Mn的溶解, 而VC可减少负极Mn的沉积, 因此提高了电池在60 ℃的电化学性能[102, 103]. Han等[104]阐明了二者的作用机理, TMSPi易氧化, 但较难被还原(还原电位低于EC), 因此不会在负极发生强烈副反应, 与VC复合, 发挥了各自的优点.一个新的研究发现, TMSPi在常温下便会与LiPF6自发地反应而被消耗, 同时其分子中TMS基团的存在, 导致被氧化形成的CEI组分中LixPFyOz含量较低, 降低了电池的电化学性能, 因此添加TMSPi的电解液不宜储存时间过长[105].
其它含磷添加剂的研究大多以P原子为中心, 在P-O键基础上改变取代基的种类. He等[106]对比了TEP和TTFP (37, 图 9), 前者无F取代, 后者是-CF3取代.结果表明, 相比于钝化表面活性位点, F取代对提高离子电导率的作用更大, 因为TEP虽然能氧化分解形成保护膜, 但这层膜的离子电导率低, 而F的引入使得TTFP的氧化产物一价磷不会继续氧化成二价或三价磷(高价态磷与Li+作用较强, 使得Li+不易解离), 因而CEI膜离子电导率较高[107]. Aspern等[108]研究了5F-TPrP、HFiP及THFPP (45~47, 图 9)三种氟代磷酸, 表明如果取代基为线性, 则形成较厚且蓬松的膜, 反之, 取代基有支链(如HFiP), 则形成较致密的膜.通过GC-MS分析, HFiP在循环过程中产生CF3CH(OH)CF3, 其生成会导致循环效率降低.除了F取代, 乙烯基取代的含磷添加剂如TAP[109, 110]在2.7 V附近(优先于EC)发生电化学交联聚合, 可改善SEI膜结构, 减少产气, 但过多的TAP也会增加负极阻抗. Mai等[111]认为, TPP (49, 图 9)中的苯基使得成膜较稳定, 但离子导电性差, 而TMP中P-O键有利于锂离子的传导, 基于此提出综合了二者优点的DMPP (50, 图 9). Wagner等[112]提出与LixPFyOz结构类似的DMFP和DEFP (51~52, 图 9), 二者与Li+络合后氧化电位降低, 有利于提高4.6 V下NMC111/Li的循环性能(详见第4.3节).此外, 环状含磷添加剂EEP (48, 图 9)[113], 兼具较好的阻燃性和成膜性.
综上, TMSP和TMSPi是含磷添加剂中研究最为深入的两种, 其P-O-Si键是活性官能团, 且Si-O键断裂比P-O键更有利.然而, 含磷添加剂对石墨等负极兼容性不好, 一般其作为共溶剂或者添加剂使用都需要另外加入其它负极成膜添加剂, TMSPi和VC的协同作用就是一个很明显的例子.其它含磷添加剂主要通过改变取代基, 希望能在电极表面形成更加稳定且离子电导率较高的保护膜, F取代是一个很好的选择.再者, 与LixPFyOz结构类似的含磷添加剂是一个新的研究方向, 如LiPO2F2, CEI膜中LixPFyOz含量增加, 相应电化学性能也提高.然而, LiPF6的分解产物纷繁复杂, 而且诸如LiPO2F2, 其在电解液与电极界面的作用机理还未知, 值得更加深入地研究.
B是一种相对原子质量轻且资源丰富的元素, 具有广阔的应用前景.如图 11所示, 由于B的空p轨道可以接受电子, 因此三价硼的化合物包括硼酸酯等都是路易斯酸.当B原子与强吸电子基团如氰基相连, 其路易斯酸性增强, 有利于锂盐的解离.硼酸酯类化合物实际上是一种阴离子受体, 与PF6-或F-络合, 可以提高Li+迁移数.此外, 硼酸酯还可以与路易斯碱如EC络合, 提高EC电解液的氧化稳定性[8].
TMSB (53, 图 12)是研究较多的一种添加剂, 其结构与TMSPi类似, 但中心原子B处于缺电子态, 使得TMSB显酸性, 可以络合PF6-或F-, 从而提高锂盐的解离度, 尤其是可以降低电极表面LiF的含量[114, 115].然而, 另外一些研究认为, TMSB是一种成膜添加剂, 在较低电压下优先氧化形成CEI膜, 改善高电压下循环性能[116~118], 还可抑制高电压下的自放电[119, 120], 但还需深入研究. Han等[121]认为, TMSB自身分解反应是热力学不利的, 即使在阴离子态下也很困难, 但它却极易与HF和LiF反应. Imholt等[122]研究了三种金属中心(B、Al及Ti)的硅氧烷, 发现只有TMSB的效率最低, 而且CEI膜厚度最小.他们认为, TMSB很可能在化成时直接与电解液反应, 而不是被氧化形成较厚的钝化膜. TMSB更多地是一种阴离子受体, 而不是成膜添加剂.
另外, Han等[123]根据物质氧化还原电位以及其与F-的结合能, 通过DFT计算, 从33种含硼化合物筛选出了5种性能优于TMSB的添加剂. Birrozzi等[124]研究了14种常见添加剂(见表 3), 发现只有PS和TMSB可以同时提高富锂材料的循环性能和效率.此外, Wang等[125]研究LCO/水系电解液时, 首次发现TMSB能在LCO表面成膜, 抑制高电压下的氧析出, 与有机电解液体系不同, TMSB没有与HF和LiF发生反应.
下载:
导出CSV
| Tested compound | Abbreviation | Concentration of additive/wt% of electrolyte | Influence on capacity retention | Influence on efficiency |
| 1, 3-Propane sultone | PS | 4 | + | + |
| Bis(trimethylsilyl)malonate | BTM | 4 | - | - |
| Diethyl(trimethylsil)amine | DTA | 4 | - | - |
| Fluoroethylene carbonate | FEC | 5 | - | + |
| Glutaric anhydride | GA | 4 | - | - |
| Lithium bis(oxalate)borate | LiBOB | 4 | - | - |
| Lithium difluoro (oxalate)borate | LiDFBOB | 4 | 0 | - |
| Methylphenyl carbonate | MPC | 4 | - | - |
| Pentafluorostyrene | PFS | 4 | - | - |
| Succinic anhydride | SA | 4 | 0 | + |
| Trimethylboroxine | TMB | 2 | - | - |
| Tris(trimethylsilyl) borate | SB | 4 | + | + |
| Tris(trimethylsilyl)phosphite | TTP | 2 | - | - |
| Vinylene carbonate | VC | 2 | 0 | 0 |
关于TMOBX (54, 图 12)的研究也较多, 而且发现其中B原子的最佳取代基是CH3O.研究表明[126], TMOBX可以降低全电池阻抗, 且与VC复合使用时能明显提高电池性能.对Graphite/Li研究发现[127], TMOBX含量越高, 碳负极的寄生反应越剧烈, 负极阻抗越大, 因此TMOBX必须与其它添加剂复合使用, 提高其对负极的兼容性. Petibon等[128]进一步发现, TMOBX能有效降低正极阻抗, 二者具有明显的协同效应.此时, TMOBX在CEI膜中占主导地位, 而VC则在SEI膜中占主导地位[129]. Ping等[130]研究了TMOBX对LCO/Li、NMC111/Li和NCA/Li三种体系的影响, 电压降曲线表明, 添加TMOBX后, 所有正极表面电解液氧化和穿梭反应速率都加剧了, 尤其是在60 ℃时.但TMOBX本身在首次循环过程中并没有发生剧烈氧化, 说明其在这些正极表面是稳定的.
PBF (55, 图 12)是一种新型高电压电解液添加剂, 包含有机碱吡啶-Py和-BF3两种活性基团.其中, Py能够和电解液中的Mn离子配位, 抑制其在负极表面被还原, 同时还能中和电解液中的酸性物质, 如PF5、HF及CO2等; BF3则是硼路易斯酸, 作为阴离子受体可溶解LiF, 降低界面阻抗[131, 132].实验表明[132]: NMC442/G在4.4 V和55 ℃时, PBF的循环性能远好于低电压下性能最好的复合添加剂如PES211 (详见第10节). Nie等[131]鉴于含硫添加剂如甲烷二磺酸亚甲酯(MMDS, 详见第7节)可降低阻抗, 提出PBF+MMDS/DTD等二元混合添加剂, 实验表明:复合后其阻抗比PBF更低.
为了进一步研究PBF中-Py和-BF3两种官能团对电池性能的影响, Nie等[133]增加PBF结构中-BF3的数量, 结果表明:电池阻抗降低, 但电化学性能如循环、充电容量均变差. Nie等[134]通过引入-PF5、-SO3及-BF3, 改变了路易斯酸官能团的种类, 结果发现-BF3具有最好的循环和最小的极化(充电-静置-放电, 充电至4.6 V), 随后依次是PF5、SO3.这说明改变BF3的数量或者种类均不能进一步提高PBF的性能.而且, 在PBF、TMP-BF (56, 图 12)和TPPO-BF (57, 图 12)三者中, 它们路易斯碱的极性依次减弱, 而极性最大的PBF的电化学性能最好.这是因为其化成时在负极被还原, 形成钝化膜导致初始化成阻抗增加, 因而SEI膜中的LiF、LiOR及Li2O的形成被抑制, 降低了后续循环和存储过程的阻抗[135].
另一种多氟取代的含硼添加剂TPFPB (61, 图 12)作为阴离子受体可以溶解电极与电解液界面膜中的LiF以及减少LiPF6的分解, 从而降低界面阻抗, 但其含量超过3%会促使PF6-分解产生PF5, 引发副反应, 对电池性能不利[136, 137].其它含硼添加剂大多是改变三价硼原子上的取代基, 如TMB、TMBi及TEB (58~60, 图 12)等[138~142], 均被证明可作为高电压电解液添加剂, 它们在较低电位时被氧化, 在正极表面形成一层较薄、阻抗低的保护膜, 但具体作用机理未明确.
总体而言, 含硼添加剂与含磷添加剂如TMSP的主要区别在于缺电子B中心, 使得其可作为阴离子受体, 提高锂盐的解离度及锂离子的迁移数.其作用机理更多是在表面发生化学反应, 降低体相及电极界面阻抗, 而不是一种成膜添加剂, 发生氧化还原反应.而且, 环状TMOBX相比TMSB有更多的BO3结构, 对正极表面阻抗降低明显, 而且可以和VC等负极成膜添加剂复合使用.综上, PBF是一种非常有潜力的添加剂, 其结构中包含了路易斯酸BF3和有机碱Py, 有利于抑制高电压下阻抗的增加, 并且与其它添加剂联用还可以进一步提高其性能.
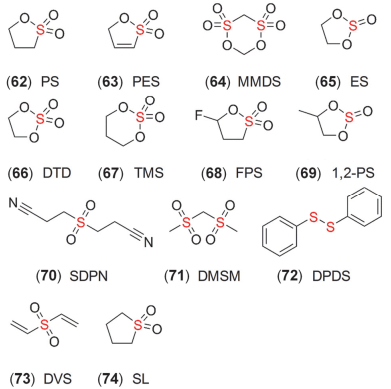
含硫有机物的种类繁多, 根据氧原子的数量、种类不同, 可分为硫化物、亚砜、砜、亚硫酸盐、硫酸盐及磺酸盐等.硫化物和亚砜极不稳定, 因此应用较少, 而砜类物质氧化稳定性较强, 在高电压溶剂中研究较多, 但其黏度较大, 一般需要其它溶剂作为共溶剂[8].磺酸内酯和硫酸盐是两类研究较多的添加剂, 代表性的有机物包括PS、PES、MMDS、ES、DTD及TMS (62~67, 图 13)等, 本小节阐述了这几类物质在三元材料中的应用及机理.
PS的研究始于其可以优化石墨负极SEI膜, 提高电解液与负极的兼容性[143, 144].深入分析发现[145], PS的还原产物包括Li2SO3、(CH-CH2-CH2-OSO3Li)2等, 这些物质组成的钝化层极性强, 对负极的黏着力好, 不易脱落, 而且优异的空间结构利于Li+传输.其中, Li+嵌入石墨电极包含几个连续的过程:首先是Li+在保护层的迁移扩散, 随后在表面层和电极界面电荷转移的作用下, 嵌入导电碳, 最终Li+通过固态扩散进入石墨.正因为这些富锂固态离子化合物存在空隙位点, 不仅可以作为保护层, 而且还可以加快锂离子的扩散, 降低极化, 抑制Li+在石墨负极表面沉积, 并促进嵌锂过程形成LiC6[146].基于此, PS还可以加入到离子液体电解液中, 提高其离子电导率[147]. Zhang等[148]认为PS还原产物RSO3Li锂离子电导率较高, 而VC的还原聚合产物热稳定性较好, 结果表明, VC+PS可提高高温电化学性能, 尤其可以抑制高温产气.这是由于溶剂EC本身在还原过程会产生C2H4, 而且VC在形成聚合物过程中也会产生CO2, 但PS的还原过程没有气体.此外, Jung等[149]发现F取代的FPS (68, 图 13)具有较高的氧化稳定性、成膜性以及热稳定性. Xu等[150]对经过“活化-高温存储”添加PS的电池进行TGA分析, 发现负极比正极失重更多, 这说明负极表面有机物更多, 印证了PS在负极优异的成膜性.但随后进行循环测试, 负极含S有机物显著降低, 这是因为高温存储时SEI膜结构被破坏, 而正极表面有机物增加, 无机物如LiF减少, 因此, PS对正极CEI膜的修饰作用是高温存储后电池循环性能提高的主要原因. Kang等[151]研究高镍NMC622正极时, 发现添加2% PS后, NMC622表面富含RSOSR和RSO2SR, 高温下的循环性能也得到提高.而Kim等[152]发现, 在EC基电解液中, 电解液还原产生的醇锂会引发生成可溶性的乙二醇双烷基碳酸酯类物质, 而它们会进一步与电解液组分反应, 生成低聚物, 导致SEI膜阻抗急剧增大, 而相比VC和FEC, PS对抑制这类物质的生成效果较差.此外, PS还可显著提高高电压富锂材料的电化学性能[124, 153, 154].
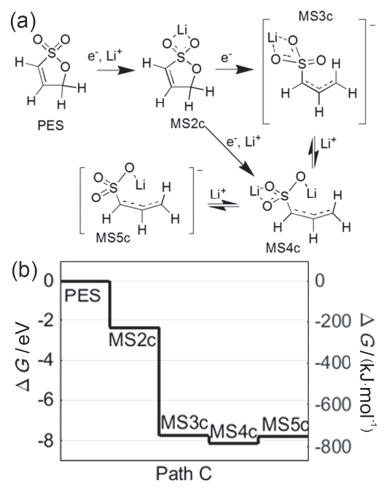
PES在PS结构基础上增加了双键, 理论计算表明[155, 156], PES更容易被还原, 添加3% PES后电池的综合电化学性能优于6% PS.与VC相比, PES在较低电压时(<4.3 V), 提高PES的含量可以降低高温产气[81].而且60 ℃存储时, PES比VC可以减少约90%的气体总量, 但是阻抗也随之增大[157, 158].深入分析发现[159]: NMC111/G产气主要集中在化成阶段, PES和VC均可抑制EC被还原产生C2H4, 而且VC还可抑制PES自身还原产生1-丙烯. Self等[160]将产气分为三个过程, 首先是低电压(3.7 V)负极界面产气, 如C2H4; 其次, 气体部分被消耗(详见第3.3节); 最后, 高电压时(4.3 V)正极界面再次产气, 如CO2.然而, 在无EC电解液FEC/TFEC中, 添加PES可以减少存储过程产气, 但会增加阻抗, 而且长时间循环及“充电-静置”过程产气仍然存在[46]. Self等[161]对PES理论计算结果表明:其氧化电位约为6.7 V, 但在充电至4.7 V时就发生分解, 这是因为正极表面在形成岩盐相时, 催化PES电化学氧化过程. Petibon等[162]分析发现PES在4.27 V时就开始被氧化, 其氧化产物会扩散至负极表面被还原, 使得负极SEI膜阻抗增加.这说明, SEI膜的性质会受正极氧化产物扩散的影响[163].另一方面, PES在负极经过两单电子步骤被还原, 形成Li2PES (MS4c)的过程, ∆G最低, 如图 14所示. GC-MS证实, MS4c还可与H•和CH3•生成丙烯、Li2SO3, 而且亲核性的MS4c还会与亲电性的PES、EC及EMC等发生反应生成有机硫酸盐, 参与形成SEI膜[161]. Han等[164]进一步研究PES的还原过程, 如图 15所示, PES只能通过O-C和S-C键断裂, 生成RSO3Li和ROSO2Li, 因为Li+参与形成稳定的七元环阻碍了其S-O键断裂, 表明Li+对PES的还原分解具有重要作用.
Li等[11]研究了高镍NMC811表面岩盐相结构形成, 与VC一样, PES可以明显抑制电极表面岩盐相结构生成, 但循环性没有明显改变, 说明容量衰减不仅仅是表面相转变导致, 电解液氧化等其它因素也是一个重要原因.
MMDS和PS、PES具有相同的官能团-SO3, 但其结构中-SO3的数量更多. MMDS在存储和循环过程中电池的阻抗更小, 因此, MMDS+VC比PS+VC电化学性能更佳[165].从Wang等[22]研究可知, MMDS单独作为添加剂效果不如VC, 因而关于它的研究并不多.少量报道认为其可改善高电压下NMC532及LCO表面CEI膜, 降低阻抗, 抑制后续电解液分解[166, 167].随后, Xia等[168]证明MMDS主要作用于NMC111表面, 存储过程中可以降低NMC111表面电解液氧化速率, 减少自放电.值得注意的是[165], 相比于PS, 添加MMDS的电池效率更高, 且MMDS与VC复合后, 效率和循环进一步提高, 而PS与VC复合则无明显提高.
其它含-SO4和-SO3基团的添加剂包括ES、DTD、TMS及1, 2-PS (69, 图 13)等, 作为单一添加剂, 只有DTD与VC性能相当[79, 169], 但它们与VC或PES复合使用时却能获得不错的效果(详见第10节).此外, SDPN (70, 图 13)可优先氧化形成CEI膜, 在4.6 V及55 ℃时具有优良的性能[170, 171].类似的还有DMSM、DPDS及DVS (71~73, 图 13)等[172~174].另外, 砜类SL (74, 图 13)作为添加剂可以提高EC基电解液的氧化稳定性[175].而且, SL替代溶剂EC, 加入VC等成膜添加剂后可以获得与EC基电解液相当的性能[169, 176].
总体而言, 含S添加剂的一个特点是其分解产物如RSO3Li等离子电导率较高.然而, S的价态多、结构多, 使得含S添加剂的分解路径与产物不同, 对正负极界面膜的组成和性质有较大影响, 还有待深入研究.一方面, 如何设计结构合理的含S添加剂, 使得化成及循环过程中的界面阻抗最低, 产气和电解液副反应最少, 是一个值得深入研究的方向.另一方面, 含S物质也常用于复合添加剂中, 抑制VC或FEC产气以及界面阻抗等问题, 但在高电压下停留时间过长, 界面膜不稳定.因此, 探索含S物质与其它添加剂的协同规律, 进行多元复合也是一种方向.此外, 砜类如SL等具有较高的氧化稳定性, 结合其它功能添加剂, 有望替代传统EC基电解液.
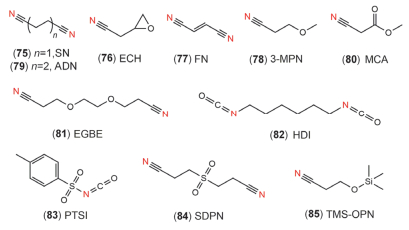
腈类物质的介电常数高, 同时黏度也较低, 因而具有较高的电导率.其中, 乙腈是除碳酸酯之外应用最多的溶剂, 其电导率(>30 mS/cm, 25 ℃)远大于碳酸酯溶剂(<10 mS/cm), 氧化稳定性较好.而腈类物质的缺点在于本身电化学窗口窄、还原稳定性较差[3, 43].因此实际应用中, 通常在其结构中引入乙烯基、烷氧基及硅氧基等官能团, 或结合其它成膜添加剂如LiBOB, VC或FEC等使用[177].
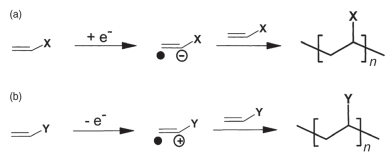
Santner等[178]在2003年提出乙烯基取代腈的还原机理主要是电化学聚合, 如图 16所示, 其中X为吸电子基团如-CN时, 为电化学还原聚合, 而当Y为强给电子基团, 则为氧化聚合.此外, 研究者发现, SN (75, 图 17)[179]、ECH (76, 图 17)[180]及FN (77, 图 17)[181]结构中强电负性的CN可与LCO、NMC等金属氧化物表面发生强络合作用形成CN-Co键, 抑制电解液直接接触正极, 从而提高了正极/电解液界面的热稳定性[182].其中ECH结构中引入烷氧基, 进一步提高其离子电导率.随后, 一系列烷氧基取代腈也被相继提出, 如3MPN、AND (78~79, 图 17)[43]、MCA (80, 图 17)[183]及EGBE (81, 图 17)[184~186]等, 它们的熔沸点和离子电导率较高、价格低廉, 但还原稳定性较差, 因此对石墨和Li负极的兼容性较差, 添加FEC或VC可提高石墨兼容性.其中, 添加5% VC可以抑制一元腈与金属Li反应生成可溶性二聚体、三聚体及低聚物.对于二元腈, 则无类似的副反应, 而是其相互间发生交联反应, 在Li表面形成不溶性的钝化膜[187]. Kim等[188]认为, 在4.2 V时, 腈类与VC相比, 循环和存储性能不具备优势; 但在4.5 V和60 ℃时, 电导率较高的短链腈如SN可以降低NMC442表面电解液氧化速率, 从而提高循环寿命.
向腈类结构引入其它官能团, 可提高腈类电解液的适用性. Liu等[189]将-CN替换为异氰酸, 得到HDI (82, 图 17), 其在4.6 V时原位在NMC111表面形成一层稳定的CEI膜, 提高电池的循环和倍率性能.此外, 在PTSI (83, 图 17)[190]和SDPN (84, 图 13)中引入-S=O, 可以提高界面离子电导率, 降低NMC532界面阻抗.而Pohl等[78]提出TMS-OPN (85, 图 17), 结合了腈的高氧化稳定性和高离子电导率, 以及硅烷的热稳定性和环境友好性能.总之, 腈类添加剂由于-CN的强吸电子效应, 使其氧化稳定性和离子电导率较高.通过在其结构中引入特定官能团或者与其它功能添加剂复合, 可进一步提高电解液的综合性能.
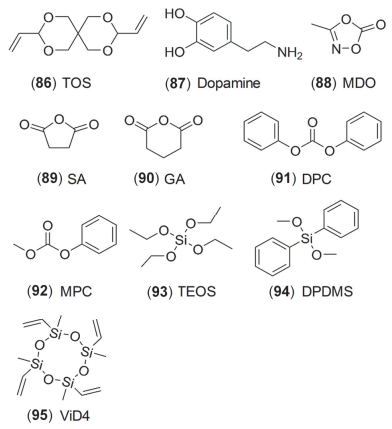
最近文献报道的其它的添加剂, 主要包括杂环化合物、酸酐及硅烷等. Qin等[191]提出TOS (86, 图 18)可以提高富锂NMC111/MCMB在55 ℃的循环性, 其主要作用在MCMB表面. Lee等[192]认为多巴胺(87, 图 18)可在NMC111正极表面形成聚合物, 添加0.1%便可明显提高4.5 V时的循环性能.其它类似的还有含N杂环化合物如MDO (88, 图 18)[193]等.鉴于EC基电解液带来的诸多问题(详见第3.3节), Xia等[194]研究EMC基电解液时发现: SA (89, 图 18)与VC相比, 电化学性能类似, 但产气量却极大降低. Peebles等[195]添加氧化电位较低的GA (90, 图 18), 不但可以降低正极表面LiF含量, 还可以增加LixPOyFz含量, 有利于电池的电化学性能.此外, 其还可抑制由于过渡金属催化而引起的NMC表面EMC酯交换反应.然而, Qiu等[196]发现, 很多适用于NMC111和NMC442的添加剂, 如PES211、PES及VC, 对于高镍材料如NMC811并不适用. DPC和MPC (91~92, 图 18)则对高镍材料适用性较好, 在石墨负极没有明显的还原反应, 说明其对SEI膜更多是修饰作用, 而没有参与成膜, 而且降低了正极表面副反应.
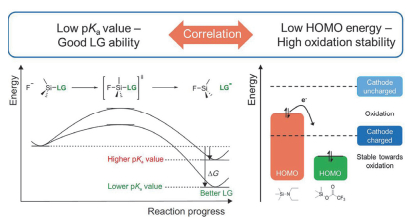
硅烷类有机物早期已经被应用到PC基电解液中, 其通过交联聚合反应来抑制PC对石墨负极的插层反应[197, 198].近年来, 一些硅烷也被应用到CEI膜的修饰中, 如TEOS[199]、DPDMS[200]及ViD4[201] (93~95, 图 18)等均可在正极界面形成稳定、低阻抗的CEI膜, 提高电池性能.此外, 如图 19所示, Gallus等[202]将离去基团(LG)能力和分子的氧化稳定性分别用pKa值和HOMO能级来表示:其中pKa值越小, LG的酸性越强, 越容易接受负电荷, TMSi与HF之间越容易发生双分子亲核取代反应(SN2@Si); 分子的HOMO能级越低, 其氧化稳定性越强.因此, 低pKa值可以提高添加剂与HF的反应活性, 同时, 低HOMO能级又能增强其氧化稳定性.这两个条件可以保证消除HF添加剂在高电压下的有效性.
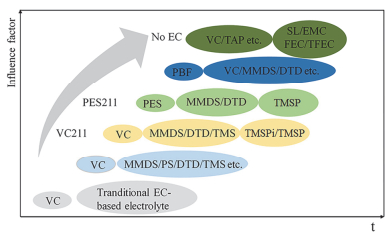
在锂离子电池的实际应用中, 单一的电解液添加剂很难获得满意的电化学性能, 或多或少都存在一些缺点.复合添加剂利用了各个物质间的协同作用, 是一种提高电池综合性能最简单有效的方法. Dahn课题组[22]在这方面做了大量研究, 如图 20所示, 其研究思路大体上是从LCO到NMC, 低镍到高镍, 低电压到高电压, 窄温差到宽温差.具体是从单一VC添加剂开始, 到VC二元复合, 再到VC、PES三元复合, 最后到PBF复合及无EC溶剂电解液.
Xia等[203]研究了含-SO4基团的DTD、TMS及PLS, 以及含-SO3的MMDS.它们单独使用时, 只有DTD性能与VC相当, 但化成阶段产气仍然较多. TMS、DTD及MMDS与VC复合后, NMC111/G在4.2 V时的电化学性能却优于2% VC, 尤其TMS+VC使得化成阶段几乎没有气体产生, 这可能是由于TMS中三碳原子环使得其结构更加稳定. Madec等[79]通过XPS与GC-MS得到类似上述结论.不同的是[204], 在LCO/G体系中, 电化学性能大小顺序为: VC+MMDS>2% VC>VC+DTD.
基于VC和TTSPi间的协同作用(详见第5.1节), Ma等[205]在二元复合添加剂的基础上, 提出VC211三元复合添加剂, 即2% VC+1% MMDS/DTD/TMS+1% TTSPi, 在2.8~4.2 V时, 各项电化学性能优于2% VC及二元复合添加剂, 其中含MMDS和DTD的复合添加剂在55 ℃时循环性明显优于含TMS.随后, Wang等[206]进一步提出4.2 V和40 ℃时的最优组合, 即2% VC+1% DTD+0.5% TTSP+0.5% TTSPi.
然而, 在更高电压(>4.2 V)和温度(>40 ℃)下, 上述含VC、DTD的复合添加剂在化成及循环过程中大量产气, 限制了其进一步应用. PES已经被证明产气比VC少, 其在NMC和石墨负极表面形成含S的保护膜, 可抑制产气和阻抗增大[23, 207].因而PES替代VC形成添加剂组合PES211, 在电压大于4.3 V时无明显的放热及阻抗变化, 而VC211的寄生反应热和阻抗随时间不断增加[208, 209]. Ma等[210]证明PES211提高了嵌锂态石墨负极的热稳定性, 在55 ℃下具有优异的电化学性能.然而, PES211对高镍正极如NMC811仍然不适用, 这说明过量的Ni与电解液添加剂之间有重要联系[211].同时, 表面包覆层与添加剂并不一定是协同作用, 如对于Al2O3包覆的NMC622/G, 加入PES211后, 电化学性能反而变差[212].在一种更苛刻的“充电-电压保持-放电”模式中, PES211表现出较大的容量损失、阻抗增加及产气量, 这说明在高电压阶段停留时间过长, 原先形成的含S钝化膜变得不稳定[23].随后, Nelson等[213]将PES211中的MMDS替换成DTD, 发现能降低高电压停留阶段的阻抗, 并可改善循环性能.此外, 他们还提出PES222 (2% PES+2% DTD+2% TTSPi), NMC442/G在“充电-电压保持-放电”过程中, 比LaPO4包覆产气更少, 阻抗更低[214].
鉴于VC及PES的复合添加剂在高电压和高温时应用受限.一方面, Nie等[131]提出了一种新的PBF+MMDS/DTD二元复合添加剂, 其在4.5 V及55 ℃下的循环性能比PES211更好.这得益于PBF中的BF3和有机碱Py[215].值得注意的是, 电解液氧化和阻抗是影响NMC/G高电压电化学性能的两个重要因素, 虽然PBF+MMDS/DTD降低了阻抗, 但是并没有抑制电解液氧化.另一方面, 由于EC是导致高电压下电解液剧烈分解的重要成分, 因此研究无EC电解液也是一种方向.其中砜类和氟代溶剂是研究较多的两个体系, Xia等[216]提出SL/EMC+1% VC电解液, 虽然优于EC/EMC, 但在4.5 V下仍然不如PES+EC/EMC, 这说明SL基电解液在高电压下依然存在问题.随后, Xia等[176]又提出SL/EMC+VC+TAP, 在4.5 V下电化学性能比PES211更好, 但R仍然较大.此外, 氟代溶剂体系如FEC/TFEC, 与PES211等相比, 高温高压下, 阻抗和产气更大, 这可能与FEC不稳定有关(详见第3.1节).因此, 从复合添加剂的研究历程看, 想要获得高温高电压下性能优异的电解液, 必须从新型溶剂、添加剂及合适的包覆材料三方面综合考虑[217].
电解液添加剂的研究始终伴随着材料的发展, 而三元层状氧化物作为目前最受关注的一种正极材料, 有着其独特的结构性质.可以看到, 电解液添加剂的研究体系也从LCO逐步过渡到NMC, 从低镍三元到高镍三元, 从低电压到高电压, 从低温到高温, 从单一到复合.总体而言, 虽然添加剂的含量相比电解液总量微小, 但其独特的化学结构能够发生化学或电化学反应, 对改善电池性能行之有效.与溶剂EC结构类似的VC, 其中C-C改变为C=C, 成膜性变好; 而F取代EC得到FEC, 不但氧化稳定性增强, 而且保留了EC特性; 新型不含F或热稳定性较高的锂盐, 避免添加剂及LiPF6分解产生HF; 含P、含B、含S及腈类等添加剂能够提高电极/电解液界面离子电导率、稳定性; 复合添加剂则利用各物质间的协同作用, 可极大地提升电池的综合电化学性能.此外, 各类新型结构添加剂也陆续出现, 可提高电池在高温、高电压等恶劣条件下的电化学性能.
虽然目前研究的添加剂种类繁多且性能较好, 但仍有一些问题值得关注:第一, 不同Ni含量的三元正极材料有着各异的表面物理化学性质, 尤其是Ni、Co与添加剂之间的催化或络合反应机理有较大异同; 第二, 正负极与电解液界面性质共同决定了电池的性能, 而添加剂对CEI和SEI膜形成机理不同, 而且二者之间还存在交联反应, 因此筛选添加剂时, 需同时考虑其氧化和还原性质, 而且有必要提出更加丰富具体的筛选条件, 如酸度系数pKa[202]、性能指数FOM[22]等; 第三, 在更高温度和电压下, 电解液氧化和阻抗增大是导致电池容量衰减的主要因素[214], 因此新型电解液添加剂的研究, 应该从物质结构与上述两个因素之间的关联出发.总之, 未来新型电解液添加剂的发展方向, 需要将电极材料、电解液, 甚至隔膜结合起来[218, 219], 设计结构与性质紧密联系的物质, 从而推动锂离子电池的进一步发展.
Zhang, S. S. J. Power Sources 2006, 162, 1379. doi: 10.1016/j.jpowsour.2006.07.074
Tan, S.; Ji, Y. J.; Zhang, Z. R.; Yang, Y. ChemPhysChem 2014, 15, 1956. doi: 10.1002/cphc.v15.10
Xu, K. Chem. Rev. 2014, 114, 11503. doi: 10.1021/cr500003w
Liu, W.; Oh, P.; Liu, X.; Lee, M.-J.; Cho, W.; Chae, S.; Kim, Y.; Cho, J. Angew. Chem.-Int. Ed. 2015, 54, 4440. doi: 10.1002/anie.201409262
Nitta, N.; Wu, F.; Lee, J. T.; Yushin, G. Mater. Today 2015, 18, 252. doi: 10.1016/j.mattod.2014.10.040
Hou, P.; Yin, J.; Ding, M.; Huang, J.; Xu, X. Small (Weinheim an der Bergstrasse, Germany) 2017, DOI: 10.1002/smll.20170180210.1002/smll.201701802.
李想, 葛武杰, 王昊, 瞿美臻, 无机材料学报, 2017, 32, 113. doi: 10.1360/N032017-00052?slug=abstractLi, X.; Ge, W.-J.; Wang, H.; Qu, M.-Z. J. Inorg. Mater. 2017, 32, 113. doi: 10.1360/N032017-00052?slug=abstract
Ue, M. ; Sasaki, Y. ; Tanaka, Y. ; Morita, M. Electrolytes For Lithium and Lithium-Ion Batteries, Spinger, New York, 2014.
Burns, J. C.; Petibon, R.; Nelson, K. J.; Sinha, N. N.; Kassam, A.; Way, B. M.; Dahn, J. R. J. Electrochem. Soc. 2013, 160, A1668. doi: 10.1149/2.031310jes
El Ouatani, L.; Dedryvere, R.; Siret, C.; Biensan, P.; Reynaud, S.; Iratcabal, P.; Gonbeau, D. J. Electrochem. Soc. 2009, 156, A103. doi: 10.1149/1.3029674
Li, J.; Liu, H.; Xia, J.; Cameron, A. R.; Nie, M.; Botton, G. A.; Dahn, J. R. J. Electrochem. Soc. 2017, 164, A655. doi: 10.1149/2.0651704jes
Burns, J. C.; Sinha, N. N.; Coyle, D. J.; Jain, G.; VanElzen, C. M.; Lamanna, W. M.; Xiao, A.; Scott, E.; Gardner, J. P.; Dahn, J. R. J. Electrochem. Soc. 2012, 159, A85. doi: 10.1149/2.028202jes
Downie, L. E.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A1782. doi: 10.1149/2.0301412jes
Lee, W. J.; Prasanna, K.; Jo, Y. N.; Kim, K. J.; Kim, H. S.; Lee, C. W. PCCP 2014, 16, 17062. doi: 10.1039/C4CP02075H
Xia, J.; Aiken, C. P.; Ma, L.; Kim, G. Y.; Burns, J. C.; Chen, L. P.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A1149. doi: 10.1149/2.108406jes
Madec, L.; Petibon, R.; Tasaki, K.; Xia, J.; Sun, J. P.; Hill, I. G.; Dahn, J. R. PCCP 2015, 17, 27062. http://www.ncbi.nlm.nih.gov/pubmed/26412322
Burns, J. C.; Sinha, N. N.; Jain, G.; Ye, H.; VanElzen, C. M.; Lamanna, W. M.; Xiao, A.; Scott, E.; Choi, J.; Dahn, J. R. J. Electrochem. Soc. 2012, 159, A1095. doi: 10.1149/2.077207jes
Deshpande, R. D.; Ridgway, P.; Fu, Y.; Zhang, W.; Cai, J.; Battaglia, V. J. Electrochem. Soc. 2015, 162, A330.
Qian, Y.; Schultz, C.; Niehoff, P.; Schwieters, T.; Nowak, S.; Schappacher, F. M.; Winter, M. J. Power Sources 2016, 332, 60. doi: 10.1016/j.jpowsour.2016.09.100
Peng, H. J.; Urbonaite, S.; Villevieille, C.; Wolf, H.; Leitner, K.; Novak, P. J. Electrochem. Soc. 2015, 162, A7072. doi: 10.1149/2.0061513jes
Qian, Y.; Niehoff, P.; Boerner, M.; Gruetzke, M.; Moennighoff, X.; Behrends, P.; Nowak, S.; Winter, M.; Schappacher, F. M. J. Power Sources 2016, 329, 31. doi: 10.1016/j.jpowsour.2016.08.023
Wang, D. Y.; Xia, J.; Ma, L.; Nelson, K. J.; Harlow, J. E.; Xiong, D.; Downie, L. E.; Petibon, R.; Burns, J. C.; Xiao, A.; Lamanna, W. M.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A1818. doi: 10.1149/2.0511412jes
Madec, L.; Ma, L.; Nelson, K. J.; Petibon, R.; Sun, J.-P.; Hill, I. G.; Dahn, J. R. J. Electrochem. Soc. 2016, 163, A1001. doi: 10.1149/2.1051606jes
马国强, 王莉, 张建君, 陈慧闯, 何向明, 丁元胜, 化学进展, 2016, 28, 1299. http://manu56.magtech.com.cn/progchem/CN/abstract/abstract11767.shtmlMa, G.; Wang, L.; Zhang, J.; Chen, H.; He, X.; Ding, Y. Prog. Chem. 2016, 28, 1299. http://manu56.magtech.com.cn/progchem/CN/abstract/abstract11767.shtml
Kim, K.; Park, I.; Ha, S.-Y.; Kim, Y.; Woo, M.-H.; Jeong, M.-H.; Shin, W. C.; Ue, M.; Hong, S. Y.; Choi, N.-S. Electrochim. Acta 2017, 225, 358. doi: 10.1016/j.electacta.2016.12.126
Wang, L.; Ma, Y.; Qu, Y.; Cheng, X.; Zuo, P.; Du, C.; Gao, Y.; Yin, G. Electrochim. Acta 2016, 191, 8. doi: 10.1016/j.electacta.2016.01.032
Streich, D.; Gueguen, A.; Mendez, M.; Chesneau, F.; Novak, P.; Berg, E. J. J. Electrochem. Soc. 2016, 163, A964. doi: 10.1149/2.0801606jes
Xiong, D. J.; Petibon, R.; Nie, M.; Ma, L.; Xia, J.; Dahn, J. R. J. Electrochem. Soc. 2016, 163, A546. doi: 10.1149/2.0951603jes
Xiong, D. J.; Ellis, L. D.; Petibon, R.; Hynes, T.; Liu, Q. Q.; Dahn, J. R. J. Electrochem. Soc. 2017, 164, A340. doi: 10.1149/2.1091702jes
Ma, L.; Xia, J.; Xia, X.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A1495. doi: 10.1149/2.0091410jes
Xu, C.; Lindgren, F.; Philippe, B.; Gorgoi, M.; Bjorefors, F.; Edstrom, K.; Gustafsson, T. Chem. Mater. 2015, 27, 2591. doi: 10.1021/acs.chemmater.5b00339
Klett, M.; Gilbert, J. A.; Trask, S. E.; Polzin, B. J.; Jansen, A. N.; Dees, D. W.; Abraham, D. P. J. Electrochem. Soc. 2016, 163, A875. doi: 10.1149/2.0271606jes
Klett, M.; Gilbert, J. A.; Pupek, K. Z.; Trask, S. E.; Abraham, D. P. J. Electrochem. Soc. 2017, 164, A6095. doi: 10.1149/2.0131701jes
Hall, D. S.; Glazier, S. L.; Dahn, J. R. PCCP 2016, 18, 11383. doi: 10.1039/C6CP01309K
Lee, Y.-M.; Nam, K.-M.; Hwang, E.-H.; Kwon, Y.-G.; Kang, D.-H.; Kim, S.-S.; Song, S.-W. J. Phys. Chem. C 2014, 118, 10631.
Nguyen, D.-T.; Kang, J.; Nam, K.-M.; Paik, Y.; Song, S.-W. J. Power Sources 2016, 303, 150. doi: 10.1016/j.jpowsour.2015.10.089
Wang, C.; Tang, S.; Zuo, X.; Xiao, X.; Liu, J.; Nan, J. J. Electrochem. Soc. 2015, 162, A1997. doi: 10.1149/2.0211510jes
Wang, C.; Zuo, X.; Zhao, M.; Xiao, X.; Yu, L.; Nan, J. J. Power Sources 2016, 307, 772. doi: 10.1016/j.jpowsour.2016.01.047
Tornheim, A.; He, M.; Su, C.-C.; Zhang, Z. J. Electrochem. Soc. 2017, 164, A6366. doi: 10.1149/2.0471701jes
Zheng, X. Z.; Huang, T.; Pan, Y.; Wang, W. G.; Fang, G. H.; Ding, K. N.; Wu, M. X. ACS Appl. Mater. Interfaces 2017, 9, 18758. doi: 10.1021/acsami.7b03014
Xia, J.; Petibon, R.; Xiong, D.; Ma, L.; Dahn, J. R. J. Power Sources 2016, 328, 124. doi: 10.1016/j.jpowsour.2016.08.015
Ma, L.; Glazier, S. L.; Petibon, R.; Xia, J.; Peters, J. M.; Liu, Q.; Allen, J.; Doig, R. N. C.; Dahn, J. R. J. Electrochem. Soc. 2017, 164, A5008. doi: 10.1149/2.0191701jes
Gmitter, A. J.; Plitz, I.; Amatucci, G. G. J. Electrochem. Soc. 2012, 159, A370. doi: 10.1149/2.016204jes
Downie, L. E.; Hyatt, S. R.; Dahn, J. R. J. Electrochem. Soc. 2016, 163, A35. doi: 10.1149/2.0081602jes
Liu, Q. Q.; Xiong, D. J.; Petibon, R.; Du, C. Y.; Dahn, J. R. J. Electrochem. Soc. 2016, 163, A3010. doi: 10.1149/2.0711614jes
Xia, J.; Nie, M.; Burns, J. C.; Xiao, A.; Lamanna, W. M.; Dahn, J. R. J. Power Sources 2016, 307, 340. doi: 10.1016/j.jpowsour.2015.12.132
Xia, J.; Petibon, R.; Xiao, A.; Lamanna, W. M.; Dahn, J. R. J. Power Sources 2016, 330, 175. doi: 10.1016/j.jpowsour.2016.09.012
Im, J.; Lee, J.; Ryou, M.-H.; Lee, Y. M.; Cho, K. Y. J. Elec-trochem. Soc. 2017, 164, A6381. doi: 10.1149/2.0591701jes
Chretien, F.; Jones, J.; Damas, C.; Lemordant, D.; Willmann, P.; Anouti, M. J. Power Sources 2014, 248, 969. doi: 10.1016/j.jpowsour.2013.09.092
任彤, 庄全超, 郝玉婉, 崔永丽, 化学学报, 2016, 74, 833. doi: 10.11862/CJIC.2016.110Ren, T.; Zhuang, Q.; Hao, Y.; Cui, Y. Acta Chim. Sinica 2016, 74, 833. doi: 10.11862/CJIC.2016.110
吴宇平, 戴晓兵, 马军旗, 程预江, 锂离子电池-应用与实践, 化学工业出版社, 北京, 2004, p. 222.Wu, Y. P. ; Dai, X. B. ; Ma, J. Q. ; Cheng, Y. J. Lithium Ion Batteries-Application and Practice, Chemical Industry Press, Beijing, 2004, p. 222.
Xu, W.; Angell, C. A. Electrochem. Solid State Lett. 2001, 4, L3. doi: 10.1149/1.1347858
Jiang, J.; Fortier, H.; Reimers, J. N.; Dahn, J. R. J. Electrochem. Soc. 2004, 151, A609. doi: 10.1149/1.1667520
Lu, W.; Chen, Z.; Joachin, H.; Prakash, J.; Liu, J.; Amine, K. J. Power Sources 2007, 163, 1074. doi: 10.1016/j.jpowsour.2006.09.010
Taeubert, C.; Fleischhammer, M.; Wohlfahrt-Mehrens, M.; Wietelmann, U.; Buhrmester, T. J. Electrochem. Soc. 2010, 157, A721. doi: 10.1149/1.3374666
Kim, G.-Y.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A1394. doi: 10.1149/2.0951409jes
Zhang, L.; Ma, Y.; Cheng, X.; Zuo, P.; Cui, Y.; Guan, T.; Du, C.; Gao, Y.; Yin, G. Solid State Ionics 2014, 263, 146. doi: 10.1016/j.ssi.2014.06.001
Li, C.; Hou, Q.; Li, S.; Tang, F.; Wang, P. J. Alloys Compd. 2017, 723, 887. doi: 10.1016/j.jallcom.2017.06.151
Xiang, H.; Shi, P.; Bhattacharya, P.; Chen, X.; Mei, D.; Bowden, M. E.; Zheng, J.; Zhang, J.-G.; Xu, W. J. Power Sources 2016, 318, 170. doi: 10.1016/j.jpowsour.2016.04.017
Abraham, D. P.; Furczon, M. M.; Kang, S. H.; Dees, D. W.; Jansen, A. N. J. Power Sources 2008, 180, 612. doi: 10.1016/j.jpowsour.2008.02.047
Qin, Y.; Chen, Z.; Liu, J.; Amine, K. Electrochem. Solid State Lett. 2010, 13, A11. doi: 10.1149/1.3261738
Mun, J.; Lee, J.; Hwang, T.; Lee, J.; Noh, H.; Choi, W. J. Electroanal. Chem. 2015, 745, 8. doi: 10.1016/j.jelechem.2015.02.034
Shkrob, I. A.; Zhu, Y.; Marin, T. W.; Abraham, D. P. J. Phys. Chem. C 2013, 117, 23750. doi: 10.1021/jp407714p
Lee, S. J.; Han, J.-G.; Lee, Y.; Jeong, M.-H.; Shin, W. C.; Ue, M.; Choi, N.-S. Electrochim. Acta 2014, 137, 1. doi: 10.1016/j.electacta.2014.05.136
Cha, J.; Han, J.-G.; Hwang, J.; Cho, J.; Choi, N.-S. J. Power Sources 2017, 357, 97. doi: 10.1016/j.jpowsour.2017.04.094
Wu, F.; Zhu, Q.; Li, L.; Chen, R.; Chen, S. J. Mater. Chem. A 2013, 1, 3659. doi: 10.1039/c3ta01182h
Liu, M.; Dai, F.; Ma, Z.; Ruthkosky, M.; Yang, L. J. Power Sources 2014, 268, 37. doi: 10.1016/j.jpowsour.2014.05.109
Park, K.; Yu, S.; Lee, C.; Lee, H. J. Power Sources 2015, 296, 197. doi: 10.1016/j.jpowsour.2015.07.052
Yan, G.; Li, X.; Wang, Z.; Guo, H.; Peng, W.; Hu, Q. J. Solid State Electrochem. 2015, 20, 507. doi: 10.1007/s10008-015-3069-3
Scheers, J.; Johansson, P.; Jacobsson, P. J. Electrochem. Soc. 2008, 155, A628. doi: 10.1149/1.2943214
Hayamizu, K.; Matsuo, A.; Arai, J. J. Electrochem. Soc. 2009, 156.
Chen, Z.; Ren, Y.; Jansen, A. N.; Lin, C.-k.; Weng, W.; Amine, K. Nat. Commun. 2013, 4.
Park, K.; Yu, S.; Lee, C.; Lee, H. J. Power Sources 2015, 296, 197. doi: 10.1016/j.jpowsour.2015.07.052
Forestier, C.; Grugeon, S.; Davoisne, C.; Lecocq, A.; Marlair, G.; Armand, M.; Sannier, L.; Laruelle, S. J. Power Sources 2016, 330, 186. doi: 10.1016/j.jpowsour.2016.09.005
Wang, D. Y.; Xiao, A.; Wells, L.; Dahn, J. R. J. Electrochem. Soc. 2015, 162, A169.
Miao, R.; Yang, J.; Xu, Z.; Wang, J.; Nuli, Y.; Sun, L. Sci. Rep. 2016, 6. http://www.ncbi.nlm.nih.gov/pubmed/26878890
Petibon, R.; Aiken, C. P.; Ma, L.; Xiong, D.; Dahn, J. R. Electrochim. Acta 2015, 154, 287. doi: 10.1016/j.electacta.2014.12.093
Pohl, B.; Gruenebaum, M.; Drews, M.; Passerini, S.; Winter, M.; Wiemhoefer, H.-D. Electrochim. Acta 2015, 180, 795. doi: 10.1016/j.electacta.2015.09.001
Madec, L.; Xia, J.; Petibon, R.; Nelson, K. J.; Sun, J.-P.; Hill, I. G.; Dahn, J. R. J. Phys. Chem. C 2014, 118, 29608. doi: 10.1021/jp509731y
Yang, G.; Shi, J.; Shen, C.; Wang, S.; Xia, L.; Hu, H.; Luo, H.; Xia, Y.; Liu, Z. RSC Adv. 2017, 7, 26052. doi: 10.1039/C7RA03926C
Li, B.; Wang, Y.; Tu, W.; Wang, Z.; Xu, M.; Xing, L.; Li, W. Electrochim. Acta 2014, 147, 636. doi: 10.1016/j.electacta.2014.09.151
Kim, K.-E.; Jang, J. Y.; Park, I.; Woo, M.-H.; Jeong, M.-H.; Shin, W. C.; Ue, M.; Choi, N.-S. Electrochem. Commun. 2015, 61, 121. doi: 10.1016/j.elecom.2015.10.013
Milien, M. S.; Tottempudi, U.; Son, M.; Ue, M.; Lucht, B. L. J. Electrochem. Soc. 2016, 163, A1369. https://www.sciencedirect.com/science/article/pii/B9780128095973002455
Murmann, P.; Streipert, B.; Kloepsch, R.; Ignatiev, N.; Sartori, P.; Winter, M.; Cekic-Laskovic, I. PCCP 2012, 17, 9352. http://www.ncbi.nlm.nih.gov/pubmed/25760031
Murmann, P.; Schmitz, R.; Nowak, S.; Ignatiev, N.; Sartori, P.; Celdc-Laskovic, I.; Winter, M. J. Electrochem. Soc. 2015, 162, A1738. doi: 10.1149/2.0261509jes
Xu, C.; Renault, S.; Ebadi, M.; Wang, Z.; Bjorklund, E.; Guyomard, D.; Brandell, D.; Edstrom, K.; Gustafsson, T. Chem. Mater. 2017, 29, 2254. doi: 10.1021/acs.chemmater.6b05247
Li, Q.; Jiao, S.; Luo, L.; Ding, M. S.; Zheng, J.; Cartmell, S. S.; Wang, C.-M.; Xu, K.; Zhang, J.-G.; Xu, W. ACS Appl. Mater. Interfaces 2017, 9, 18826. doi: 10.1021/acsami.7b04099
Wagner, R.; Streipert, B.; Kraft, V.; Jimenez, A. R.; Roeser, S.; Kasnatscheew, J.; Gallus, D. R.; Boerner, M.; Mayer, C.; Arlinghaus, H. F.; Korth, M.; Amereller, M.; Cekic-Laskovic, I.; Winter, M. Adv. Mater. Interfaces 2016, 3, 1. https://www.sciencedirect.com/science/article/pii/S0013468617305625
Yan, G.; Li, X.; Wang, Z.; Guo, H.; Wang, C. J. Power Sources 2014, 248, 1306. doi: 10.1016/j.jpowsour.2013.10.037
Liao, X.; Zheng, X.; Chen, J.; Huang, Z.; Xu, M.; Xing, L.; Liao, Y.; Lu, Q.; Li, X.; Li, W. Electrochim. Acta 2016, 212, 352. doi: 10.1016/j.electacta.2016.07.026
Rong, H.; Xu, M.; Xie, B.; Huang, W.; Liao, X.; Xing, L.; Li, W. J. Power Sources 2015, 274, 1155. doi: 10.1016/j.jpowsour.2014.10.123
Borodin, O.; Behl, W.; Jow, T. R. J. Phys. Chem. C 2013, 117, 8661. doi: 10.1021/jp400527c
Delp, S. A.; Borodin, O.; Olguin, M.; Eisner, C. G.; Allen, J. L.; Jow, T. R. Electrochim. Acta 2016, 209, 498. doi: 10.1016/j.electacta.2016.05.100
Mai, S.; Xu, M.; Liao, X.; Hu, J.; Lin, H.; Xing, L.; Liao, Y.; Li, X.; Li, W. Electrochim. Acta 2014, 147, 565. doi: 10.1016/j.electacta.2014.09.157
Han, J.-G.; Lee, S. J.; Lee, J.; Kim, J.-S.; Lee, K. T.; Choi, N.-S. ACS Appl. Mater. Interfaces 2015, 7, 8319. doi: 10.1021/acsami.5b01770
Yim, T.; Woo, S.-G.; Lim, S. H.; Cho, W.; Song, J. H.; Han, Y.-K.; Kim, Y.-J. J. Mater. Chem. A 2015, 3, 6157. doi: 10.1039/C4TA06531J
Zhu, Y.; Luo, X.; Xu, M.; Zhang, L.; Yu, L.; Fan, W.; Li, W. J. Power Sources 2016, 317, 65. doi: 10.1016/j.jpowsour.2016.03.090
Song, Y.-M.; Kim, C.-K.; Kim, K.-E.; Hong, S. Y.; Choi, N.-S. J. Power Sources 2016, 302, 22. doi: 10.1016/j.jpowsour.2015.10.043
Kim, D. Y.; Park, H.; Choi, W. I.; Roy, B.; Seo, J.; Park, I.; Kim, J. H.; Park, J. H.; Kang, Y. S.; Koh, M. J. Power Sources 2017, 355, 154. doi: 10.1016/j.jpowsour.2017.04.062
Han, Y.-K.; Yoo, J.; Yim, T. RSC Adv. 2017, 7, 20049. doi: 10.1039/C6RA28268G
Peebles, C.; Sahore, R.; Gilbert, J. A.; Garcia, J. C.; Tornheim, A.; Bareño, J.; Iddir, H.; Liao, C.; Abraham, D. P. J. Electrochem. Soc. 2017, 164, A1579. doi: 10.1149/2.1101707jes
Sinha, N. N.; Burns, J. C.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A1084. doi: 10.1149/2.087406jes
Koo, B.; Lee, J.; Lee, Y.; Kim, J. K.; Choi, N.-S. Electrochim. Acta 2015, 173, 750. doi: 10.1016/j.electacta.2015.05.129
Han, Y.-K.; Yoo, J.; Yim, T. J. Mater. Chem. A 2015, 3, 10900. doi: 10.1039/C5TA01253H
Qi, X.; Tao, L.; Hahn, H.; Schultz, C.; Gallus, D. R.; Cao, X.; Nowak, S.; Roeser, S.; Li, J.; Cekic-Laskovic, I.; Rad, B. R.; Winter, M. RSC Adv. 2016, 6, 38342. doi: 10.1039/C6RA06555D
He, M.; Su, C.-C.; Peebles, C.; Feng, Z.; Connell, J. G.; Liao, C.; Wang, Y.; Shkrob, I. A.; Zhang, Z. ACS Appl. Mater. Interfaces 2016, 8, 11450. doi: 10.1021/acsami.6b01544
Wang, L.; Ma, Y. L.; Li, Q.; Cui, Y. Z.; Wang, P. P.; Cheng, X. Q.; Zuo, P. J.; Du, C. Y.; Gao, Y. Z.; Yin, G. P. Electrochim. Acta 2017, 243, 72. doi: 10.1016/j.electacta.2017.05.008
von Aspern, N.; Röser, S.; Rezaei Rad, B.; Murmann, P.; Streipert, B.; Mönnighoff, X.; Tillmann, S. D.; Shevchuk, M.; Stubbmann-Kazakova, O.; Röschenthaler, G.-V.; Nowak, S.; Winter, M.; Cekic-Laskovic, I. J. Fluorine Chem. 2017, 198, 24. doi: 10.1016/j.jfluchem.2017.02.005
Xia, J.; Madec, L.; Ma, L.; Ellis, L. D.; Qiu, W.; Nelson, K. J.; Lu, Z.; Dahn, J. R. J. Power Sources 2015, 295, 203. doi: 10.1016/j.jpowsour.2015.06.151
Liu, Q. Q.; Petibon, R.; Du, C. Y.; Dahn, J. R. J. Electrochem. Soc. 2017, 164, A1173. doi: 10.1149/2.1081706jes
Mai, S.; Xu, M.; Liao, X.; Xing, L.; Li, W. J. Power Sources 2015, 273, 816. doi: 10.1016/j.jpowsour.2014.09.171
Wagner, R.; Korth, M.; Streipert, B.; Kasnatscheew, J.; Gallus, D. R.; Brox, S.; Arnereller, M.; Cekic-Laskovic, I.; Winter, M. ACS Appl. Mater. Interfaces 2016, 8, 30871. doi: 10.1021/acsami.6b09164
Gao, D.; Xu, J. B.; Lin, M.; Xu, Q.; Ma, C. F.; Xiang, H. F. RSC Adv. 2015, 5, 17566. doi: 10.1039/C4RA15899G
Zuo, X.; Fan, C.; Liu, J.; Xiao, X.; Wu, J.; Nan, J. J. Power Sources 2013, 229, 308. doi: 10.1016/j.jpowsour.2012.12.056
Yan, C.; Xu, Y.; Xia, J.; Gong, C.; Chen, K. J. Energy Chem. 2016, 25, 659. doi: 10.1016/j.jechem.2016.04.010
Rong, H.; Xu, M.; Xie, B.; Liao, X.; Huang, W.; Xing, L.; Li, W. Electrochim. Acta 2014, 147, 31. doi: 10.1016/j.electacta.2014.09.105
Li, J.; Xing, L.; Zhang, R.; Chen, M.; Wang, Z.; Xu, M.; Li, W. J. Power Sources 2015, 285, 360. doi: 10.1016/j.jpowsour.2015.03.113
Wang, K.; Xing, L.; Zhu, Y.; Zheng, X.; Cai, D.; Li, W. J. Power Sources 2017, 342, 677. doi: 10.1016/j.jpowsour.2016.12.112
Liao, X.; Huang, Q.; Mai, S.; Wang, X.; Xu, M.; Xing, L.; Liao, Y.; Li, W. J. Power Sources 2014, 272, 501. doi: 10.1016/j.jpowsour.2014.08.117
Liao, X.; Sun, P.; Xu, M.; Xing, L.; Liao, Y.; Zhang, L.; Yu, L.; Fan, W.; Li, W. Appl. Energy 2016, 175, 505. doi: 10.1016/j.apenergy.2016.03.114
Han, Y.-K.; Yoo, J.; Yim, T. Electrochim. Acta 2016, 215, 455. doi: 10.1016/j.electacta.2016.08.131
Imholt, L.; Roeser, S.; Boerner, M.; Streipert, B.; Rad, B. R.; Winter, M.; Cekic-Laskovic, I. Electrochim. Acta 2017, 235, 332. doi: 10.1016/j.electacta.2017.03.092
Han, Y.-K.; Lee, K.; Yoo, J.; Huh, Y. S. Theor. Chem. Acc. 2014, 133.
Birrozzi, A.; Laszczynski, N.; Hekmatfar, M.; von Zamory, J.; Giffin, G. A.; Passerini, S. J. Power Sources 2016, 325, 525. doi: 10.1016/j.jpowsour.2016.06.054
Wang, F.; Lin, Y.; Suo, L.; Fan, X.; Gao, T.; Yang, C.; Han, F.; Qi, Y.; Xu, K.; Wang, C. Energ. Environ. Sci. 2016, 9, 3666. doi: 10.1039/C6EE02604D
Burns, J. C.; Sinha, N. N.; Jain, G.; Ye, H.; VanElzen, C. M.; Lamanna, W. M.; Xiao, A.; Scott, E.; Choi, J.; Dahn, J. R. J. Electrochem. Soc. 2012, 159, A1105. doi: 10.1149/2.078207jes
Burns, J. C.; Xia, X.; Dahn, J. R. J. Electrochem. Soc. 2013, 160, A383. https://www.dal.ca/diff/dahn/publications.html
Petibon, R.; Aiken, C. P.; Sinha, N. N.; Burns, J. C.; Ye, H.; VanElzen, C. M.; Jain, G.; Trussler, S.; Dahn, J. R. J. Electrochem. Soc. 2013, 160, A117. doi: 10.1149/2.041308jes
Petibon, R.; Sinha, N. N.; Burns, J. C.; Aiken, C. P.; Ye, H.; VanElzen, C. M.; Jain, G.; Trussler, S.; Dahn, J. R. J. Power Sources 2014, 251, 187. doi: 10.1016/j.jpowsour.2013.11.054
Ping, P.; Xia, X.; Wang, Q. S.; Sun, J. H.; Dahn, J. R. J. Electro-chem. Soc. 2013, 160, A426. doi: 10.1149/2.041308jes
Nie, M.; Xia, J.; Dahn, J. R. J. Electrochem. Soc. 2015, 162, A1693. doi: 10.1149/2.0171509jes
Nie, M.; Xia, J.; Dahn, J. R. J. Electrochem. Soc. 2015, 162, A1186. doi: 10.1149/2.0271507jes
Nie, M.; Xia, J.; Ma, L.; Dahn, J. R. J. Electrochem. Soc. 2015, 162, A2066. doi: 10.1149/2.0411510jes
Nie, M.; Ma, L.; Xia, J.; Xiao, A.; Lamanna, W. M.; Smith, K.; Dahn, J. R. J. Electrochem. Soc. 2016, 163, A2124. doi: 10.1149/2.0041610jes
Nie, M.; Madec, L.; Xia, J.; Hall, D. S.; Dahn, J. R. J. Power Sources 2016, 328, 433. doi: 10.1016/j.jpowsour.2016.08.048
Pang, C.; Xu, G.; An, W.; Ding, G.; Liu, X.; Chai, J.; Ma, J.; Liu, H.; Cui, G. Energy Technol. 2017, 5, 1979. doi: 10.1002/ente.201700118
Chen, Z. H.; Amine, K. J. Electrochem. Soc. 2006, 153, A1221. doi: 10.1149/1.2194633
Wang, Z.; Xing, L.; Li, J.; Li, B.; Xu, M.; Liao, Y.; Li, W. Electrochim. Acta 2015, 184, 40. doi: 10.1016/j.electacta.2015.10.044
Yu, Q.; Chen, Z.; Xing, L.; Chen, D.; Rong, H.; Liu, Q.; Li, W. Electrochim. Acta 2015, 176, 919. doi: 10.1016/j.electacta.2015.07.058
Li, J.; Xing, L.; Chen, J.; Zhou, H.; Xu, M.; Li, W. J. Electrochem. Soc. 2016, 163, A2258. doi: 10.1149/2.0631610jes
Li, J.; Zhang, L.; Yu, L.; Fan, W.; Wang, Z.; Yang, X.; Lin, Y.; Xing, L.; Xu, M.; Li, W. J. Phys. Chem. C 2016, 120, 26899. doi: 10.1021/acs.jpcc.6b09097
Wang, Z.; Xing, L.; Li, J.; Xu, M.; Li, W. J. Power Sources 2016, 307, 587. doi: 10.1016/j.jpowsour.2015.11.091
Zuo, X.; Xu, M.; Li, W.; Su, D.; Liu, J. Electrochem. Solid-State Lett. 2006, 9, A196. doi: 10.1149/1.2170462
Lee, H.; Choi, S.; Choi, S.; Kim, H.-J.; Choi, Y.; Yoon, S.; Cho, J.-J. Electrochem. Commun. 2007, 9, 801. doi: 10.1016/j.elecom.2006.11.008
Leggesse, E. G.; Jiang, J.-C. RSC Adv. 2012, 2, 5439. doi: 10.1039/c2ra20200j
Park, G.; Nakamura, H.; Lee, Y.; Yoshio, M. J. Power Sources 2009, 189, 602. doi: 10.1016/j.jpowsour.2008.09.088
Kim, J. H.; Bae, S.-y.; Min, J.-H.; Song, S.-W.; Kim, D.-W. Electrochim. Acta 2012, 78, 11. doi: 10.1016/j.electacta.2012.05.161
Zhang, B.; Metzger, M.; Solchenbach, S.; Payne, M.; Meini, S.; Gasteiger, H. A.; Garsuch, A.; Lucht, B. L. J. Phys. Chem. C 2015, 119, 11337. doi: 10.1021/acs.jpcc.5b00072
Jung, H. M.; Park, S.-H.; Jeon, J.; Choi, Y.; Yoon, S.; Cho, J.-J.; Oh, S.; Kang, S.; Han, Y.-K.; Lee, H. J. Mater. Chem. A 2013, 1, 11975. doi: 10.1039/c3ta12580g
Xu, M.; Li, W.; Lucht, B. L. J. Power Sources 2009, 193, 804. doi: 10.1016/j.jpowsour.2009.03.067
Kang, K. S.; Choi, S.; Song, J.; Woo, S.-G.; Jo, Y. N.; Choi, J.; Yim, T.; Yu, J.-S.; Kim, Y.-J. J. Power Sources 2014, 253, 48. doi: 10.1016/j.jpowsour.2013.12.024
Kim, H.; Grugeon, S.; Gachot, G.; Armand, M.; Sannier, L.; Laruelle, S. Electrochim. Acta 2014, 136, 157. doi: 10.1016/j.electacta.2014.05.072
Yim, T.; Kim, S. H.; Woo, S.-G.; Lee, K.; Song, J. H.; Cho, W.; Kim, K. J.; Kim, J.-S.; Kim, Y.-J. RSC Adv. 2014, 4, 19172. doi: 10.1039/c4ra01441c
Pires, J.; Timperman, L.; Castets, A.; Peña, J. S.; Dumont, E.; Levasseur, S.; Dedryvère, R.; Tessier, C.; Anouti, M. RSC Adv. 2015, 5, 42088. doi: 10.1039/C5RA05650K
Li, B.; Xu, M.; Li, T.; Li, W.; Hu, S. Electrochem. Commun. 2012, 17, 92. doi: 10.1016/j.elecom.2012.02.016
Li, B.; Xu, M.; Li, B.; Liu, Y.; Yang, L.; Li, W.; Hu, S. Electrochim. Acta 2013, 105, 1. doi: 10.1016/j.electacta.2013.04.142
Nelson, K. J.; Xia, J.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A1884. doi: 10.1149/2.0791412jes
Xia, J.; Ma, L.; Aiken, C. P.; Nelson, K. J.; Chen, L. P.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A1634. doi: 10.1149/2.0541410jes
Madec, L.; Petibon, R.; Xia, J.; Sun, J. P.; Hill, I. G.; Dahn, J. R. J. Electrochem. Soc. 2015, 162, A2635. doi: 10.1149/2.0741512jes
Self, J.; Aiken, C. P.; Petibon, R.; Dahn, J. R. J. Electrochem. Soc. 2015, 162, A796. doi: 10.1149/2.0081506jes
Self, J.; Hall, D. S.; Madec, L.; Dahn, J. R. J. Power Sources 2015, 298, 369. doi: 10.1016/j.jpowsour.2015.08.060
Petibon, R.; Madec, L.; Rotermund, L. M.; Dahn, J. R. J. Power Sources 2016, 313, 152. doi: 10.1016/j.jpowsour.2016.02.054
Ellis, L. D.; Xia, J.; Louli, A. J.; Dahn, J. R. J. Electrochem. Soc. 2016, 163, A1686. doi: 10.1149/2.0851608jes
Han, Y.-K.; Yoo, J.; Jung, J. J. Phys. Chem. C 2016, 120, 28390. doi: 10.1021/acs.jpcc.6b07525
Xia, J.; Harlow, J. E.; Petibon, R.; Burns, J. C.; Chen, L. P.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A547. doi: 10.1149/2.049404jes
Zuo, X.; Fan, C.; Xiao, X.; Liu, J.; Nan, J. ECS Electrochem. Lett. 2012, 1, A50. doi: 10.1149/2.006203eel
Zuo, X.; Fan, C.; Xiao, X.; Liu, J.; Nan, J. J. Power Sources 2012, 219, 94. doi: 10.1016/j.jpowsour.2012.07.026
Xia, J.; Sinha, N. N.; Chen, L. P.; Kim, G. Y.; Xiong, D. J.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A84. https://www.dal.ca/diff/dahn/publications.html
Ding, Z.; Li, X.; Wei, T.; Yin, Z.; Li, X. Electrochim. Acta 2016, 196, 622. doi: 10.1016/j.electacta.2016.02.205
Huang, T.; Zheng, X.; Pan, Y.; Wang, W.; Fang, G.; Wu, M. Electrochim. Acta 2015, 156, 328. doi: 10.1016/j.electacta.2015.01.006
Zheng, X.; Huang, T.; Pan, Y.; Wang, W.; Fang, G.; Ding, K.; Wu, M. J. Power Sources 2016, 319, 116. doi: 10.1016/j.jpowsour.2016.04.053
Zheng, X.; Huang, T.; Pan, Y.; Wang, W.; Fang, G.; Wu, M. J. Power Sources 2015, 293, 196. doi: 10.1016/j.jpowsour.2015.05.061
Yim, T.; Kang, K. S.; Mun, J.; Lim, S. H.; Woo, S.-G.; Kim, K. J.; Park, M.-S.; Cho, W.; Song, J. H.; Han, Y.-K.; Yu, J.-S.; Kim, Y.-J. J. Power Sources 2016, 302, 431. doi: 10.1016/j.jpowsour.2015.10.051
Zuo, X.; Zhao, M.; Ma, X.; Xiao, X.; Liu, J.; Nan, J. Electrochim. Acta 2017, 245, 705. doi: 10.1016/j.electacta.2017.05.155
Cai, H.; Jing, H.; Zhang, X.; Shen, M.; Wang, Q. J. Electrochem. Soc. 2017, 164, A714. doi: 10.1149/2.0801704jes
Xia, J.; Dahn, J. R. J. Power Sources 2016, 324, 704. doi: 10.1016/j.jpowsour.2016.06.008
夏兰, 余林颇, 胡笛, 陈政, 化学学报, 2017, 75, 1183. doi: 10.6023/A17060284Xia, L.; Yu, L.; Hu, D.; George, C. Z. Acta Chim. Sinica 2017, 75, 1183. doi: 10.6023/A17060284
Santner, H. J.; Moller, K. C.; Ivanco, J.; Ramsey, M. G.; Netzer, F. P.; Yamaguchi, S.; Besenhard, J. O.; Winter, M. J. Power Sources 2003, 119, 368. doi: 10.1007/1-4020-4812-2_28
Kim, Y.-S.; Kim, T.-H.; Lee, H.; Song, H.-K. Energ. Environ. Sci. 2011, 4, 4038. doi: 10.1039/c1ee01272j
Nurpeissova, A.; Park, D. I.; Kim, S. S.; Sun, Y. K. J. Elec-trochem. Soc. 2015, 163, A171. doi: 10.1149/2.0431602jes
Wang, X.; Zheng, X.; Liao, Y.; Huang, Q.; Xing, L.; Xu, M.; Li, W. J. Power Sources 2017, 338, 108. doi: 10.1016/j.jpowsour.2016.10.103
Kim, Y.-S.; Lee, H.; Song, H.-K. ACS Appl. Mater. Interfaces 2014, 6, 8913. doi: 10.1021/am501671p
Brox, S.; Roeser, S.; Husch, T.; Hildebrand, S.; Fromm, O.; Korth, M.; Winter, M.; Cekic-Laskovic, I. ChemSusChem 2016, 9, 1704. doi: 10.1002/cssc.201600369
Hong, P.; Xu, M.; Zheng, X.; Zhu, Y.; Liao, Y.; Xing, L.; Huang, Q.; Wan, H.; Yang, Y.; Li, W. J. Power Sources 2016, 329, 216. doi: 10.1016/j.jpowsour.2016.07.111
Hong, P.; Xu, M.; Chen, D.; Chen, X.; Xing, L.; Huang, Q.; Li, W. J. Electrochem. Soc. 2017, 164, A137. doi: 10.1149/2.0531702jes
Wang, C.; Yu, L.; Fan, W.; Liu, J.; Ouyang, L.; Yang, L.; Zhu, M. ACS Appl. Mater. Interfaces 2017, 9, 9630. doi: 10.1021/acsami.6b16220
Rohan, R.; Kuo, T.-C.; Lin, J.-H.; Hsu, Y.-C.; Li, C.-C.; Lee, J.-T. J. Phys. Chem. C 2016, 120, 6450. doi: 10.1021/acs.jpcc.6b00980
Kim, G.-Y.; Dahn, J. R. J. Electrochem. Soc. 2015, 162, A437. doi: 10.1149/2.0651503jes
Liu, Y.; Qin, Y.; Peng, Z.; Zhou, J.; Wan, C.; Wang, D. J. Mater. Chem. A 2015, 3, 8246. doi: 10.1039/C4TA07055K
Dong, P.; Wang, D.; Yao, Y.; Li, X.; Zhang, Y.; Ru, J.; Ren, T. J. Power Sources 2017, 344, 111. doi: 10.1016/j.jpowsour.2017.01.116
Qin, Y.; Chen, Z.; Lu, W.; Amine, K. J. Power Sources 2010, 195, 6888. doi: 10.1016/j.jpowsour.2010.04.040
Lee, H.; Han, T.; Cho, K. Y.; Ryou, M.-H.; Lee, Y. M. ACS Appl. Mater. Interfaces 2016, 8, 21366. doi: 10.1021/acsami.6b06074
Röser, S.; Lerchen, A.; Ibing, L.; Cao, X.; Kasnatscheew, J.; Glorius, F.; Winter, M.; Wagner, R. Chem. Mater. 2017, 29, 7733. doi: 10.1021/acs.chemmater.7b01977
Xia, J.; Liu, Q.; Hebert, A.; Hynes, T.; Petibon, R.; Dahn, J. R. J. Electrochem. Soc. 2017, 164, A1268. doi: 10.1149/2.1341706jes
Peebles, C.; He, M.; Feng, Z.; Su, C.-C.; Zeng, L.; Bedzyk, M. J.; Fenter, P.; Wang, Y.; Zhang, Z.; Liao, C. J. Electrochem. Soc. 2017, 164, A173. doi: 10.1149/2.0721702jes
Qiu, W.; Xia, J.; Chen, L.; Dahn, J. R. J. Power Sources 2016, 318, 228. doi: 10.1016/j.jpowsour.2016.03.105
Qin, X.-Y.; Wang, J.-L.; Tang, D.-P.; Mai, Y.-J.; Zhang, L.-Z. J. Zhejiang Univ.-Sci. A 2013, 14, 514. doi: 10.1631/jzus.A1300026
秦雪英, 汪靖伦, 张灵志, 化学进展, 2012, 24, 810. http://manu56.magtech.com.cn/progchem/CN/abstract/abstract10844.shtmlQin, X.; Wang, J.; Zhang, L. Prog. Chem. 2012, 24, 810. http://manu56.magtech.com.cn/progchem/CN/abstract/abstract10844.shtml
Wang, Z.; Huang, Y.; Wang, X.; Jia, D.; Guo, Z.; Miao, M. Solid State Ionics 2013, 232, 19. doi: 10.1016/j.ssi.2012.11.017
Deng, B.; Wang, H.; Ge, W.; Li, X.; Yan, X.; Chen, T.; Qu, M.; Peng, G. Electrochim. Acta 2017, 236, 61. doi: 10.1016/j.electacta.2017.03.155
Wang, H.; Sun, D.; Li, X.; Ge, W.; Deng, B.; Qu, M.; Peng, G. Electrochim. Acta 2017, 254, 112. doi: 10.1016/j.electacta.2017.09.111
Gallus, D. R.; Wagner, R.; Wiemers-Meyer, S.; Winter, M.; Cekic-Laskovic, I. Electrochim. Acta 2015, 184, 410. doi: 10.1016/j.electacta.2015.10.002
Xia, J.; Sinha, N. N.; Chen, L. P.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A264. https://www.dal.ca/diff/dahn/publications.html
Xia, J.; Petibon, R.; Sinha, N. N.; Dahn, J. R. J. Electrochem. Soc. 2015, 162, A2227. doi: 10.1149/2.0151512jes
Ma, L.; Wang, D. Y.; Downie, L. E.; Xia, J.; Nelson, K. J.; Sinha, N. N.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A1261. doi: 10.1149/2.0541409jes
Wang, D. Y.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A1890. doi: 10.1149/2.0841412jes
Xia, J.; Ma, L.; Dahn, J. R. J. Power Sources 2015, 287, 377. doi: 10.1016/j.jpowsour.2015.04.070
Downie, L. E.; Hyatt, S. R.; Wright, A. T. B.; Dahn, J. R. J. Phys. Chem. C 2014, 118, 29533. doi: 10.1021/jp508912z
Ma, L.; Xia, J.; Dahn, J. R. J. Electrochem. Soc. 2014, 161, A2250. doi: 10.1149/2.1041414jes
Ma, L.; Xia, J.; Dahn, J. R. J. Electrochem. Soc. 2015, 162, A1170. doi: 10.1149/2.0181507jes
Ma, L.; Self, J.; Nie, M.; Glazier, S.; Wang, D. Y.; Lin, Y.-S.; Dahn, J. R. J. Power Sources 2015, 299, 130. doi: 10.1016/j.jpowsour.2015.08.084
Arumugam, R. S.; Ma, L.; Li, J.; Xia, X.; Paulsen, J. M.; Dahn, J. R. J. Electrochem. Soc. 2016, 163, A2531. doi: 10.1149/2.0171613jes
Nelson, K. J.; d'Eon, G. L.; Wright, A. T. B.; Ma, L.; Xia, J.; Dahn, J. R. J. Electrochem. Soc. 2015, 162, A1046. doi: 10.1149/2.0831506jes
Nelson, K. J.; Abarbanel, D. W.; Xia, J.; Lu, Z.; Dahn, J. R. J. Electrochem. Soc. 2016, 163, A272. doi: 10.1149/2.0691602jes
Xiong, D. J.; Ellis, L. D.; Nelson, K. J.; Hynes, T.; Petibon, R.; Dahn, J. R. J. Electrochem. Soc. 2016, 163, A3069. doi: 10.1149/2.1031614jes
Xia, J.; Self, J.; Ma, L.; Dahn, J. R. J. Electrochem. Soc. 2015, 162, A1424. doi: 10.1149/2.0121508jes
Xia, J.; Ma, L.; Nelson, K. J.; Nie, M.; Lu, Z.; Dahn, J. R. J. Electrochem. Soc. 2016, 163, A2399. doi: 10.1149/2.1211610jes
黄佳琦, 孙滢智, 王云飞, 张强, 化学学报, 2017, 75, 173. doi: 10.7503/cjcu20160462Huang, J.; Sun, Y.; Wang, Y.; Zhang, Q. Acta Chim. Sinica 2017, 75, 173. doi: 10.7503/cjcu20160462
向兴德, 卢艳莹, 陈军, 化学学报, 2017, 75, 154. http://sioc-journal.cn/Jwk_hxxb/EN/abstract/abstract345659.shtmlXiang, X.; Lu, Y.; Chen, J. Acta Chim. Sinica 2017, 75, 154. http://sioc-journal.cn/Jwk_hxxb/EN/abstract/abstract345659.shtml

图 1 VC、PC及氟代溶剂或添加剂的分子结构
Figure 1 Molecular structures of VC, PC, and fluoro-substituted solvent and additives


图 3 电池充放电末容量差随时间的变化图, 红色表示添加1% VC的电解液, 蓝色表示对照组电解液[18]
Figure 3 Figure of difference value between charge and discharge endpoint capacity for cells against cycling time, with 1 wt% VC additive in the electrolyte (red) and with baseline electrolyte (blue)[18]
Capacity was measured at the rate of C/10 for 2 cycles after every 40 cycles at the rate of C/2[18]


图 6 含不同电解液组分的NMC442/Graphite电池在量热仪中第一阶段充电至3.8 V(顶部), 第二阶段3.8~4.5 V(中间)以及循环过程中的产气(底部)[44]
Figure 6 Gas produced during the first formation process (to 3.8 V, top), during the second formation process (between 3.8~4.5 V, middle), and after all cycling process was completed in the calorimeter (bottom) for NMC442/graphite cells containing different electrolyte compositions[44]


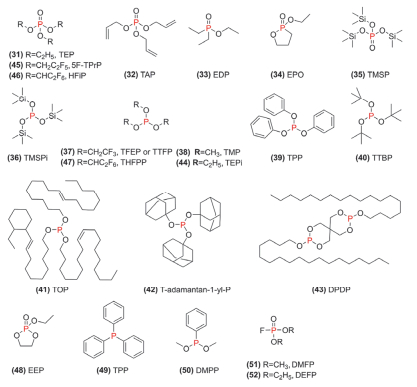
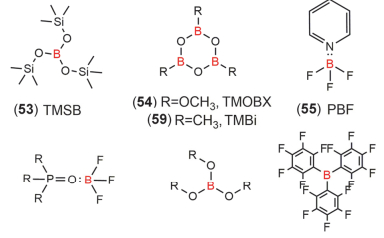
图 9 一些含磷添加剂的化学结构式
Figure 9 Chemical structural formula of some phosphorus-containing additives



图 12 一些含硼添加剂的化学结构式
Figure 12 Chemical structural formula of some boron-containing additives

图 13 一些含硫添加剂的化学结构式
Figure 13 Chemical structural formula of some sulfur-containing additives



图 16 (a) 在阴极, 乙烯的α位置取代基X为吸电子基团时, 促进其发生还原聚合; (b)在阳极, 乙烯的α位置取代基Y为给电子基团时, 促进其发生氧化聚合[178]
Figure 16 (a) Cathodic, when the functional group ''X'' in α-position to the vinylene group is electron-withdrawing, reduction induced polymerization of vinylene monomers is facilitated; (b) anodic, when the functional group "Y" in α-position to the vinylene group is electron-pushing, oxidation induced polymerization of vinylene monomers is promoted[178]

表 1 不同电解液组分EC、VC、ES及EMC在有无Li存在情况下的还原能(∆Ered, kcal•mol-1)和溶剂化能(∆Esolv, kcal•mol-1)[16]
Table 1. DFT Calculation of reduction energy (∆Ered, kcal•mol-1) and solvation energy (∆Esolv, kcal•mol-1) in presence of Li or not for different electrolytes components (EC, VC, ES, EMC)[16]
| EC | VC | ES | EMC | |
| ΔEred, without Li*/(kcal"mol-1) | 26.9 | 26.4 | -0.7 | 34.9 |
| ΔEred, with Li**/(kcal"mol-1) | -89.5 | -88.1 | -131.8 | -91.5 |
| ΔEsolv/(kcal"mol-1) | -47.1 | -43.9 | -42.7 | -42.6 |
| The calculation of ∆Ered and ∆Esolv is based on the MP2/6-311+G(2d, p) level in gas phase. The geometry was optimized at the B3LYP/6-311+G(2d, p) level[16]. | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 量化(GC)计算得到的还原电位(vs. Li/Li+)[93]
Table 2. Calculated reduction potentials vs. Li/Li+ from Quantum chemistry (GC)[93]
| Complex | G4MP2 | M05-2X/6-31+G** |
| Li+(EC) | 0.48~0.61 | 0.45~0.61 |
| Li+(DMC)(cis-cis) | 0.22~0.4 | 0.33 |
| Li+(DMC)(cis-trans) | 0.6 | |
| Li+(FEC) | 0.9 | 0.92 |
| Li+(FEC)(Li-F formed) | 2.25 | 1.7 |
| Li+(VC) | 0.80 | 0.23 |
| Li+(PS) | 0.46 | 0.04 |
| Li+(TMSP) | 0.64 | 0 |
| LiDFOB | 1.57 | 1.71 |
| (LiDFOB)2 | 2.12 | 2.18 |
| LiTDi | 0.75 | 0.72 |
| Li2TDi | 1.10 | 1.11 |
| TFSI- | 1.4 | |
| (Li+)2 TFSI-(LiF formed) | 2.1~2.9 | |
| (LiPF6)2(LiF formed) | 1.61 | 1.7 |
| (PF6-)(Li+)3(PF6-) | 2.1 | 2.2 |
| (LiBF4)2 | 0.0 | -0.4 |
| (BF4-) (Li+)3(BF4-) | 0.2~0.3 | -0.1 |
下载: 导出CSV
表 3 不同物质作为添加剂对正极电化学性能的影响, (+)、(−)和(0)分别表示正面影响、负面影响以及无影响[124]
Table 3. List of electrolyte additives on the electrochemical performances of the cathodes, among which (+), (−) and (0) indicate a positive, negative and a null effect, respectively[124]
| Tested compound | Abbreviation | Concentration of additive/wt% of electrolyte | Influence on capacity retention | Influence on efficiency |
| 1, 3-Propane sultone | PS | 4 | + | + |
| Bis(trimethylsilyl)malonate | BTM | 4 | - | - |
| Diethyl(trimethylsil)amine | DTA | 4 | - | - |
| Fluoroethylene carbonate | FEC | 5 | - | + |
| Glutaric anhydride | GA | 4 | - | - |
| Lithium bis(oxalate)borate | LiBOB | 4 | - | - |
| Lithium difluoro (oxalate)borate | LiDFBOB | 4 | 0 | - |
| Methylphenyl carbonate | MPC | 4 | - | - |
| Pentafluorostyrene | PFS | 4 | - | - |
| Succinic anhydride | SA | 4 | 0 | + |
| Trimethylboroxine | TMB | 2 | - | - |
| Tris(trimethylsilyl) borate | SB | 4 | + | + |
| Tris(trimethylsilyl)phosphite | TTP | 2 | - | - |
| Vinylene carbonate | VC | 2 | 0 | 0 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们