 图1
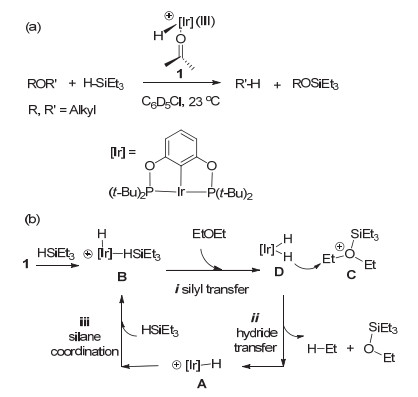
(a) Ir(Ⅲ)催化剂1催化的还原反应和(b)可能的反应机理(Path Ⅰ)
Figure1.
(a) Ir(Ⅲ) pincer complex 1 catalyzed reduction reaction and (b) the proposed mechanism (Path Ⅰ) for Figure 1a
图1
(a) Ir(Ⅲ)催化剂1催化的还原反应和(b)可能的反应机理(Path Ⅰ)
Figure1.
(a) Ir(Ⅲ) pincer complex 1 catalyzed reduction reaction and (b) the proposed mechanism (Path Ⅰ) for Figure 1a
引用本文:
张琪, 刘奥, 于海珠, 傅尧. Ir(Ⅲ)螯合物催化醚的硅氢化反应中负氢来源的机理研究[J]. 化学学报,
2018, 76(2): 113-120.
doi:
10.6023/A17070328

Citation: Zhang Qi, Liu Ao, Yu Haizhu, Fu Yao. Hydride Source in Ethers Hydrosilylation Reaction Catalyzed by Brookhart's Ir(Ⅲ) Pincer Complex[J]. Acta Chimica Sinica, 2018, 76(2): 113-120. doi: 10.6023/A17070328
Citation: Zhang Qi, Liu Ao, Yu Haizhu, Fu Yao. Hydride Source in Ethers Hydrosilylation Reaction Catalyzed by Brookhart's Ir(Ⅲ) Pincer Complex[J]. Acta Chimica Sinica, 2018, 76(2): 113-120. doi: 10.6023/A17070328
Ir(Ⅲ)螯合物催化醚的硅氢化反应中负氢来源的机理研究
摘要:
采用DFT方法对Ir(Ⅲ)螯合物催化乙醚硅氢化生成乙烷和乙基硅醚的反应展开理论研究.反应中[H-Ir-H],[H-Ir-Si],[Ir(HSiEt3)]和[Et3Si-Ir-(H)3]化合物均为可能的负氢来源.理论研究表明[H-Ir-H]化合物是最优势的负氢来源.通过扭曲-相互作用能分析,发现其他三种可能的负氢来源不优势的原因在于HSiEt3或SiEt3基团对铱中心的络合.更为重要的,我们发现[H-Ir-H]化合物中适中的Ir-H键解离能,小位阻以及SiEt3对醚的络合而产生的促进作用共同使得[H-Ir-H]化合物上的负氢转移相对优势.
English
Hydride Source in Ethers Hydrosilylation Reaction Catalyzed by Brookhart's Ir(Ⅲ) Pincer Complex
Abstract:
The hydrosilylative reduction with silane is a popular defunctionalization strategy to convert biomass into chemicals and energies because of the mild reaction conditions. Among these, the reduction of C-O bond is particularly important because of its application in sugar biomass reduction. The (C6F5)3 B/silane catalytic system has been frequently used in the reduction of C-O bonds in the past years. However, Brookhart et al. reported alkyl ethers reduction by using Ir(Ⅲ) pincer catalyst and reductant HSiEt3. This work provides a novel hydrosilylation catalyst for C-O reduction and an effective method for sugar biomass deoxygenation. According to the previous mechanistic proposals on similar Ir catalysed hydrosilylation reactions, the iridium dihydride complex, iridium silyl hydride complex, silane adduct iridium complex and iridium silyl trihydride complex might possibly act as the hydride source. We carried out the theoretical study on Brookhart's Ir(Ⅲ) Pincer Complex/HSiEt3 catalyzed hydrosilylation reaction of EtOEt yielding ethane and EtOSiEt3. The density functional theory (DFT) calculations in our study indicate that the iridium dihydride complex is the best hydride source. Our calculation result is consistent well with experimental observations in Brookhart's experiment. For example, the phenomenon that adding iridium dihydride complex into the reaction system increases the reaction rate is understandable because the complex is involved in the rate-determining step. From the Distortion/Interaction analysis, we found that hydride transfer steps on the other three possible hydride sources are disfavoured by the HSiEt3/-SiEt3 group (derived from HSiEt3) bonded with Ir center. The iridium silyl hydride complex is unfavourable because the Ir-H bond is strengthened and the pincer ligand is distorted. For the silane adduct iridium complex, the coordination of HSiEt3 destabilizes iridium complex intermediate for entropy increases and trans effect, and destabilizes the related transition state by damaging its pincer ligand. Further, the corresponding hydride transfer transition state from iridium silyl trihydride is highly unstable and Si-H bond always reform automatically. What's more important, the moderate bond dissociation energy of Ir-hydride, small steric hindrance and the promotion effect of SiEt3 group coordination with ether all facilitate the hydride transfer on the iridium dihydride complex.
-
Key words:
- hydrosilylation
- / iridium
- / hydride
- / pincer
- / DFT
-
1 引言
随着化石资源的减少, 通过生物质资源去官能化获得化学品和能源的途径逐渐引起广泛关注[1].在这一背景下, 硅烷参与的硅氢化还原反应, 由于其温和的反应条件而成为一种重要的去官能化策略[2].通过硅氢化还原方法能够实现卤代物中的C—X键[3], 醇和醚中的C—O键[4], 醛或酮中的羰基[5], 酯类和羧酸的酯基和羧基[6], 二氧化碳[7], 以及亚胺和烯烃[8]的还原.其中, C—O键的硅氢化还原由于能够应用于糖类等生物质的脱氧去官能化, 而引起广泛的研究兴趣[9].硅烷还原剂与吸电子基团取代的硼烷(如(C6F5)3B)催化剂组成催化体系, 能够高效地实现C—O键的还原.例如, Gevorgyan和Yamamoto课题组在(C6F5)3B催化剂/HSiEt3还原剂条件下实现一级, 二级和三级醚的硅氢化还原, 得到硅醚[4a, 4b].采用同样的催化剂, Piers课题组以PhMe2SiH作为还原剂实现了烯醇硅醚的还原[4c]. McRae课题组采用正丁基硅烷n-BuSiH3和二乙基硅烷Et2SiH2还原一级、二级和三级醇生成相应的烷烃[4d]. Njardarson课题组采用(C6F5)3B/HSiEt3体系实现了不饱和环醚的选择性C—O键还原[4e].此外, Brookhart课题组采用螯合的Ir(Ⅲ)催化剂1/HSiEt3还原剂体系同样实现了烷基醚的还原(图 1a).该工作为C—O键的硅氢化还原以及糖类生物质的脱氧提供了一种新型的催化体系[10a].
图1
(a) Ir(Ⅲ)催化剂1催化的还原反应和(b)可能的反应机理(Path Ⅰ)
Figure1.
(a) Ir(Ⅲ) pincer complex 1 catalyzed reduction reaction and (b) the proposed mechanism (Path Ⅰ) for Figure 1a
在该工作中, Brookhart小组通过实验方法对乙醚的硅氢化还原反应进行了机理研究(图 1b)[10].认为反应的机理可能是原始催化剂1先与硅烷反应生成活性催化剂B, 在B的催化下引发反应循环: (ⅰ)硅基迁移:乙醚对B中的硅烷亲核进攻, 硅基转移到乙醚上得到阳离子C和双负氢铱中间体D; (ⅱ)负氢迁移: D作为负氢来源进攻C, 将H-转移到乙基上, 生成还原产物乙烷, 乙基硅醚和单负氢铱中间体A; (ⅲ)硅烷活化:单负氢铱中间体A络合一分子硅烷重新生成活性催化剂B, 进而完成一个催化循环.该循环中的负氢来源是双负氢铱中间体D (Path Ⅰ, 图 2). D可与体系中的硅烷络合, 得到硅烷络合中间体E通过Si—H键的氧化加成和随后的H—H键还原消除, 生成中性的硅基单负氢铱中间体G. Brookhart等认为G也可以作为负氢来源(Path Ⅱ)参与负氢迁移步骤, 但是其效率低于双负氢铱中间体D (Path Ⅰ).近期, Oestreich课题组在研究同样催化体系(1/HSiEt3)下羰基化合物还原反应时发现, 单独的中间体C和D并不发生反应, 继续加入硅烷才能得到产物[11].由此Oestreich等认为负氢迁移步骤中的负氢来源可能是硅烷络合的双负氢铱中间体E (Path Ⅲ, 图 2), 或者Si—H键氧化加成所得到的硅基三负氢Ir(Ⅴ)中间体F (Path Ⅳ, 图 2).
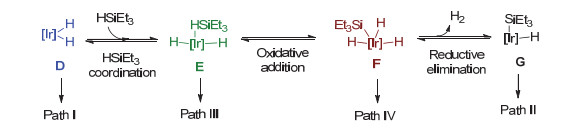 图2
四种可能的负氢来源
Figure2.
Four possible hydride source
图2
四种可能的负氢来源
Figure2.
Four possible hydride source
基于上述讨论, 在硅氢化还原乙醚的体系中, 四种不同类型的负氢铱化合物(即D, E, F, G)均可能作为负氢来源.为了阐明优势的负氢来源, 我们通过理论计算对1/HSiEt3催化乙醚的硅氢化还原反应展开详细的机理研究[12].计算结果表明双负氢铱中间体D是最优势的负氢来源, Path Ⅰ是生成产物乙烷和EtOSiEt3的优势路径.由于金属中心周围的大位阻, E, F和G化合物上的负氢转移均不优势.相比之下Path Ⅰ优势的原因在于中等强度的Ir—H键, 较低的空间位阻以及SiEt3基团与醚络合而产生的促进作用.基于此, 我们认为具有强反式效应以及低空间位阻的配体能够促进硅氢化反应的进行.希望我们的研究结果能够帮助理解硅氢化反应的机理, 进而促进新的还原策略的发现.
2 结果
2.1 双负氢中间体D(4)作为负氢来源(Path Ⅰ)
如图 3所示, 原始催化剂1中的丙酮迅速被还原而生成单负氢化合物2[13].这里我们采用2作为能量零点. 2络合一分子硅烷HSiEt3得到活性催化剂3.硅烷与Ir(Ⅲ)中心的络合使得σ(Si—H)键的部分电子进入Ir(Ⅲ)的空的d轨道, 同时也形成了Ir(dπ)到硅烷反键轨道(σ*)的反馈键[14, 15].上述电子流动过程使得中间体3中Si—H键被削弱(硅烷中的Si—H键长度从自由硅烷HSiEt3中的1.494 增长到1.568 , Si—H键的键级从0.594减小到0.584).基于此, 对于随后的硅基迁移步骤, 我们首先考察Si—H键自动断裂的机理.如图 3所示, Si—H键直接断裂生成双负氢铱中间体4(对应图 2中的D)和硅烷阳离子Et3Si+, 该过程需要吸收43.5 kcal/mol的能量.因此高能量需求排除了Si—H键自动离解的可能性.基于上述结果, 我们另外考察了EtOEt参与的协同机理.在该机理中, Si—H键的断裂和EtOEt的亲核进攻同时进行, 得到双负氢铱中间体4和氧鎓离子5[16~18].相应的硅基转移协同过渡态TS1的吉布斯自由能为27.7 kcal/mol, 硅基转移步骤吸收能量15.3 kcal/mol (2→4+5).基于此, 协同的硅基迁移机理较为优势.
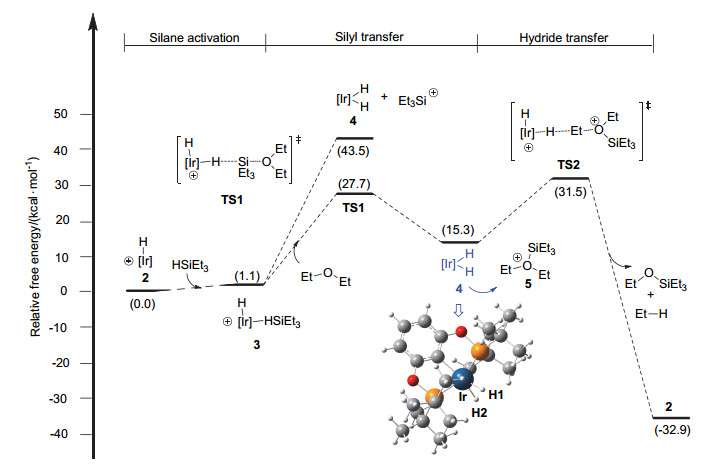 图3
Path Ⅰ的能量曲线
Figure3.
The energy profiles of Path Ⅰ
图3
Path Ⅰ的能量曲线
Figure3.
The energy profiles of Path Ⅰ
随后, 双负氢铱中间体4作为负氢来源转移其中一个负氢到阳离子5中的Et基团上, 得到脱氧产物乙烷和EtOSiEt3, 并重新生成单负氢铱催化剂2.该过程经历负氢转移过渡态TS2, 其中Et—H键的形成和Ir—H, Et—O键的裂解同时发生.该步骤能垒为31.5 kcal/mol (2→TS2), 负氢迁移步骤释放能量32.9 kcal/mol.由图 3可知, Path Ⅰ机理中负氢迁移是决速步骤, 总能垒为31.5 kcal/mol.
2.2 硅基负氢铱中间体G(9)作为负氢来源(Path Ⅱ)
如引言所述, 双负氢铱中间体4可能先后经历硅烷络合, Si—H氧化加成及H—H还原消除而生成另一个可能的负氢来源9(对应图 2中的络合物G, Path Ⅱ)[10a].
如图 4所示, 一分子硅烷HSiEt3与4络合的过程使得4中的H—Ir—H角从59.6°增加到175.4°, 得到两个负氢位于反式位的中间体6.络合过程吸收能量12.8 kcal/mol.随后, 中间体6经历Si—H键对Ir(Ⅲ)中心氧化加成过渡态TS3, 得到硅基三负氢Ir(Ⅴ)中间体7.该过程能垒为32.7 kcal/mol (2→TS3).接着, 中间体7经历H—H键还原消除过渡态TS4得到H2络合的Ir(Ⅲ)中间体8, 该步骤的能垒为35.2 kcal/mol (2→TS4). 8解离一分子H2生成中性的硅基负氢中间体9. 9随后作为负氢来源进攻阳离子5中的Et基团.相应的负氢迁移过渡态为TS5, 吉布斯自由能为54.8 kcal/mol.最后, 得到产物乙烷, EtOSiEt3和Ir(Ⅲ)化合物10, 路径Path Ⅱ释放能量4.2 kcal/mol.
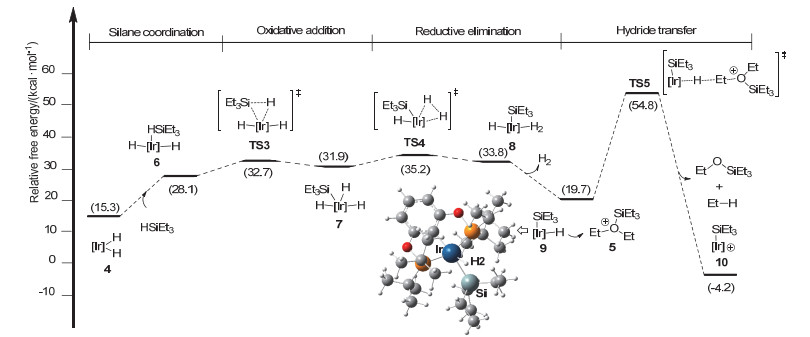 图4
Path Ⅱ的能量曲线
Figure4.
The energy profiles of Path Ⅱ
图4
Path Ⅱ的能量曲线
Figure4.
The energy profiles of Path Ⅱ
基于此, 对于硅基负氢铱中间9作为负氢来源的机理Path Ⅱ, 总能垒为54.8 kcal/mol (2→TS5).
2.3 硅烷络合双负氢铱中间体E(6)作为负氢来源(Path Ⅲ)
在Path Ⅲ中, 硅烷与中间体4络合后生成的硅烷络合双负氢铱中间体6(对应图 2中的化合物E)作为负氢来源参与负氢迁移步骤.
如图 5所示, 中间体6经历负氢迁移过渡态TS6将负氢转移到阳离子5的Et基团上, 该过程的能垒为48.0 kcal/mol (2→TS6).在TS6中Ir—H(1)和Ir…H(2)…C(Et)键处于反位, HSiEt3处于配体上叔丁基所组成的凹槽中(图 4).考虑到Ir…H(2)…C(Et)结构与配体上的叔丁基之间存在位阻, 我们尝试将Ir…H(2)…C(Et)置于凹槽.然而对应过渡态TS7的能量反而升高(54.3 kcal/mol).通过对比过渡态TS6和TS7(图 6), 我们发现正在断裂的Ir—H(2)键分别与H(1)-和C(Ph)-处于对位.由于H-的反位效应大于C(Ph)-, 因此TS6中的Ir—H(2)键较容易断裂[19]. TS6中Ir—H(2)键的键长大于TS7中Ir—H(2)键的键长(1.775 vs. 1.716 )也支持这一推论.基于此, 硅烷络合双负氢铱中间体6作为负氢来源的机理Path Ⅲ, 总能垒为48.0 kcal/mol (2→TS6).
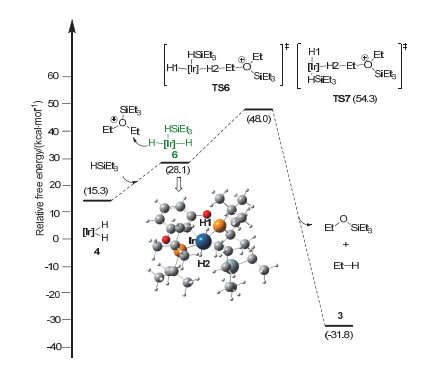 图5
Path Ⅲ的能量曲线
Figure5.
The energy profiles of Path Ⅲ
图5
Path Ⅲ的能量曲线
Figure5.
The energy profiles of Path Ⅲ
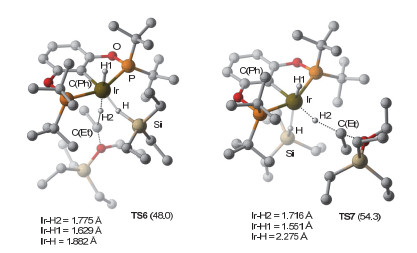 图6
TS6和TS7的结构
Figure6.
TS6 and TS7 in Path Ⅲ
图6
TS6和TS7的结构
Figure6.
TS6 and TS7 in Path Ⅲ
2.4 硅基三负氢Ir(Ⅴ)中间体F(7)作为负氢来源(Path Ⅳ)
在Path Ⅳ中, 硅基三负氢Ir(Ⅴ)中间体7(对应图 2的F)作为负氢来源参与负氢迁移步骤.如图 7所示, 中间体7中与铱中心成键的三个H-均可以进攻阳离子5中的乙基.我们首先考虑位阻最小的H-参与的负氢迁移过渡态TS8.在过渡态优化过程中, 我们发现Ir(Ⅴ)中心上相邻的SiEt3和—H会重新还原消除形成Si—H键, 从而形成硅烷络合的过渡态TS6(Path Ⅲ).同样的现象也存在于另外两种H-参与的负氢迁移过渡态中.这一现象的原因可能是Ir(Ⅴ)中心极度缺电子且空间位阻过于拥挤, 因此容易还原为更为稳定的Ir(Ⅲ)中心.另外, 由于Si—H键氧化加成产物7的能量已经高于Path Ⅰ中过渡态TS2的能量(31.9 kcal/mol vs. 31.5 kcal/mol), 我们排除Ir(Ⅴ)络合物7作为负氢来源的可能性(Path Ⅳ).
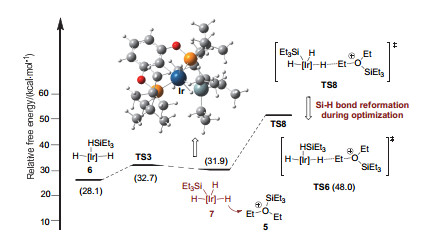 图7
Path Ⅳ的能量曲线
Figure7.
The energy profiles of Path Ⅳ
图7
Path Ⅳ的能量曲线
Figure7.
The energy profiles of Path Ⅳ
3 讨论
3.1 四种负氢迁移路径的对比
图 8所示是四种负氢来源参与的亲核进攻步骤总能量曲线.双负氢铱中间体4, 硅基负氢铱中间体9以及硅烷络合双负氢铱中间体6分别是Path Ⅰ, Ⅱ和Ⅲ中的负氢来源.三种机理的总能垒分别为31.5 kcal/mol (2→TS2)[20], 54.8 kcal/mol (2→TS5)及48.0 kcal/mol (2→TS6).硅基三负氢铱中间体7作为负氢来源的Path Ⅳ中, 过渡态TS8无法稳定存在, 而在优化后自动形成TS6.因此Path Ⅰ是最为优势的路径, 即双负氢铱中间体4是体系中的负氢来源.上述结论为Brookhart课题组的机理猜测提供了理论依据, 同时也解释了相关实验现象:
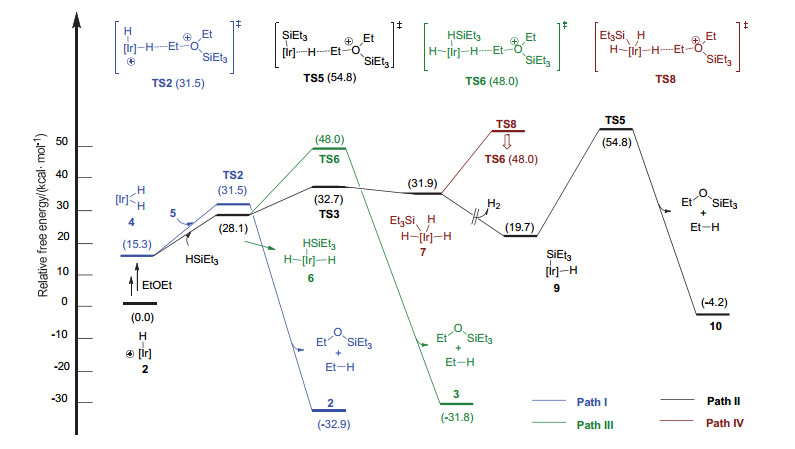 图8
四种负氢迁移过程的能量曲线
Figure8.
The energy profiles of four hydride transfer paths
图8
四种负氢迁移过程的能量曲线
Figure8.
The energy profiles of four hydride transfer paths
(1) 实验中向体系加入双负氢中间体4能够提高反应速率.基于计算结果, 这是因为负氢来源4参与优势机理Path Ⅰ的决速步骤(负氢迁移).
(2) 实验中发现4与过量硅烷(220当量)在室温条件下反应生成氢气和硅基负氢中间体9.计算结果显示, 这一变化过程的能垒为17.4 kcal/mol (4→TS3), 因此能够在室温下进行.且由于这一过程仅轻微吸收能量4.4 kcal/mol, 因此实验中需要大大过量的硅烷.
(3) 实验中增加硅烷的量会使反应速率下降.根据我们的计算结果, 需要硅烷参与的路径Path Ⅱ, Ⅲ和Ⅳ都是Path Ⅰ的竞争路径, 过量的硅烷使得参加Path Ⅰ的中间体4减少, 因此速率会下降.
(4) 实验发现在-40 ℃的低温下4和5反应, 不生成产物, 而是得到反应物乙醚和催化剂3.根据计算结果, 4和5生成产物的能垒和得到反应物的能垒分别为16.2 kcal/mol (4+5→TS2)和12.4 (4+5→TS1).反应在低温下受动力学控制, 进而经历逆反应得到反应物.
3.2 四种负氢迁移路径的讨论
在本节中, 我们对路径Path Ⅰ~Ⅲ的负氢迁移步骤进行扭曲/相互作用分析, 以阐明化合物4 (Path 1)为最优势负氢来源的原因[21~23].扭曲能Edist指的是负氢来源和阳离子5从自由状态到过渡态中的结构TS-Ir-X和TS-5-X (X=2, 5和6)的能量变化.相互作用能Eint指的是分离的TS-Ir-X和TS-5-X合并为过渡态TSX的能量变化.活化能∆E≠是Edist和Eint两部分的加和.
对比Path Ⅰ和Path Ⅱ的负氢转移步骤(图 9, 表 1), 可知两者的相互作用能Eint相近(-12.4 vs. -15.2 kcal/mol), 然而Path Ⅱ的扭曲能Edist较高(15.7 vs. 33.3 kcal/mol), 这是导致Path Ⅱ不优势的原因.扭曲能主要体现在以下两个方面: (1) Ir—H(2)键的解离.根据计算结果, 4和9的Ir—H(2)键解离能分别为65.4和89.7 kcal/mol[24].中间体9中存在Ir—Si—H(2)的3c-2e键, 这说明H-不仅与Ir中心成键, 同时与SiEt3基团成键.因此中间9中的Ir—H键更难以断裂[13]. (2)螯合配体的扭曲.如图 10所示, 4和9均为具有三角双锥结构的五配位化合物, 4中的C(Ph), H(1), H(2)原子, 9中的C(Ph), H(1), Si原子分别构成三角面.在负氢迁移步骤中, 两者均变为四角锥结构.在TS2中, H(1)位于轴向位置, 而在TS5中, 大体积的SiEt3基团占据轴向位置. SiEt3基团和螯合配体之间存在明显的空间位阻, 9中的二面角C(Ph)—Ir—P—O由2.9°增大到18.7° (9→TS5).而4→TS2过程中二面角则无明显变化.综上所述, Path Ⅱ中SiEt3基团一方面使得负氢来源9中Ir—H(2)键难以断裂, 另一方面使得TS5中的螯合配体平面扭曲, 从而导致Path Ⅱ不优势, 9则为不优势的负氢来源.
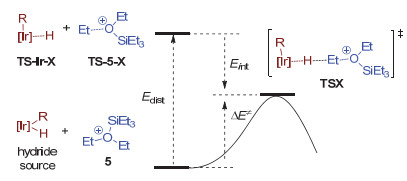 图9
扭曲/相互作用分析
Figure9.
Edist,
Eint and ∆E≠ in Distortion/Interaction Analysis
图9
扭曲/相互作用分析
Figure9.
Edist,
Eint and ∆E≠ in Distortion/Interaction Analysis
 表 1
扭曲/相互作用分析
Table 1.
Distortion/Interaction Analysis (kcal/mol)
表 1
扭曲/相互作用分析
Table 1.
Distortion/Interaction Analysis (kcal/mol)
Hydride transfer step Edist Eint ∆E≠ Ed[Ir—H(2)]a 4+5→TS2 (Path Ⅰ) 15.7 -12.4 3.3 65.4 9+5→TS5 (Path Ⅱ) 33.3 -15.2 18.2 89.7 6+5→TS6 (Path Ⅲ) 20.2 -15.2 5.2 52.6 a The Ir-H(2) bond dissociation energy (kcal/mol). 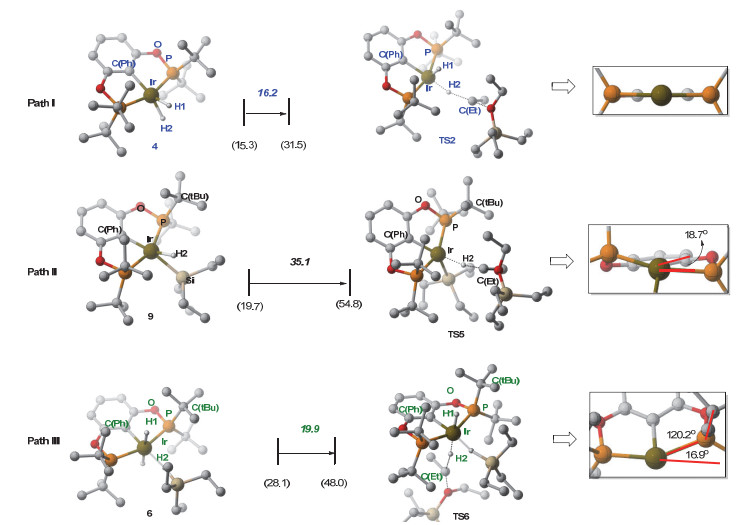 图10
Path Ⅰ~Ⅲ中的负氢迁移步骤
Figure10.
Hydride transfer steps of Path Ⅰ~Ⅲ
图10
Path Ⅰ~Ⅲ中的负氢迁移步骤
Figure10.
Hydride transfer steps of Path Ⅰ~Ⅲ
对比Path Ⅲ和Path Ⅰ路径负氢迁移步骤, 我们发现: (1)负氢来源6的自由能比4高出12.8 kcal/mol.这一方面归因于在4到6的转化过程的熵增; 另一方面, HSiEt3对中间体4的配位使得两个负氢[H(1)和H(2)]位于反式位置, H(1)和H(2)之间相互的反式效应使得6不稳定.
(2) Path Ⅲ中负氢迁移的基元步骤能垒比Path Ⅰ高出4.8 kcal/mol (19.9 kcal/mol vs 16.2 kcal/mol).这一差别由Path Ⅲ和Path Ⅰ的扭曲能Edist差异(20.2 vs. 15.7 kcal/mol)造成.这里的Edist同样体现在Ir—H(2)键解离和螯合配体扭曲两个方面.然而, 由于6的Ir—H(2)键解离能低于4 (52.6 vs. 65.4 kcal/mol), 因此螯合配体扭曲占主导作用.在TS6中, HSiEt3对Ir中心的络合使得Ir…H(2)…C(Et)和螯合配体之间存在较大的位阻.如图 10所示, 为了适应Ir…H(2)…C(Et)结构, 螯合配体的叔丁基向上移动. Ir—P—C(tBu)角从120.2o增大到TS6中的126.1°, 螯合平面的二面角C(Ph)—Ir—P—O从6.2°增大到TS6中的16.9°[23].螯合配体的平面结构破坏导致TS6不稳定.
根据上述比较和讨论, 可知Path Ⅰ相对优势的原因体现在以下三个方面:第一, 中等程度的Ir—H(2)键解离能(65.4 kcal/mol); 第二, 小位阻, Ir…H(2)…C(Et)处于配体的凹槽中, 螯合配体平面在负氢迁移步骤中扭曲程度小; 第三, SiEt3基团对乙醚的络合有利于Et—O键的断裂, 部分电荷由乙醚到SiEt3的转移减弱了C—O键, 因此中间体5中C—O键的解离能明显低于自由的EtOEt (57.3 vs. 66.6 kcal/mol).由于第三个现象也出现在其他的路径中, 因此前两个方面占据主导.基于上述结论, 我们提出一些可能有助于提高催化效率的策略.首先, 高反式效应和低空间位阻的配体有益于Ir—H键解离, 从而有利于负氢迁移步骤.另外, 硅烷中硅原子上的吸电取代基会降低Et2OSiR3+中Et—O键的电子密度, 从而削弱Et—O键, 利于负氢迁移步骤发生.然而, 吸电子取代基不利于甲硅烷基转移步骤中的Si—H键断裂(Path Ⅰ的第一步, 图 2), 因此该策略应谨慎使用.
4 结论
我们通过理论计算对Brookhart课题组报道的铱催化醚的硅氢化还原反应进行了详细的机理研究, 阐明体系中优势的负氢来源.考察了双负氢铱化合物, 硅基单负氢铱化合物, 硅烷络合双负氢铱化合物和硅基三负氢铱化合物分别作为负氢来源的机理(Path Ⅰ~Ⅳ). Path Ⅰ~Ⅲ的总能垒分别为31.5, 54.8和48.0 kcal/mol. Path Ⅳ中的过渡态高度不稳定, Si—H键自动重新成键.因此, Path Ⅰ是最优势的机理, 双负氢铱化合物是最优势的负氢来源.对比Path Ⅰ~Ⅲ路径中的负氢迁移步骤显示, Path Ⅱ中的硅基SiEt3阻碍了硅基负氢化合物中Ir—H(2)键的解离, 同时使得负氢迁移步骤中螯合配体产生扭曲. Path Ⅲ中硅烷HSiEt3对金属中心的络合产生了熵增效应, 反式效应, 同时破坏螯合配体平面, 从而导致该路径不优势.相比之下, Path Ⅰ的优势归因于Ir—H的中等强度键解离能, 小位阻以及SiEt3基团对乙醚的络合.
5 计算方法
本文中所有的计算均采用Gaussian09程序完成[25].运用M06[26~28]/GEN2的密度泛函理论方法对文中提到的所有结构进行非限制性几何构型全优化(GEN2: 6-31g* for C, H, O, N, P, Si and SDD for Ir).在同一计算方法下对这些优化结构进行频率分析, 以获得焓值的热力学矫正, 转动和振动对熵效应的贡献, 以及确定势能面上驻点的性质(中间体的虚频个数为0, 过渡态有唯一虚频).对于文中的每一个过渡态, 采用内禀反应坐标(IRC) [29, 30]对其进行反应路径分析, 以确定其连接正确的反应物和产物.在优化构型的基础上, 采用M06/GEN1方法和SMD[31]模型计算各化合物的液相单点能量(GEN1: 6-311++G*/ for C, H, O, N, P and Si; SDD for Ir), (氯苯与Brookhart实验中使用的5-氘代氯苯极性相同, 因此采用氯苯作为溶剂).对Ir添加极化函数[ζ(f)=0.938][32, 33].本文中所有的能量均为气相自由能校正和液相单点能之和.
-
-
[1]
(a) Kerr, R. A. ; Service, R. F. Science 2005, 309, 101. (b) Ragauskas, A. J. ; Williams, C. K. ; Davison, B. H. ; Britovsek, G. ; Cairney, J. ; Eckert, C. A. ; Fredrick, W. J. J. ; Hallett, J. P. ; Leak, D. J. ; Liotta, C. L. ; Mielenz, J. R. ; Murphy, R. ; Templer, R. ; Tschaplinski, T. Science 2006, 311, 484.
-
[2]
(a) Douvris, C. ; Ozerov, O. V. Science 2008, 321, 1188. (b) Dioumaev, V. K. ; Bullock, R. M. Nature 2003, 424, 530. (c) Fernandez-Alvarez, F. J. ; Aitani A. M. ; Oro, L. A. Catal. Sci. Technol. 2014, 4, 611. (d) Sui, Y. -Z. ; Zhang, X. -C. ; Wu, J. -W. ; Li, S. ; Zhou, J. -N. ; Li, M. ; Fang, W. ; Chan A. S. C. ; Wu, J. Chem. -Eur. J. 2012, 18, 7486. (e) Addis, D. ; Das, S. ; Junge K. ; Beller, M. Angew. Chem., Int. Ed. 2011, 50, 6004. (f) Malacea, R. ; Poli, R. ; Manoury, E. Coord. Chem. Rev. 2010, 254, 729. (g) Pan, Z. ; Liu, M. ; Zheng, C. ; Gao, D. ; Huang, W. Chin. J. Chem. 2017, 35, 1227. (h) Hu, X. ; Hu, F. ; Zhang, M. ; Liao, Y. ; Xu, X. ; Yuan, W. ; Zhang, X. Chin. J. Org. Chem. 2016, 36, 1895. (扈晓艳, 胡方芝, 张敏敏, 廖益均, 徐小英, 袁伟成, 张晓梅, 有机化学, 2016, 36, 1895. ) (i) Zaranek, M. ; Marciniec, B. ; Pawluć, P. Org. Chem. Front. 2016, 3, 1337. (j) Hu, X. ; Tian, C. ; Maxim, B. ; Nie, W. Acta Chim. Sinica 2015, 73, 1025. (胡茜, 田冲, Borzov Maxim, 聂万丽, 化学学报, 2015, 73, 1025.
-
[3]
(a) Yang, J. ; Brookhart, M. Adv. Synth. Catal. 2009, 351, 175. (b) Caputo, C. B. ; Stephan, D. W. Organometallics 2012, 31, 27. (c) Scott, V. J. ; Çelenligil-Çetin, R. ; Ozerov, O. V. J. Am. Chem. Soc. 2005, 127, 2852.
-
[4]
(a) Gevorgyan, V. ; Liu, J. -X. ; Rubin, M. ; Benson S. ; Yamamoto, Y. Tetrahedron Lett. 1999, 40, 8919. (b) Gevorgyan, V. ; Rubin, M. ; Benson, S. ; Liu, J. -X. ; Yamamoto, Y. J. Org. Chem. 2000, 65, 6179. (c) Blackwell, J. M. ; Morrison, D. J. ; Piers, W. E. Tetrahedron 2002, 58, 8247. (d) Nimmagadda, R. D. ; McRae, C. Tetrahedron Lett. 2006, 47, 5755. (e) Mack, D. J. ; Guo, B. ; Njardarson, J. T. Chem. Commun. 2012, 48, 7844. (f) Chojnowski, J. ; Rubinsztajn, S. ; Cella, J. A. ; Fortuniak, W. ; Cypryk, M. ; Kurjata, J. ; Kaźmierski, K. Organometallics 2005, 24, 6077.
-
[5]
(a) Park, S. ; Brookhart, M. Organometallics 2010, 29, 6057. (b) Parks, D. J. ; Piers, W. E. J. Am. Chem. Soc. 1996, 118, 9440. (c) Skjel, M. K. ; Houghton, A. Y. ; Kirby, A. E. ; Harrison, D. J. ; McDonald, R. ; Rosenberg, L. Org. Lett. 2010, 12, 376. (d) Chandrasekhar, S. ; Reddy, C. R. ; Babu, B. N. J. Org. Chem. 2002, 67, 9080.
-
[6]
(a) Bézier, D. ; Park, S. ; Brookhart, M. Org. Lett. 2013, 15, 496. (b) Gevorgyan, V. ; Rubin, M. ; Liu, J. -X. ; Yamamoto, Y. J. Org. Chem. 2001, 66, 1672. (c) Cheng, C. ; Brookhart, M. Angew. Chem. , Int. Ed. 2012, 51, 9422.
-
[7]
(a) Park, S. ; Bézier, D. ; Brookhart, M. J. Am. Chem. Soc. 2012, 134, 11404. (b) Berkefeld, A. ; Piers, W. E. ; Parvez, M. J. Am. Chem. Soc. 2010, 132, 10660.
-
[8]
(a) Blackwell, J. M. ; Sonmor, E. R. ; Scoccitti, T. ; Piers, W. E. Org. Lett. 2000, 2, 3921. (b) Rubin, M. ; Schwier, T. ; Gevorgyan, V. J. Org. Chem. 2002, 67, 1936. (c) Ding, S. ; Song, L. -J. ; Chung, L. W. ; Zhang, X. ; Sun, J. ; Wu, Y. -D. J. Am. Chem. Soc. 2013, 135, 13835.
-
[9]
(a) McLaughlin, M. P. ; Adduci, L. L. ; Becker, J. J. ; Gagné, M. R. J. Am. Chem. Soc. 2013, 135, 1225. (b) Robert, T. ; Oestreich, M. Angew. Chem. , Int. Ed. 2013, 52, 5216. (c) Adduci, L. L. ; McLaughlin, M. P. ; Bender, T. A. ; Becker, J. J. ; Gagné, M. R. Angew. Chem. , Int. Ed. 2014, 53, 1646.
-
[10]
(a) Yang, J. ; White, P. S. ; Brookhart, M. J. Am. Chem. Soc. 2008, 130, 17509. (b) Yang, J. ; Brookhart, M. J. Am. Chem. Soc. 2007, 129, 12656. (c) Park, S. ; Brookhart, M. Chem. Commun. 2011, 47, 3643.
-
[11]
Mets nen, T. T.; Hrobárik, P.; Klare, H. F. T.; Kaupp, M.; Oestreich, M. J. Am. Chem. Soc. 2014, 136, 6912. doi: 10.1021/ja503254f
-
[12]
Herein we examine the first cleavage of EtOEt. The cleavage of the silyl ether could also be achieved (the second cleavage) if lengthen the reaction time.
-
[13]
Yang, J.; White, P. S.; Schauer, C. K.; Brookhart, M. Angew. Chem., Int. Ed. 2008, 47, 4141. doi: 10.1002/(ISSN)1521-3773
-
[14]
Perutz, R. N.; Sabo-Etienne, S. Angew. Chem., Int. Ed. 2007, 46, 2578. doi: 10.1002/(ISSN)1521-3773
-
[15]
(a) Lin, Z. Chem. Soc. Rev. 2002, 31, 239. (b) Chung, L. W. ; Lee, H. G. ; Lin, Z. ; Wu, Y. -D. J. Org. Chem. 2006, 71, 6000. (c) Lee, T. Y. ; Dang, L. ; Zhou, Z. ; Yeung, C. H. ; Lin, Z. ; Lau, C. P. Eur. J. Inorg. Chem. 2010, 5675.
-
[16]
Rendler, S.; Oestreich, M. Angew. Chem., Int. Ed. 2008, 47, 5997. doi: 10.1002/anie.v47:32
-
[17]
(a) Wang, W. ; Gu, P. ; Wang, Y. ; Wei, H. Organometallics 2014, 33, 847. (b) Sakata, K. ; Fujimoto, H. J. Org. Chem. 2013, 78, 12505.
-
[18]
Cheng, Y.-H.; Zhao, X.; Song, K.-S.; Liu, L.; Guo, Q.-X. J. Org. Chem. 2002, 67, 6638. doi: 10.1021/jo020085h
-
[19]
Anderson, K. M.; Orpen, A. G. Chem. Commun. 2001, 24, 2682. http://www.ncbi.nlm.nih.gov/pubmed/12240289
-
[20]
The DFT method leads to an overestimation of entropy, resulting in an overestimation of the free energy. This phenomenon has also appeared in previous theoretical calculations. For example: (a) Ding, L. ; Ishida, N. ; Murakami, M. ; Morokuma, K. J. Am. Chem. Soc. 2014, 136, 169. (b) Sugiyama, A. ; Ohnishi, Y. -Y. ; Nakaoka, M. ; Nakao, Y. ; Sato, H. ; Sakaki, S. ; Nakao, Y. ; Hiyama, T. J. Am. Chem. Soc. 2008, 130, 12975. (c) Yu, Z. -X. ; Houk, K. N. J. Am. Chem. Soc. 2003, 125, 13825. (d) Hermans, J. ; Wang, L. J. Am. Chem. Soc. 1997, 119, 2707. (e) Tanaka, R. ; Yamashita, M. ; Chung, L. W. ; Morokuma, K. ; Nozaki, K. Organometallics 2011, 30, 6742. (f) Dub, P. A. ; Ikariya, T. J. Am. Chem. Soc. 2013, 135, 2604. (g) Strajbl, M. ; Sham, Y. Y. ; Villà, J. ; Chu, Z. -T. ; Warshel, A. J. Phys. Chem. B 2000, 104, 4578. (h) Hermans, J. ; Wang, L. J. Am. Chem. Soc. 1997, 119, 2707.
-
[21]
Gorelsky, S. I.; Lapointe, D.; Fagnou, K. J. Am. Chem. Soc. 2008, 130, 10848. doi: 10.1021/ja802533u
-
[22]
张琪, 于海珠, 石景, 物理化学学报, 2013, 29, 2321. doi: 10.3866/PKU.WHXB201310082Zhang, Q.; Yu, H.-Z.; Shi, J. Acta Phys.-Chim. Sin. 2013, 29, 2321. doi: 10.3866/PKU.WHXB201310082
-
[23]
Zhang, Q.; Yu, H.-Z.; Fu, Y. Organometallics 2016, 35, 2473. doi: 10.1021/acs.organomet.6b00347
-
[24]
Hydride is dissociated in the calculation of Ir-H(2) bond dissociation energy. See reference herein: Qi, X. -J. ; Liu, L. ; Fu, Y. ; Guo, Q. -X. Organometallics 2006, 25, 5879.
-
[25]
Gaussian 09, revision D. 01; Gaussian, Inc., Wallingford, CT, 2013.
-
[26]
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215. doi: 10.1007/s00214-007-0310-x
-
[27]
(a) Kundu, S. ; Choi, J. ; Wang, D. Y. ; Choliy, Y. ; Emge, T. J. ; Krogh-Jespersen, K. ; Goldman, A. S. J. Am. Chem. Soc. 2013, 135, 5127. (b) Haibach, M. C. ; Guan, C. ; Wang, D. Y. ; Li, B. ; Lease, N. ; Steffens, A. M. ; Krogh-Jespersen, K. ; Goldman, A. S. J. Am. Chem. Soc. 2013, 135, 15062. (c) Tian, Y. ; Fu, Y. ; Zhang, Q. ; Yu, H. -Z. ; Shi, J. Acta Chim. Sinica 2014, 72, 935. (田燕, 傅尧, 张琪, 于海珠, 石景, 化学学报, 2014, 72, 935. ) (d) Liu, D. -J. ; Yu, H. -Z. ; Fu, Y. Acta Chim. Sinica 2013, 71, 1385. (刘丁嘉, 于海珠, 傅尧, 化学学报, 2013, 71, 1385.
-
[28]
Zhang, Q.; Yu, H.-Z.; Fu, Y. Org. Chem. Front. 2014, 1, 614. doi: 10.1039/C4QO00036F
-
[29]
Fukui, K. J. Phys. Chem. 1970, 74, 4161. doi: 10.1021/j100717a029
-
[30]
Fukui, K. Acc. Chem. Res. 1981, 14, 363. doi: 10.1021/ar00072a001
-
[31]
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378. doi: 10.1021/jp810292n
-
[32]
Ehlers, A. W.; Bohme, M.; Dapprich, S.; Gobbi, A.; Hollwarth, A.; Jonas, V.; Kohler, K. F.; Stegmann, R.; Veldkamp, A.; Frenking, G. Chem. Phys. Lett. 1993, 208, 111. doi: 10.1016/0009-2614(93)80086-5
-
[33]
Hollwarth, A.; Bohme, M.; Dapprich, S.; Ehlers, A. W.; Gobbi, A.; Jonas, V.; Kohler, K. F.; Stegmann, R.; Veldkamp, A.; Frenking, G. Chem. Phys. Lett. 1993, 208, 237. doi: 10.1016/0009-2614(93)89068-S
-
[1]
-

图 1 (a) Ir(Ⅲ)催化剂1催化的还原反应和(b)可能的反应机理(Path Ⅰ)
Figure 1 (a) Ir(Ⅲ) pincer complex 1 catalyzed reduction reaction and (b) the proposed mechanism (Path Ⅰ) for Figure 1a
表 1 扭曲/相互作用分析
Table 1. Distortion/Interaction Analysis (kcal/mol)
Hydride transfer step Edist Eint ∆E≠ Ed[Ir—H(2)]a 4+5→TS2 (Path Ⅰ) 15.7 -12.4 3.3 65.4 9+5→TS5 (Path Ⅱ) 33.3 -15.2 18.2 89.7 6+5→TS6 (Path Ⅲ) 20.2 -15.2 5.2 52.6 a The Ir-H(2) bond dissociation energy (kcal/mol).  下载: 导出CSV
下载: 导出CSV
-











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 3
- 文章访问数: 4092
- HTML全文浏览量: 556
 下载:
下载:
