Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Collaborative Innovation Center of Chemistry for Energy Materials, Department of Chemistry, Fudan University, Shanghai 200433
b.
Shanghai Key Laboratory of Materials Protection and Advanced Materials in Electric Power, College of Environmental and Chemical Engineering, Shanghai University of Electric Power, Shanghai 200090
Received Date:
02 November 2017 Available Online:
15 April 2018
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21473039, 21733004), Science and Technology Commission of Shanghai Mu-nicipality (No. 17520711200) and 973 program (No. 2015CB932303)
Abstract:
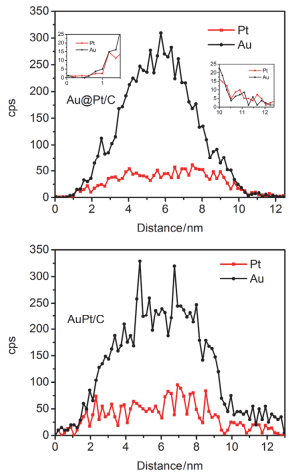
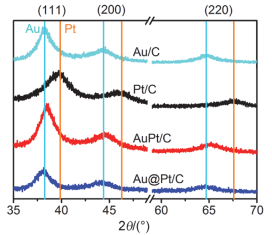
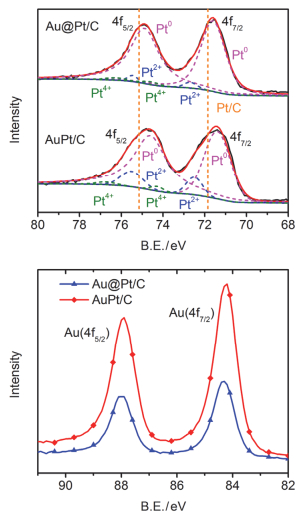
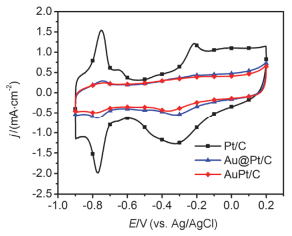
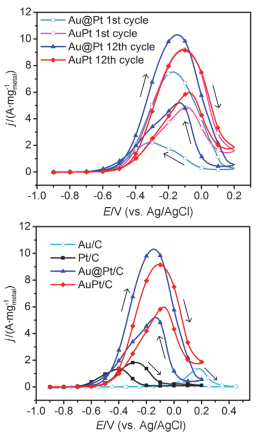
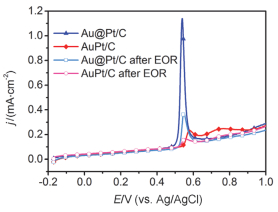
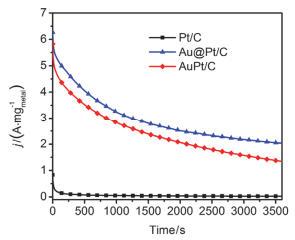
Ethanol oxidation reaction (EOR) is a common anode process for direct ethanol fuel cell (DEFC) and ethanol reforming electrolyzer. Au@Pt and AuPt alloy are widely used bimetallic catalysts, yet no comparative study has been reported of electrocatalysis of EOR on these two differently structured catalysts. The present work aims to synthesize and characterize carbon supported Au@Pt and AuPt with controlled composition and size, and compare their electrocatalytic activities and stabilities toward EOR in alkaline media. For the synthesis of Au@Pt/C, a 5-nm Au colloid was first obtained by adding excessive amount of sodium borohydride to a chloroauric acid precursor containing sodium citrate with a mixed ice-water bath. CO gas was bubbled into the Au colloidal solution at 60℃ under strong stirring to reduce a desired amount of potassium tetrachloroplatinate(Ⅱ) to terminate Pt quasi-monolayer shell on Au nanoparticle core. A sonicated carbon black (Vulcan XC-72) aqueous slurry was then dropwise added to the above Au@Pt colloid, and the mixture was kept stirring for 48 h to ensure the exhaustive loading of Au@Pt nanoparticles onto the carbon support. For the synthesis of AuPt/C with the same Au:Pt molar ratio and metal loading as that for Au@Pt/C, coreduction of the Au(Ⅲ) and Pt(Ⅱ) species was attained by using sodium borohydride as the reducing agent with the rest procedures being same as the above mentioned. X-ray diffractometry (XRD) revealed that the diffraction peaks for Au@Pt/C were virtually same as those for Au/C, consistent with a Pt quasi-monolayer, while the diffraction peaks for AuPt/C located in between those for Au/C and Pt/C. X-ray photoelectron spectroscopy (XPS) results were consitent with the different structures of the two catalysts, and the Pt core level shift suggested an upshift of Pt d-band center for both bimetallic catalysts. Cyclic voltammetry and chronoamperometry revealed markedly increased EOR current on Au@Pt/C and AuPt/C, as compared to that of Pt/C and Au/C. CO-stripping voltammetry on Au@Pt/C and AuPt/C indicated that surface reconstruction occurred by potential cycling, resulting in a decrease of exposed Pt sites but not the electrocatalytic activities. 1H NMR analysis confirmed the C2 pathway is predominant. Nevertheless, Au@Pt/C outperformed AuPt/C and Pt/C with a lower onset oxidation potential and a higher peak current for EOR, as well as a slightly higher selectivity toward C1 pathway. Although the synergetic effect of Au-Pt bimetallic interface for EOR is not well understood, the enhanced adsorption of ethanol, OH, acetyl and CO on Pt sites may be accountable for the observed results.
Li, H. H.; Zhao, S.; Gong, M.; Cui, C. H.; He, D.; Liang, H. W.; Liang, W.; Yu, S. H. Angew. Chem., Int. Ed. 2013, 52, 7472. doi: 10.1002/anie.201302090
[2]
Coutanceau, C.; Baranton, S. WIREs Energy Environ. 2016, 5, 388. doi: 10.1002/wene.193
[3]
de Lucas-Consuegra, A.; Ana, R.; Calcerrada, A. B.; Linares, J. J.; Horwat, D. J. Power Sources 2016, 321, 248. doi: 10.1016/j.jpowsour.2016.05.004
[4]
Chen, H. M.; Xing, Z. L.; Zhu, S. Q.; Zhang, L. L.; Chang, Q. W.; Huang, J. L.; Cai, W. B.; Kang, N.; Zhong, C. J.; Shao, M. H. J. Power Sources 2016, 321, 264. doi: 10.1016/j.jpowsour.2016.04.072
[5]
Lamy, C.; Lima, A.; LeRhun, V.; Delime, F.; Coutanceau, C.; Léger, J. M. J. Power Sources 2002, 105, 283 doi: 10.1016/S0378-7753(01)00954-5
Adzic, R. R.; Zhang, J.; Sasaki, K.; Vukmirovic, M. B.; Shao, M.; Wang, J. X.; Nilekar, A. U.; Mavrikakis, M.; Valerio, J. A.; Uribe, F. Top. Catal. 2007, 46, 249. doi: 10.1007/s11244-007-9003-x
[12]
Mulvaney, S. P.; Keating, C. D. Anal. Chem. 2000, 72, 145. doi: 10.1021/a10000155
[13]
Enache, D. I.; Edwards, J. K.; Landon, P.; Solsona-Espriu, B.; Carley, A. F.; Herzing, A. A.; Watanabe, M.; Kiely, C. J.; Knight, D. W.; Hutchings, G. J. Science 2006, 311, 362. doi: 10.1126/science.1120560
[14]
Song, H. M.; Anjum, D. H.; Sougrat, R.; Hedhili, M. N.; Khashab, N. M. J. Mater. Chem. 2012, 22, 25003. doi: 10.1039/c2jm35281h
Bayer, D.; Berenger, S.; Joos, M.; Cremers, C.; Tübke, J. Int. J. Hydrogen Energ. 2010, 35, 12660. doi: 10.1016/j.ijhydene.2010.07.102
[40]
Teng, X. In Materials and Processes for Energy: Communicating Current Research and Technological Developments, Atrazhev, V. V. ; Burlatsky, S. F., Formatex Research Center, Durham, 2013, pp. 473~484.
[41]
Wang, S. Y.; Kristian, N.; Jiang, S. P.; Wang, X. Nanotechnology 2008, 20, 025605.
[42]
Wang, H.; Jiang, K.; Chen, Q.; Xie, Z.; Cai, W. B. Chem. Commun. 2016, 52, 374. doi: 10.1039/C5CC06551H
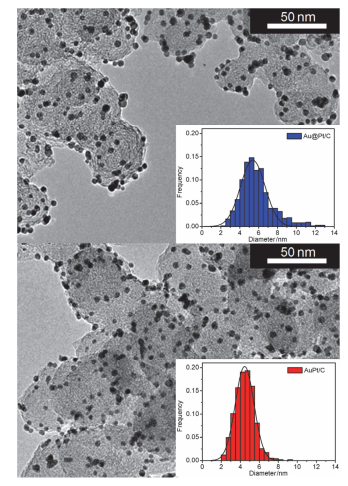
图 1
Au@Pt/C (上)和AuPt/C (下)催化剂TEM图及其粒径分布
Figure 1
TEM image and size distribution histogram of Au@Pt/C (upper panel) and AuPt/C (lower panel) catalysts

 下载:
下载:

 下载:
下载:









